

Abstract
Background
Bone Morphogenetic Protein (BMP) signaling has multiple roles in the development and function of the blood vessels. In humans, mutations in BMP type 2 receptors (BMPR2), a key component of BMP signaling, have been identified in the majority of patients with familial pulmonary arterial hypertension (PAH). However, only a small subset of individuals with BMPR2 mutation develops PAH, suggesting that additional modifiers of BMPR2 function play an important role in the onset and progression of PAH.
Methods
We utilized a combination of studies in zebrafish embryos and genetically engineered mice lacking endothelial expression of Vegfr3 to determine the interaction between VEGFR3 and BMPR2. Additional in vitro studies were performed using human endothelial cells, including primary endothelial cells from subjects with PAH.
Results
Attenuation of Vegfr3 in zebrafish embryos abrogated Bmp2b-induced ectopic angiogenesis. Endothelial cells (ECs) with disrupted VEGFR3 expression failed to respond to exogenous BMP stimulation. Mechanistically, VEGFR3 is physically associated with BMPR2 and facilitates ligand-induced endocytosis of BMPR2 to promote phosphorylation of SMADs and transcription of ID genes. Conditional, endothelial specific deletion of Vegfr3 in mice resulted in impaired BMP signaling responses, and significantly worsened hypoxia-induced pulmonary hypertension (PH). Consistent with this data, we found significant decrease in VEGFR3 expression in pulmonary arterial endothelial cells (PAECs) from human PAH subjects, and reconstitution of VEGFR3 expression in PAH PAECs restored BMP signaling responses.
Keywords: Pulmonary arterial hypertension, BMPR2, VEGFR3, endothelial cells
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a rare disease with the hallmark of vascular remodeling of the pulmonary arterioles, associated with formation of plexiform and concentric vascular lesions comprised of hyperproliferating endothelial and vascular smooth muscle cells.1 A key signaling paradigm that has been extensively demonstrated to underlie both the clinical context and experimental models is that mediated by Bone Morphogenetic Protein Receptor Type 2 (BMPR2). The majority of patients with familial PAH, as well as many with idiopathic PAH, have BMPR2 mutations.2 Familial PAH is considered to be a disease transmitted in an autosomal dominant fashion, and while heterozygous BMPR2 mutations can cause disease, their incomplete penetrance is highlighted by the fact that the majority of individuals with BMPR2 mutations (~80%) do not progress to develop PAH.2–6 These findings suggest the presence of genetic or environmental modifiers that are critical determinants of disease pathogenesis. Unfortunately, these potential modifiers of BMPR2 mediated signaling remain incompletely defined in the context of the pulmonary vasculature. In addition, the regulators of BMPR2 signaling in specific cell types involved in PAH, including endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and others, remain to be fully investigated.
Vascular endothelial growth factor receptors (VEGFRs) are receptor tyrosine kinases (RTKs) which bind to and transduce signaling from the VEGF ligand family. To date, three VEGFRs have been identified; VEGFR1 (FLT1), VEGFR2 (KDR), and VEGFR3 (FLT4).7 During developmental and pathological angiogenesis, VEGFR2, which binds to VEGF-A, appears to function as the primary receptor for VEGF signaling.8 VEGFR3, which shares structural similarity to VEGFR2, preferentially binds to other members of VEGF ligands (VEGF-C and VEGF-D) and promotes angiogenesis as well as lymphangiogenesis.9–11 In the context of PAH, components of VEGF signaling have been implicated as potential biomarkers in human disease,12 and in experimental models, a VEGFR2 antagonist (SU-5416) in conjunction with chronic hypoxia is used routinely to induce pulmonary hypertension (PH) in rodents.13 While VEGR3 is known to be highly expressed in the lungs (www.gtexportal.org/home/gene/FLT4),14, 15 its specific role in the pulmonary arterial bed has not been fully defined.
Our current studies describe convergence between the BMP and VEGF signaling pathways through a novel interaction between VEGFR3 and BMPR2. We found VEGFR3 to be highly expressed in the pulmonary arterial endothelial cells (PAECs). Moreover, the VEGFR3-BMPR2 interaction was found to regulate multiple aspects of BMP signaling, including ligand induced internalization of BMPR2 and phosphorylation of BMP target proteins. Genetic deletion of Vegfr3 in ECs led to impaired BMP signaling in the lungs, associated with exacerbation of hypoxia induced PH in mice. Finally, VEGFR3 expression was found to be significantly reduced in human PAH PAECs, and restoring its expression resulted in rescue of impaired BMP signaling in these patient cells. Overall, these findings demonstrate a key role for VEGFR3 as a molecule that can potentiate BMP signaling in the pulmonary arterial endothelium, and define its role as a potential disease modifier in the multiple hit model of PAH.
METHODS
(Additional detailed methods are included in the online supplement)
Human samples
The study was approved by the Cleveland Clinic and the Yale University School of Medicine Institutional Review Boards, and written informed consent was obtained from all participating individuals. Human lung tissues were obtained from either unused, explanted normal donor lungs or explanted lungs from confirmed subjects with PAH undergoing lung transplantation at the Cleveland Clinic. Additional PAH and normal donor lung samples were obtained from the NDRI (National Disease Research Interchange).
Mice
The protocols were approved by the Institutional Animal Care and Use Committee of Yale University. C57/Bl6 mice (Jackson Laboratories) were maintained in the Animal Research Center at Yale University. The Vegfr3:YFP, Vegfr3fl/fl, and Cdh5(Pac):CreERT2 mice have been previously described.16–18 For Cdh5(Pac):CreERT2 mice, six week old mice were injected with 2 mg tamoxifen (Sigma, T5648; 10 mg/mL dissolved in 10% ethanol with corn oil) intraperitoneally daily for five days to induce Cre activity. Vegfr3fl/fl mice lacking the Cre driver injected with the same tamoxifen regimen were used as controls. The mice were used five weeks after the completion of tamoxifen injection for the indicated experiments. For establishing PH, mice were exposed to continuous chronic hypoxia (10 % FiO2) for 3 weeks.
