

Abstract
Hepatocellular carcinoma (HCC) is one of the most common malignancies around the world. Long noncoding RNAs (lncRNAs) are greater than 200 nucleotides without protein-coding potential and play critical roles in tumorigenesis, cell differentiation, and cancer metastasis. Colon cancer-associated transcript 2 (CCAT2), a newly identified lncRNA, was shown to be dysregulated in cancers. However, the functional role of CCAT2 in HCC remains questionable. In the present study, we found a significant upregulation of CCAT2 in HCC tissues as compared to non-tumor tissues. Functional assays showed that CCAT2 promotes cell growth in vivo and in vitro. In addition, we found a positive feedback loop between CCAT2 and FOXM1. CCAT2 upregulates FOXM1 expression through interaction with, and suppression of, miR-34a, and FOXM1 activates CCAT2 transcription. We evaluated the therapeutic potential of ultrasound-targeted microbubble destruction (UTMD)-mediated siRNA delivery to specifically target CCAT2. UTMD-mediated siCCAT2 delivery significantly suppressed tumor growth in vivo. Thus, CCAT2-FOXM1 may be a novel target for the treatment of HCC.
Keywords: CCAT2, FOXM1, miR-34a
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignancies in the world. The prognosis of HCC patients is very poor in spite of advances in the surgical resection and medical treatments, making HCC the third leading cause of cancer-related mortality [1]. Therefore, it is necessary to understand the underlying molecular mechanisms contributing to HCC tumorigenesis and progression for the development of novel early diagnosis and treatment strategies. Although alterations in many oncogenes and tumor suppressor genes are known to be closely associated with HCC, the molecular and genetic basis of hepatic carcinogenesis remain largely unknown [2].
Noncoding RNA (ncRNA) transcripts are classified into two groups based on an arbitrary transcript size greater or less than 200 nucleotides. MicroRNAs (miRNAs) are 21-nucleotide ncRNAs that perform post-transcriptional regulation of gene expression through cleavage or translation inhibition of target mRNA transcripts. Approximately 60% of protein-coding genes can be targeted by miRNAs [3], and the functional role of miRNAs has been well demonstrated in liver diseases [4]. Long ncRNAs (lncRNAs) are greater than 200 nucleotides without protein-coding potential. LncRNAs play critical role in tumorigenesis, cell differentiation, and cancer metastasis through multiple mechanisms, including epigenetic silencing and interaction with miRNAs or proteins [5,6]. Some lncRNA expression is frequently dysregulated and closely correlated with tumor growth, metastasis, and unfavorable prognosis in different cancer types [7,8]. For instance, MEG3-a putative suppressor in HCC, highly up-regulated in liver cancer (HULC), and metastasis associated lung adenocarcinoma transcript-1 (MALAT-1)-functions in the regulation of alternate splicing and exhibits an oncogenic role in HCC development [9,10]. However, an enormous number of lncRNAs remains to be elucidated and characterized.
Colon cancer-associated transcript 2 (CCAT2), a newly identified lncRNA, has been reported to be abnormally expressed in colon cancer and breast cancer [11,12]. A previous study has revealed CCAT2 to promote growth of breast cancer through the regulation of WNT signaling pathway [12]. To date, the functional roles of CCAT2 in HCC remain questionable. It is essential to uncover the underlying molecular mechanism by which CCAT2 functions in HCC. Here, we found that CCAT2 promoted cell growth in vivo and in vitro. Mechanistic investigation showed that CCAT2 increased forkhead box protein M1 (FOXM1) expression through miR-34a suppression. Furthermore, FOXM1 also activated CCAT2 transcription. Taken together, these results suggest a novel positive feedback regulation between CCAT2 and FOXM1, which plays a crucial role in HCC progression.
Materials and methods
Cell culture and tissue samples
HCC cell lines, SMMC-7721, PLC/PRF/5, Huh7, SK-Hep-1, and Hep3B, were purchased from Cell Bank of Chinese Academy of Sciences. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) at 37°C in 5% CO2. Tumor tissues were obtained from The First Affiliated Hospital of Jinzhou Medical University. A written informed consent was obtained from all patients. The use of human specimens in this study was permitted by the local ethics committee at The First Affiliated Hospital of Jinzhou Medical University.
RNA isolation and real-time polymerase chain reaction (RT-PCR)
Total RNA was extracted using TRIzol Reagent (Invitrogen) according to the standard protocol. RNA was reverse-transcribed using a High Capacity RNA-to-cDNA Kit (Applied Biosystems) according to the manufacturer’s instructions. Complementary DNA (cDNA) was quantified by RT-PCR using an ABI 7500 detection System (Applied Biosystems). RT-PCR was performed using SYBR Green Mixture (Roche). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. Following primers were used for RT-RCR: GAPDH-F: 5’-GATTCCACCCATGGCAAATTC-3’, GAPDH-R: 5’-CTGGAAGATGGTGATGGGATT-3’; CCAT2-F: 5’-GGGCACTAGACTGGGAATTAG-3’, CCAT2-R: 5’-TAGGGAGCTGAGATAGGAAGAG-3’; FOXM1-F: 5’-GATTGAGGACCACTTTCCCTAC-3’, FOXM1-R: 5’-CAGAAGGAGACCTTGCCATT-3’.