Hemodynamic and morphometric analysis
All procedures were performed under anesthesia with inhaled isoflurane. For evaluation of PH severity, 6 to 8 mice were used per group. Pressure catheter (Millar Instruments) was inserted in the right jugular vein to measure right ventricular systolic pressure (RVSP) as described previously.19 The right ventricle was perfused with normal saline and the right lung was ligated, isolated and snap-frozen in liquid nitrogen for protein and RNA analysis. The left lung was perfused and fixed with 4 % paraformaldehyde overnight for immunohistochemistry. To assess right ventricular hypertrophy, hearts were dissected and the Fulton index was calculated as the weight ratio of the RV and LV + septum.
To assess pulmonary artery muscularization, lung tissues were fixed in 4% paraformaldehyde, washed in 1× PBS and embedded in OCT (Sakura Tissue-Tek) and frozen, then tissues were sectioned on a Leica CM1950 cryotome at 10 μm and incubated with anti-von Willebrand Factor (vWF) antibody (A0082, Dako), anti-smooth muscle actin (SMA) antibody conjugated to Cy3 (C6198, Sigma) overnight at 4 °C. The sections were then incubated with Alexa Fluor 488 goat anti-rabbit IgG antibody (A11008, Life technologies). DAPI (Thermo Scientific) was used to stain the nuclei. Sections were imaged using Nikon eclipse Ti confocal microscope.
Pulmonary artery muscularization was quantified by calculating the ratio of the number of muscularized peripheral pulmonary arteries to the number of total peripheral pulmonary vessels (with diameters less than 75 μm) in 10 random fields per lung (with each field at ×200 magnification). To assess the thickness of the medial smooth muscle cell layer of the pulmonary arterioles, lung tissue sections were immunostained for vWF, SMA and DAPI, then immunofluorescence images were obtained on a Nikon eclipse Ti confocal microscope. The thickness of the smooth muscle layer in the pulmonary arterioles were measured using ImageJ software in 45 ~ 50 random fields per lung (with each field at ×600 magnification). Quantifications were carried out by investigators blinded to the experimental condition. H&E staining was performed using standard methods and visualized with a light microscope (Nikon 80i). The number of obliterated vessels was counted in ten random fields per lung per mouse using H&E stained sections, and each dot in the figure represents the average value per high power field per mouse.
Cell culture and reagents
Human PAECs were isolated from normal and PAH explanted donor lungs, as described previously.20 In brief, human pulmonary arteries were dissected from the distal small arterioles in lungs, and PAECs were harvested from the isolated pulmonary arterial tree and were grown in EBM-2 containing EGM-2 supplement (Lonza). HUVECs were purchased from Yale Vascular Biology and Therapeutics (VBT) core and cultured in EBM-2 containing EGM-2 supplement. All of the cells were passaged at 80–90% confluence, and were used between passages 3 and 8. BMP6 (R&D Systems) or VEGF-C (R&D Systems) were used for stimulation using indicated doses and time points. For phosphorylation studies, HUVECs or PAECs were serum starved prior to stimulation. For blocking dynamin-mediated endocytosis, cells were treated with 80mM Dynasore (Sigma) for 30 min prior to the lysis as previously described.21
Zebrafish embryos
The research protocols were approved by the Institutional Animal Care and Use Committee of Yale University. Morpholinos (MOs) with previously confirmed efficacy were used (www.zfin.org). MOs were injected at single cell stage embryos. Injected embryos were incubated at 42°C for 30 min to induce the expression of hsp:bmp2b transgene. The degree of ectopic angiogenesis was assessed by confocal analyses as previously described.22
Statistics
All experiments were performed in triplicate (unless otherwise specified) in at least three independent experiments, and data shown are means ± S.E.M. When only 2 groups were compared, statistical differences were assessed with unpaired 2-tailed Student’s t test. To examine the statistical differences in the expression level of VEGFR3 between control and PAH patient samples which contains a number of outliers, Wilcoxon rank sum test was utilized to calculate the significance. Otherwise statistical significance was determined using 1- or 2-way ANOVA followed by Bonferroni multiple comparison test. A P value less than 0.05 was considered statistically significant.
RESULTS
VEGFR3 is required for endothelial BMP response
We have previously reported that ectopic activation of Bmp2b (zebrafish ortholog of mammalian BMP2) during zebrafish vascular development induced excessive angiogenic sprouts from the caudal vein plexus.22 We postulated that additional modulators may potentiate angiogenic responses in ECs upon BMP activation. To identify such factors, we performed a multimodal screen using in situ hybridization, bioinformatics, and MO based knockdown analyses in zebrafish. Among the candidates tested (Suppl. Table 1), we found that knockdown of Vegfr3 by MO injection substantially reduced the number of ectopic venous sprouts emanating from the caudal vein in response to Bmp2b induction (Fig. 1A–D). Interestingly, inhibition of its cognate ligand vegfc, while being statistically significant, only had minor effects on Bmp2b-induced angiogenesis compared to inhibition of vegfr3 (Fig. 1A–D), suggesting that Vegfr3, in a ligand independent manner, is essential for Bmp signaling response. Consistent with this idea, knockdown of VEGFR3 in human umbilical vein endothelial cells (HUVECs) significantly reduced phosphorylation of SMADs and induction of ID genes upon BMP ligand stimulation (Fig. 1E, F, and Suppl. Fig. 1).
Fig. 1. VEGFR3 is essential for BMP signaling and closely associated with BMPR2.
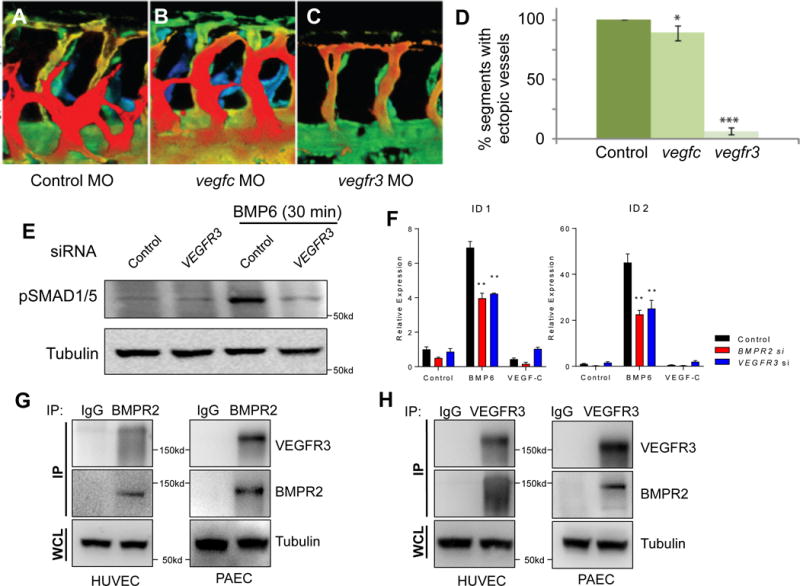
(A–C) Depth-encoded confocal images taken from the trunk region of 32 hours post fertilization (hpf) control (A), vegfc (B), or vegfr3 (C) morpholino (MO)-injected Tg(hsp70:bmp2b)fr13;Tg(kdrl:eGFP)s843 embryos upon heat shock. Overexpression of Bmp2b led to ectopic angiogenesis which was abrogated by inactivation of vegfr3. (D) Quantification on the number of somites containing ectopic angiogenic sprouts in control, vegfc, or vegfr3 MO-injected Tg(hsp70:bmp2b)fr13;Tg(kdrl:eGFP)s843 embryos upon heat shock. (E) BMP6-induced phosphorylation of SMADs is abrogated in VEGFR3 siRNA-treated HUVECs. (F) ID1 and ID2 are decreased in VEGFR3 siRNA-treated HUVECs. (G) Co-immunoprecipitation using BMPR2 antibody can pull down VEGFR3 in HUVECs and PAECs. (H) Co-immunoprecipitation using VEGFR3 antibody can pull down BMPR2 in HUVECs and PAECs. IP: immunoprecipitant; WCL: whole cell lysate. * P < 0.05, ** P < 0.01, *** P < 0.001.
VEGFR3 is physically associated with BMPR2 and regulates its intracellular trafficking
We next examined whether BMPR2 may be directly associated with VEGFR3. Immunoprecipitation experiments using antibodies against either BMPR2 or VEGFR3 were able to robustly pull down VEGFR3 or BMPR2, respectively, in both HUVECs and PAECs (Fig. 1G–1H). We found in HUVECs that BMP6 stimulation led to robust internalization of BMPR2 (Fig. 2A). Pretreatment of the cells with Dynasore, which inhibits endocytosis, led to marked inhibition of BMP6 induced BMPR2 internalization (Fig. 2A) and SMAD1/5 phosphorylation (Fig. 2B), further supporting the importance of BMPR2 internalization in downstream signaling. Interestingly, we found that BMP6 stimulation led to robust internalization of VEGFR3 in both HUVECs and PAECs, in addition to BMPR2 (Fig. 2C and Suppl. Fig. 2), suggesting that VEGFR3 is not only closely associated with BMPR2, but internalizes together with BMPR2 upon ligand activation and may act as a co-receptor. To further elucidate the functional relevance of VEGFR3 in impacting BMPR2 internalization, we examined the internalization of BMPR2 upon BMP6 treatment in the absence of VEGFR3. The amount of internalized BMPR2 was significantly reduced in VEGFR3 siRNA-treated HUVECs (Fig. 2D). Collectively, our data suggest that VEGFR3 is likely to function as a co-receptor for BMPR2 and is essential for BMP induced BMPR2 endocytosis.
Fig. 2. VEGFR3 and BMPR2 co-internalize upon ligand stimulation.
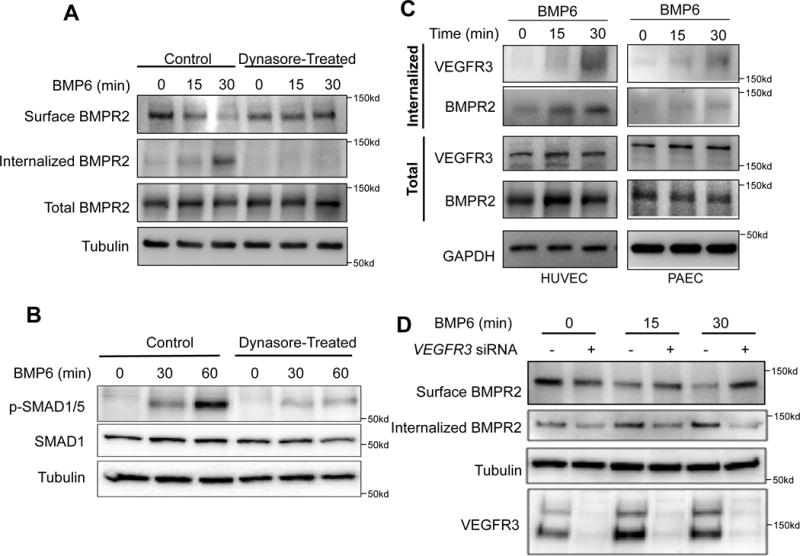
(A) Biotinylation assay showing internalization of surface BMPR2 in response to BMP6 stimulation in a time dependent manner. (B) Inhibition of Dynamin-dependent endocytosis decreases phosphorylation of SMAD 1/5. (C) Stimulation with BMP6 induces internalization of VEGFR3 and BMPR2 in both HUVECs and PAECs in a time dependent manner. (D) Lack of VEGFR3 in HUVECs abrogated ligand-dependent internalization of BMPR2 in HUVECs.