Isolation of cytoplasmic and nuclear RNA
Cytoplasmic and nuclear RNA were isolated and purified using the Cytoplasmic & Nuclear RNA Purification Kit (Norgen, Belmont, CA) according to the manufacturer’s instructions.
Cell transfection
Cells were transfected with lentiviral particles expressing full-length human CCAT2 or FOXM1 gene sequence or an empty lentiviral vector control. After 48 hours, stable clones were selected for 1 week by using puromycin.
We constructed lentivirus-based short hairpin RNA (shRNA) expression constructs into pLKO.1 vector (Addgene) to knockdown CCAT2 or FOXM1 expression. Cells were transfected with either of lentiviral vectors encoding specific shRNA sequence or the negative control vector (scramble shRNA, shCon). Following target sequences were used: shCCAT2-1: 5’-AGGTGATCAGGTGGACTTT-3’, shCCAT2-2: 5’-CCAAGAGCTAAGAGGAAAC-3’, and shFOXM1: 5’-CGGCCACTGATTCTCAAAA-3’.
RNA immunoprecipitation (RIP)
SMMC-7721 cells were cotransfected with pcDNA3.1-MS2, pcDNA3.1-CCAT2-MS2, or pcDNA3.1-CCAT2-mut-MS2 and pMS2-GFP (Addgene). After 48 hours of transfection, cells were used to perform RIP assay using a GFP antibody (Abcam) and Magna RIPTM RNA-Binding Protein Immunoprecipitation Kit (Millipore) according to the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP)
We performed ChIP using the EZ ChIPTM Chromatin Immunoprecipitation Kit (Millipore), as per the manufacturer’s instruction. Briefly, crosslinked chromatin was sonicated into 200 to 1000 bp fragments and immunoprecipitated using anti-FOXM1 antibody (Abcam). Normal rabbit immunoglobulin G (IgG) was used as a negative control. RT-PCR was conducted using SYBR Green Mixture. Primers used for CCAT2 promoter regions were F: 5’-GGTCTTGTGGAGAGGACATAAA-3’ and R: 5’-CCTGGCAGGCTATGTTTCTAT-3’.
Dual luciferase reporter assay
To construct plasmids for the dual luciferase reporter assay, wild-type CCAT2 cDNA and a mutant CCAT2 sequence (mutant in miR-34a binding site) were cloned into pmirGLO reporter vector. pmirGLO, pmirGLO-CCAT2, or pmirGLO- CCAT2-mut was cotransfected with miR-34a mimics or negative control (miR-NC) into SMMC-7721 cells using Lipofectamine reagent. The relative luciferase activity was normalized to Renilla luciferase activity 48 hours after transfection.
CCK-8 assay and colony formation
Cells were seeded at a density of 1×103 cells/well in 96-well plates. Cell proliferation was determined by the Cell Counting Kit-8 (CCK-8). Each well was incubated with WST-8 solution (Dojindo) for 1.5 hours and the absorbance of each well measured at 450 nm wavelength using a spectrophotometer. For colony formation assay, 2.0×103 cells were seeded in six-well plates. After incubation for 14 days, the number of clones was counted and analyzed.
Western blot
Cells were lysed in radioimmunoprecipitation assay (RIPA) extraction reagent (Beyotime). Total proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore). Membranes were blocked in 5% nonfat milk (Bio-Rad), followed by incubation with anti-FOXM1 (Cell Signaling) and anti-GAPDH (Cell Signaling) antibodies. After incubation, membranes were treated with anti-rabbit secondary antibodies conjugated with horseradish peroxidase (HRP, GE Healthcare Life Sciences). Immunoreactive proteins were visualized using the LumiGLO chemiluminescent substrate (Cell Signaling).
Transcriptome microarray assay
Total RNA was isolated with Trizol from shCon and shCCAT2-1 cells. Biotinylated cDNA was prepared according to the standard Affymetrix protocol from 250 ng total RNA using Ambion® WT Expression Kit. Following labeling, 5.5 μg of cDNA was hybridized for 16 hours at 45°C on GeneChip Human Transcriptome Array 2.0. GeneChips were washed, stained in the Affymetrix Fluidics Station 450, and scanned using Affymetrix® GeneChip Command Console (AGCC) installed in GeneChip® Scanner 3000 7G. Data were analyzed with Robust Multichip Analysis (RMA) algorithm using Affymetrix default analysis settings. Values presented are log2 RMA signal intensity.
siRNA-microbubble preparation
Cationic lipid microbubbles were prepared by sonicating an aqueous dispersion of 1 mg/mL polyethylene PEG2000, 2 mg/mL distearoylphophatidyl choline (DSPC), and 0.4 mg/mL 1,2-distearoyl-3-trimethyl ammonium propane (DOTAP) (all from Avanti, Germany) with perfluoropropane gas. Scramble and CCAT2 siRNA were added into cationic lipid microbubbles and the mixture was incubated on a flat rocker to facilitate siRNA-microbubble interaction for 30 minutes.