Deletion of endothelial Vegfr3 results in exacerbation of hypoxia induced PH and impairs BMP signaling
Given that mutations in BMPR2 in humans predispose to development of PAH, we sought to investigate the functional relevance of VEGFR3 in the pulmonary vasculature. Using the recently described Vegfr3:YFP reporter mouse,16 which expresses the yellow fluorescent protein (YFP) in cells that express Vegfr3, we injected a bismuth contrast agent into the right ventricle to identify the pulmonary arterial blood vessels. We found robust YFP expression in the ECs surrounding the bismuth contrast filled vascular lumen (Fig. 3A), confirming expression of Vegfr3 in the pulmonary arterial endothelial cells.
Fig. 3. Lack of VEGFR3 in endothelial cells attenuates BMP signaling in mice.
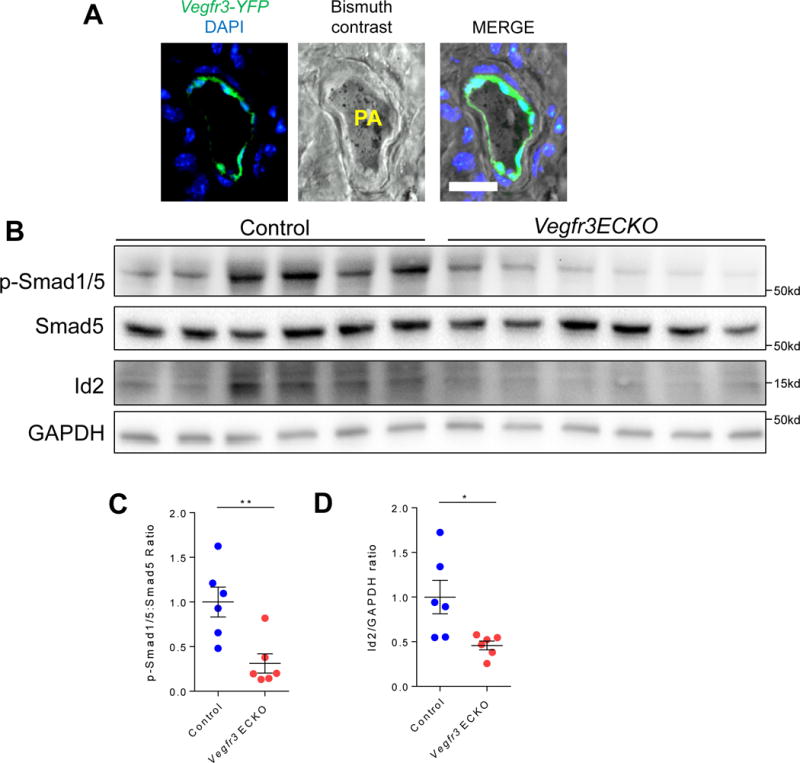
(A) Localization of YFP expression driven by the Vegfr3 promoter to the endothelial layer of bismuth contrast filled pulmonary arteriole. Scale bar = 50 μm. (B) Reduced pSMAD 1/5 levels in lung homogenates from Vegfr3 ECKO mice compared to lung homogenates from control littermates. (C) pSMAD 1/5 to total SMAD 5 ratio in control littermates and Vegfr3 ECKO mice. (D) Quantification of Id2 expression to GAPDH ratio in control and Vegf3 ECKO mice. *P < 0.05, **P < 0.01.
To further elucidate the role of VEGFR3 in regulation of BMPR2 signaling in pulmonary vasculature, we next investigated the phenotype of mice with inducible, conditional deletion of Vegfr3 in the endothelium using the Cdh5(Pac):CreERT2 mice (henceforth Vegfr3 ECKO) (Suppl. Fig. 3).18 Cre recombinase was induced with tamoxifen in adulthood to avoid any potential deleterious effects of endothelial Vegfr3 deletion in development. Consistent with our data obtained from the cell culture studies, we found a significant reduction in SMAD1/5 phosphorylation in the lung homogenates of Vegfr3 ECKO mice compared to controls (Fig. 3B and C). In addition, expression of Id2, a known downstream target of pSMAD, was markedly decreased in Vegfr3 ECKO lungs (Fig. 3B and D).
To determine whether lack of endothelial VEGFR3, at least in part via impaired BMP signaling, may contribute to the pathogenesis of PH, we evaluated the Vegfr3 ECKO mice either at baseline or after three weeks of chronic hypoxia to promote development of PH.23 While there was no significant difference in the RVSP in the Vegfr3 ECKO mice compared to control littermates at the baseline (28.4 mmHg +/− 3.15 for control vs. 28.7 mmHg +/− 4.1 for Vegfr3 ECKO, P=n.s.), there was a significantly increase in RVSP and RV hypertrophy in the Vegfr3 ECKO mice after chronic hypoxia exposure (Fig. 4A and B), indicating that Vegfr3 ECKO mice are more susceptible to developing PH in response to chronic hypoxia. Moreover, morphometric analyses of Vegfr3 ECKO mice after chronic hypoxia demonstrated significantly increased number of muscularized pulmonary arterioles and increased medial layer thickness compared to control mice (Fig. 4C–E). Lastly, we found that Vegfr3 ECKO mice had significantly increased number of obliterated pulmonary arterioles, which is rarely, if ever, seen in mice subjected to chronic hypoxia (Fig 4F and G). Taken together, the exacerbated PH phenotype in Vegfr3 ECKO mice, in conjunction with impaired BMP signaling in these mice, support our hypothesis that VEGFR3 may be a key mediator of endothelial BMPR2 mediated signaling.
Fig. 4. Deletion of Vegfr3 in endothelial cells exacerbates pulmonary hypertension.
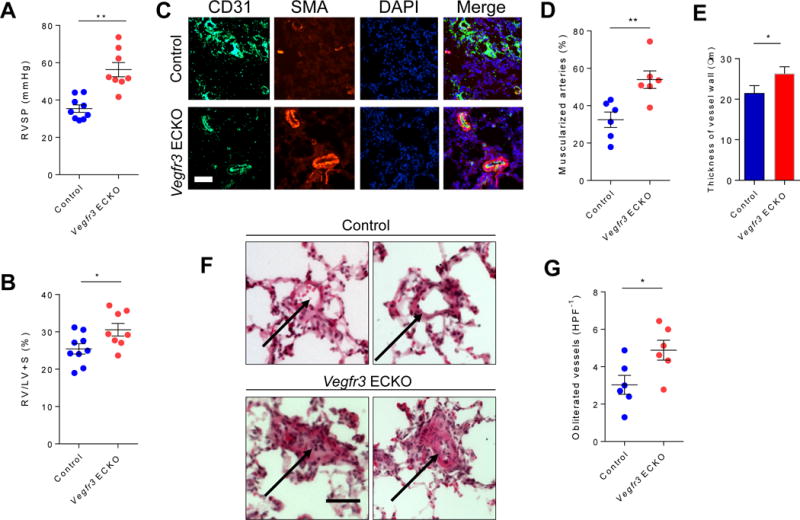
(A) Right ventricle systolic pressure (RVSP) measured in control and Vegfr3 ECKO mice. (B) Weight ratio of right ventricle to left ventricle plus septum calculated in control and Vegfr3 ECKO mice. (C) Immunofluorescent staining of lung sections from control and Vegfr3 ECKO mice for endothelial cells (CD31; green), vascular smooth muscle cells (SMA; red), and nuclei (DAPI; blue). Scale bar = 50 μm. (D) Percentage of muscularized arterioles within the lung tissue obtained from control and Vegfr3 ECKO mice. (E) Thickness of SMA positive medial layer in the muscularized arteries in control and Vegfr3 ECKO mice. (F) H&E stained lung section taken from control (top) and Vegfr3 ECKO mice (bottom). Arrows point to representative images of intact (control) and obliterated (Vegfr3 ECKO) lumens of the pulmonary arterioles. Scale bar = 50 μm. (G) Percentage of pulmonary arterioles with obliterated lumen in control and Vegfr3 ECKO mice. *P < 0.05, **P < 0.01. HPF−1: high power field.
Decreased VEGFR3 expression in human PAH PAECs
We next sought to further elucidate the role of VEGFR3 in the human PAH context. Similar to murine lung, VEGFR3 is highly expressed in pulmonary arterioles in human lung tissue (Fig. 5A). To further investigate the role of VEGFR3 in pathogenesis of PAH, we examined the VEGFR3 expression in PAH and control PAECs. Expression of VEGFR3 in the lung sections from a PAH subject was significantly decreased compared to control (Fig 5A). Moreover, both VEGFR3 mRNA and protein levels were substantially reduced in PAECs isolated from PAH subjects (Fig. 5B and C), despite the expected heterogeneity among the different patient samples. For instance, PAECs isolated from one PAH subject, CC011, displayed comparable level of VEGFR3 expression to control samples, while a number of PAECs derived from other PAH subjects demonstrated markedly reduced VEGFR3 expression (Fig. 5C). The presence or absence of BMPR2 mutations in PAECs did not appear to be a determining factor for VEGFR3 expression, as we found reduced VEGFR3 expression in both those PAECs with and without BMPR2 mutant alleles (Fig. 5C). Interestingly, we found that PAH PAECs with high VEGFR3 expression (CC011) responded robustly to BMP6 stimulation (Fig. 5D), whereas PAECs from a PAH subject without BMPR2 mutation, but with low VEGFR3 (CC016) demonstrated minimal BMP response (Fig. 5E), indicating that VEGFR3 may be an important mediator which may contribute to the BMP responsiveness in PAECs.
Fig. 5. Attenuation of VEGFR3 contributes to the pathogenesis of pulmonary arterial hypertension in humans.
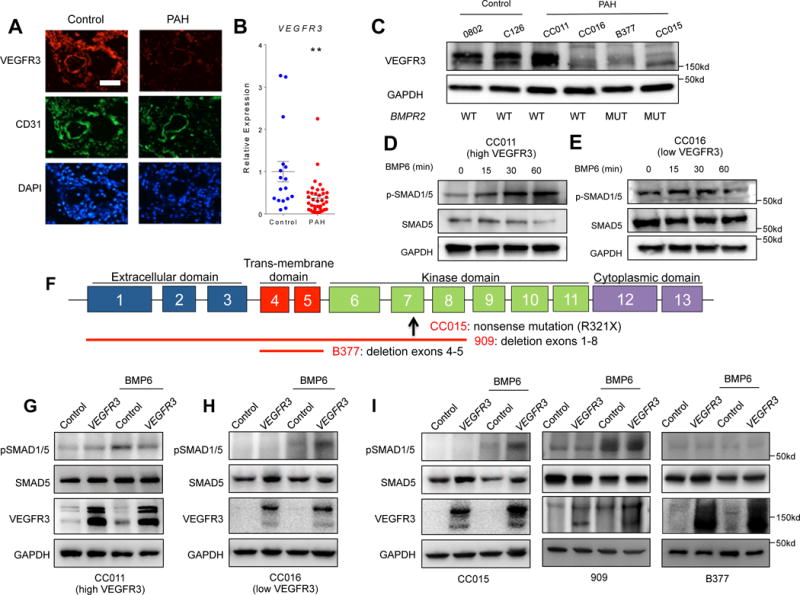
(A) Immunofluorescence staining of a normal lung section (left) and a PAH lung section (right) for endothelial cells (CD31; green), VEGFR3 (red), and nuclei (DAPI; blue) showing decreased VEGFR3 expression in PAH endothelium. Scale bar = 50 μm. (B) Relative expression level of VEGFR3 mRNA in normal and PAH PAECs. The significance was calculated using Wilcoxon rank-sum test. **P < 0.01 (C) Overall decreased but heterogeneous VEGFR3 protein expression among PAH patient-derived PAECs is not dependent on mutations in BMPR2. The mean of all the expression values in the control group were used for normalization. (D–E) Expression level of VEGFR3 influences the response of PAH PAECs to BMP6 (50 ng/mL) stimulation. (D) Upon BMP6 treatment, phosphorylation of SMAD1/5 was only increased in PAH PAECs with a relatively high level of VEGFR3. (E) Phosphorylation of SMAD 1/5 did not change in PAH PAECs with low VEGFR3 expression. (F) Schematic of BMPR2 genomic loci depicting the specific gene mutation/deletion in human PAECs tested. The up-arrow depicts location of the missense mutation, and the red lines depict the locations of gene deletion. (G–I) Effect of lentiviral-mediated over-expression of VEGFR3 on BMP-ligand mediated phosphorylation of SMAD 1/5. In PAH PAEC without BMPR2 mutations with high level of VEGFR3 expression, over-expression of VEGFR3 did not increased the phosphorylation of SMAD 1/5 upon BMP6 stimulation (G), whereas in PAH PAECs with low VEGFR3 expression, over-expression of VEGFR3 restored the phosphorylation of SMAD 1/5 upon BMP6 stimulation (H). (I) In PAH PAECs from subjects with BMPR2 mutations, efficacy of rescue with VEGFR3 overexpression in enhancing BMP6 response was variable depending on the underlying BMPR2 mutation.
In light of these findings, we set forth to further investigate the relationship between VEGFR3 and BMPR2 in PAH context by determining the effects of VEGFR3 overexpression in PAH PAECs. Using PAECs from 5 subjects with PAH, 3 of whom were heterozygotes for BMPR2 mutations (Fig. 5F), we tested the efficacy of lentiviral induced VEGFR3 overexpression on BMP signaling (Suppl. Fig. 4). First, we tested two PAEC lines derived from PAH subjects without BMPR2 mutations. While VEGFR3 overexpression did not enhance the response to BMP stimulation in PAECs with normal basal level of VEGFR3 expression (Fig. 5G), in PAH PAECs with low baseline VEGFR3 expression, lentivirus-mediated VEGFR3 overexpression restored robust induction of SMAD phosphorylation upon BMP stimulation (Fig. 5H). We next tested the effects of VEGFR3 overexpression on 3 PAEC lines derived from subjects carrying distinct BMPR2 mutations. VEGFR3 overexpression in cell line CC015, which carries a nonsense mutation in the kinase domain of BMPR2, induced robust increase in SMAD1/5 phosphorylation in response to BMP6 stimulation (Fig. 5I). We also tested two additional PAEC lines from BMPR2 mutation carriers. Line 909 had relatively higher expression of VEGFR3 at baseline, and despite having a BMPR2 allele with deletion of exons 1 through 8, responded robustly to BMP6, which was not augmented further with VEGFR3 overexpression. PAEC line B377 was derived from a BMPR2 mutant carrier with deletion of the trans-membrane domain. This cell line failed to respond to BMP6 stimulation both at baseline and with VEGFR3 overexpression (Fig. 5I).
DISCUSSION
Our data collectively present compelling evidence indicating that VEGFR3 functions within BMP signaling pathway to modulate internalization of BMPR2 upon ligand stimulation, which in turn influences the response to BMP signaling in ECs. We found that VEGFR3 is physically associated with BMPR2, and internalizes together upon BMP stimulation. The function of VEGFR3 in mediating BMP signaling appears to be independent of its own ligands, substantiating previous finding that VEGFR3 may play an important role outside the context of VEGF-C/D signaling.24
A number of membrane receptors can influence the signaling properties of the RTKs by facilitating ligand mediated endocytosis. For instance, Neuropilins, which are well characterized receptors for Semaphorins, can function as a co-receptor for VEGFR2 and VEGFR3 in ECs by providing an additional scaffolding,25, 26 and EphrinB2 can facilitate the internalization of VEGFR3 and VLDLR/ApoER2 by recruiting Clathrin-adaptors.18, 27 It has been speculated that these unusual facultative receptor-receptor interactions provide an additional regulatory step for the specific signaling cascade.28 In almost all cases, RTKs are the subjects of these unusual receptor-receptor interactions. Interestingly, such interactions have not been documented for the receptor serine/threonine kinases, which share similar biochemical properties with the RTKs. Our study provides strong evidence that the activity of BMPR2 is regulated by its interaction with VEGFR3, demonstrating that receptor serine/threonine kinases might be similarly regulated by the facultative receptor-receptor interactions.
Upon ligand stimulation, BMPR2 undergoes endocytosis, facilitated by clathrin adaptors such as Dab2 and FCHO.29, 30 The endocytosis of BMPR2 appears to be critical for the downstream activation, since its main effectors SMAD1/5 are associated with endosomes.31–33 Consistent with this idea, certain BMPR2 mutant isoforms isolated from human PAH patients fail to localize to the cell membrane and are therefore unable to undergo endocytosis in response to BMP stimulation.34 Since VEGFR3 facilitates endocytosis of BMPR2, we speculate that the interface of BMPR2 and VEGFR3 can be therapeutically targeted to augment BMP signaling activity in pathological conditions such as PAH. In support of this notion, we demonstrate rescue of BMP signaling in cultured PAECs by restoring VEGFR3 expression in a subset of PAH patient-derived PAECs. In addition to restoring BMP response in PAH PAECs without BMPR2 mutation, VEGFR3 overexpression was also able to augment BMP response in a PAH PAEC line with a nonsense mutation in the BMPR2 cytoplasmic kinase domain (Fig. 5I), but not in two additional PAH PAEC lines with multi-exon deletions spanning the BMPR2 trans-membrane domain. These variable responses in PAH PAECs to our cellular manipulations highlight the heterogeneous nature of the disease, while providing potentially important insights into the interaction between these receptors, as well as variability in PAEC response to BMP in the context of heterozygous BMPR2 mutations, as we pursue further studies to evaluate the multitude of factors that may contribute to PAH development in BMPR2 heterozygosity. Aspects of these studies would be best carried out in manners that apply precision medicine approaches, which is beginning to emerge in the PAH context.35, 36
Collectively, our current findings provide a more comprehensive understanding of how inputs of diverse signaling pathways can be integrated and coordinated. The demonstrated relevance of this interaction to the disease context of PAH provide a novel signaling paradigm that may at least in part explain the presence of incomplete penetrance of disease in those with BMPR2 mutations, and also identify strategies to modulate VEGFR3 activity, or its interaction with BMPR2, as a potential target for PAH therapy. Considering the importance of BMP signaling in diverse organs and tissues, it is difficult to selectively modulate BMPR2 activity without side-effects.37–41 Therefore, our results demonstrating the interaction between BMPR2 and VEGFR3 provide a theoretical background for a novel way to manipulate BMPR2 activity in pathological conditions.
Supplementary Material
CLINICAL PERSPECTIVE.
What’s new?
We provide evidence, using a combination of experimental animal models, human patient cells, and detailed signaling studies, that demonstrate the importance of a novel interaction between BMPR2 and VEGFR3 in regulating the robustness of endothelial BMP signaling response.
We demonstrate that this interaction is critical for promoting BMPR2 internalization in response to BMP stimulation.
We show that genetic deletion of endothelial Vegfr3 in mice results in exacerbation of chronic hypoxia induced pulmonary hypertension, and impaired BMP signaling.
What are the clinical implications?
Our findings are the first to demonstrate that VEGFR3 is a key modifier of BMPR2 function.
Given the heterogenous nature of pulmonary arterial hypertension (PAH), as well as the complexity surrounding the incomplete penetrance of PAH even in human subjects with BMPR2 mutations, these findings shed important insights into a potential disease modifier that warrants further investigation in larger human cohorts.
Acknowledgments
The authors would like to thank members of Chun and Jin laboratories, Rita Webber and Nicole Copeland for animal care, Saejeong Park and Suran Ryu for technical assistance, Sirkwoo Jin for statistical analyses, Anne Eichmann, Michael Simons, Martin Schwartz and William Sessa for helpful discussions, R. Adams for Cdh5(Pac)CreERT2 mice and K. Alitalo for Vegfr3fl/fl mice.
FUNDING SOURCES
This work was supported by grants from the NIH (HL114820 to S.W.J., HL119345 to H.J.C., and HL060917 and HL081064 to S.C.E.), Gwangju Institute of Science and Technology Research Institute (GRI) (to S.W.J.), Cell Logistics Research Center, National Research Foundation of Korea (NRF-2016R1A5A1007318) (to S.W.J.), the Ministry of Tarde, Industry and Energy, Korea (2016-10063396) (to S.W.J), and American Heart Association (Established Investigator Award to H.J.C.).
Footnotes
CONFLICT OF INTEREST DISCLOSURES
None.
References
- 1.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamid R, Cogan JD, Hedges LK, Austin E, Phillips JA, 3rd, Newman JH, Loyd JE. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum Mutat. 2009;30:649–654. doi: 10.1002/humu.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng Z, Haghighi F, Helleby L, Vanterpool K, Horn EM, Barst RJ, Hodge SE, Morse JH, Knowles JA. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am J Respir Crit Care Med. 2000;161:1055–1059. doi: 10.1164/ajrccm.161.3.9906051. [DOI] [PubMed] [Google Scholar]
- 4.International PPHC, Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 5.Lehmann K, Seemann P, Silan F, Goecke TO, Irgang S, Kjaer KW, Kjaergaard S, Mahoney MJ, Morlot S, Reissner C, Kerr B, Wilkie AO, Mundlos S. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet. 2007;81:388–396. doi: 10.1086/519697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res. 2014;115:189–202. doi: 10.1161/CIRCRESAHA.115.303404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrara N, Alitalo K. Clinical applications of angiogenic growth factors and their inhibitors. Nat Med. 1999;5:1359–1364. doi: 10.1038/70928. [DOI] [PubMed] [Google Scholar]
- 8.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 9.Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011;17:1371–1380. doi: 10.1038/nm.2545. [DOI] [PubMed] [Google Scholar]
- 10.Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012;484:110–114. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- 11.Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K. VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci USA. 2015;112:761–766. doi: 10.1073/pnas.1423278112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiede SL, Gall H, Dorr O, Troidl C, Liebetrau C, Voss S, Voswinckel R, Schermuly RT, Seeger W, Grimminger F, Zeiher AM, Dimmeler S, Mollmann H, Hamm CW, Ghofrani HA, Nef HM. New potential diagnostic biomarkers for pulmonary hypertension. Eur Respir J. 2015;46:1390–1396. doi: 10.1183/13993003.00187-2015. [DOI] [PubMed] [Google Scholar]
- 13.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–2754. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 14.Yao LC, Testini C, Tvorogov D, Anisimov A, Vargas SO, Baluk P, Pytowski B, Claesson-Welsh L, Alitalo K, McDonald DM. Pulmonary lymphangiectasia resulting from vascular endothelial growth factor-C overexpression during a critical period. Circ Res. 2014;114:806–822. doi: 10.1161/CIRCRESAHA.114.303119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pajusola K, Aprelikova O, Korhonen J, Kaipainen A, Pertovaara L, Alitalo R, Alitalo K. FLT4 receptor tyrosine kinase contains seven immunoglobulin-like loops and is expressed in multiple human tissues and cell lines. Cancer Res. 1992;52:5738–5743. [PubMed] [Google Scholar]
- 16.Calvo CF, Fontaine RH, Soueid J, Tammela T, Makinen T, Alfaro-Cervello C, Bonnaud F, Miguez A, Benhaim L, Xu Y, Barallobre MJ, Moutkine I, Lyytikka J, Tatlisumak T, Pytowski B, Zalc B, Richardson W, Kessaris N, Garcia-Verdugo JM, Alitalo K, Eichmann A, Thomas JL. Vascular endothelial growth factor receptor 3 directly regulates murine neurogenesis. Genes Dev. 2011;25:831–844. doi: 10.1101/gad.615311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haiko P, Makinen T, Keskitalo S, Taipale J, Karkkainen MJ, Baldwin ME, Stacker SA, Achen MG, Alitalo K. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol. 2008;28:4843–4850. doi: 10.1128/MCB.02214-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Luthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 19.Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, McLean DL, Park H, Comhair SA, Greif DM, Erzurum SC, Chun HJ. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med. 2013;19:74–82. doi: 10.1038/nm.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Comhair SA, Xu W, Mavrakis L, Aldred MA, Asosingh K, Erzurum SC. Human primary lung endothelial cells in culture. Am J Respir Cell Mol Biol. 2012;46:723–730. doi: 10.1165/rcmb.2011-0416TE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Wiley DM, Kim JD, Hao J, Hong CC, Bautch VL, Jin SW. Distinct signalling pathways regulate sprouting angiogenesis from the dorsal aorta and the axial vein. Nat Cell Biol. 2011;13:686–692. doi: 10.1038/ncb2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandra SM, Razavi H, Kim J, Agrawal R, Kundu RK, de Jesus Perez V, Zamanian RT, Quertermous T, Chun HJ. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2011;31:814–820. doi: 10.1161/ATVBAHA.110.219980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tammela T, Zarkada G, Nurmi H, Jakobsson L, Heinolainen K, Tvorogov D, Zheng W, Franco CA, Murtomaki A, Aranda E, Miura N, Yla-Herttuala S, Fruttiger M, Makinen T, Eichmann A, Pollard JW, Gerhardt H, Alitalo K. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol. 2011;13:1202–1213. doi: 10.1038/ncb2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, Yuan L, Mak J, Pardanaud L, Caunt M, Kasman I, Larrivee B, Del Toro R, Suchting S, Medvinsky A, Silva J, Yang J, Thomas JL, Koch AW, Alitalo K, Eichmann A, Bagri A. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J Cell Biol. 2010;188:115–130. doi: 10.1083/jcb.200903137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 27.Senturk A, Pfennig S, Weiss A, Burk K, Acker-Palmer A. Ephrin Bs are essential components of the Reelin pathway to regulate neuronal migration. Nature. 2011;472:356–360. doi: 10.1038/nature09874. [DOI] [PubMed] [Google Scholar]
- 28.Pitulescu ME, Adams RH. Regulation of signaling interactions and receptor endocytosis in growing blood vessels. Cell Adh Migr. 2014;8:366–377. doi: 10.4161/19336918.2014.970010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JD, Kang H, Larrivee B, Lee MY, Mettlen M, Schmid SL, Roman BL, Qyang Y, Eichmann A, Jin SW. Context-dependent proangiogenic function of bone morphogenetic protein signaling is mediated by disabled homolog 2. Dev Cell. 2012;23:441–448. doi: 10.1016/j.devcel.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Umasankar PK, Sanker S, Thieman JR, Chakraborty S, Wendland B, Tsang M, Traub LM. Distinct and separable activities of the endocytic clathrin-coat components Fcho1/2 and AP-2 in developmental patterning. Nat Cell Biol. 2012;14:488–501. doi: 10.1038/ncb2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartung A, Bitton-Worms K, Rechtman MM, Wenzel V, Boergermann JH, Hassel S, Henis YI, Knaus P. Different routes of bone morphogenic protein (BMP) receptor endocytosis influence BMP signaling. Mol Cell Biol. 2006;26:7791–7805. doi: 10.1128/MCB.00022-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pi X, Schmitt CE, Xie L, Portbury AL, Wu Y, Lockyer P, Dyer LA, Moser M, Bu G, Flynn EJ, 3rd, Jin SW, Patterson C. LRP1-dependent endocytic mechanism governs the signaling output of the bmp system in endothelial cells and in angiogenesis. Circ Res. 2012;111:564–574. doi: 10.1161/CIRCRESAHA.112.274597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi W, Chang C, Nie S, Xie S, Wan M, Cao X. Endofin acts as a Smad anchor for receptor activation in BMP signaling. J Cell Sci. 2007;120:1216–1224. doi: 10.1242/jcs.03400. [DOI] [PubMed] [Google Scholar]
- 34.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–1525. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- 35.Austin ED, Loyd JE. Toward Precision Medicine in Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2015;192:1272–1274. doi: 10.1164/rccm.201508-1607ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benza RL, Gomberg-Maitland M, Demarco T, Frost AE, Torbicki A, Langleben D, Pulido T, Correa-Jaque P, Passineau MJ, Wiener HW, Tamari M, Hirota T, Kubo M, Tiwari HK. Endothelin-1 Pathway Polymorphisms and Outcomes in Pulmonary Arterial Hypertension. Am J Respir Crit Care Med. 2015;192:1345–1354. doi: 10.1164/rccm.201501-0196OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS genetics. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crisan M, Kartalaei PS, Vink CS, Yamada-Inagawa T, Bollerot K, van IW, van der Linden R, de Sousa Lopes SM, Monteiro R, Mummery C, Dzierzak E. BMP signalling differentially regulates distinct haematopoietic stem cell types. Nat Commun. 2015;6:8040. doi: 10.1038/ncomms9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi T, Lyons KM, McMahon AP, Kronenberg HM. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc Natl Acad Sci USA. 2005;102:18023–18027. doi: 10.1073/pnas.0503617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, Einhorn T, Tabin CJ, Rosen V. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet. 2006;38:1424–1429. doi: 10.1038/ng1916. [DOI] [PubMed] [Google Scholar]
- 41.Wagner DO, Sieber C, Bhushan R, Borgermann JH, Graf D, Knaus P. BMPs: from bone to body morphogenetic proteins. Sci Signal. 2010;3:mr1. doi: 10.1126/scisignal.3107mr1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.