Tumor xenograft
Male nude mice (4-6-week old) were used for xenograft assays. Cells (1×106) were trypsinized, harvested in phosphate-buffered saline (PBS), and injected subcutaneously into the flank of the animals. Approximately 12 days later, tumors were detectable and tumor size was measured using a vernier caliper. Tumor volumes were calculated by the formula V=½ (L×W2), where L is the length (longest dimension) and W, the width (shortest dimension). The animal study protocol was reviewed and approved by the local ethics committee at The First Affiliated Hospital of Jinzhou Medical University. We evaluated the therapeutic potential of ultrasound-targeted microbubble destruction (UTMD)-mediated shRNA delivery that specifically targets CCAT2. Once palpable tumors were established and reached 200 mm3, mice were divided into different treatment groups as follows: G1 group injected with scramble siRNA; G2 group injected with CCAT2 siRNA; and G3 group injected with CCAT2 siRNA-microbubbles. Following injection, mice were treated with ultrasound for 20 minutes. A single-element transducer with a 1/2-inch diameter aperture with 1 MHz ultrasound was used in this study. An acoustic pressure of 1 MPa at the focus with a 50% duty cycle and a sonication intensity of 0.9 w/cm2 were employed.
Statistics
Data were analyzed using the SPSS software 19.0. Means of different treatment groups were tested for statistical difference and compared to the untreated control group using a Student’s t-test. A value of P<0.05 was considered statistically significant.
Results
CCAT2 is upregulated in HCC
We evaluated CCAT2 expression in 60 pairs of HCC and non-tumor tissues by RT-PCR and found the expression of CCAT2 transcript to be significantly higher in HCC tissues as compared with that in paired non-tumor tissues (Figure 1A). To determine whether CCAT2 expression level is related to HCC progression, we analyzed the association between CCAT2 and clinicopathological features in HCC patients. As shown in Table 1, we observed a strong correlation between CCAT2 expression and tumor size (P=0.007) and Barcelona Clinic Liver Cancer (BCLC) stage (P=0.018). Furthermore, Kaplan-Meier and log-rank test analyses suggested a positive correlation between the tumorous CCAT2 expression and a significantly reduced overall survival (OS) (P<0.001; Figure 1B).
Figure 1.
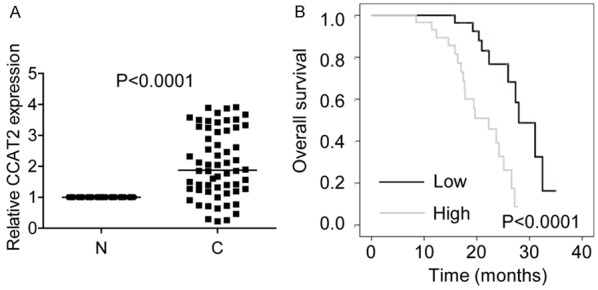
CCAT2 is upregulated in HCC. A. The relative expression of CCAT2 in 60 pairs of HCC tissues (C) and non-tumor tissues (N) was detected by RT-PCR. CCAT2 expression in non-tumor tissues was considered as control. B. Kaplan-Meier survival curve and log-rank test were used to evaluate the association between CCAT2 expression and overall survival rate. Patients were grouped into CCAT2-high and CCAT2-low group according to the median of ZFAS1 expression in OS tissues.
Table 1.
The correlation between CCAT2 expression and clinicopathological features in HCC patients
| Clinicopathological features | CCAT2 expression levels | P value | |
|---|---|---|---|
|
| |||
| Low | High | ||
| Gender | 1.000 | ||
| Male | 24 | 24 | |
| Female | 6 | 6 | |
| Age | 0.519 | ||
| 519 | 23 | 25 | |
| >55 | 7 | 5 | |
| Liver cirrhosis | 0.766 | ||
| With | 22 | 23 | |
| Without | 8 | 7 | |
| AFP (ng/mL) | 0.688 | ||
| 688 | 3 | 4 | |
| >20 | 27 | 26 | |
| Portal vein tumor thrombus | 0.519 | ||
| No | 23 | 25 | |
| Yes | 7 | 5 | |
| Tumor size (cm) | 0.007* | ||
| 00 | 16 | 6 | |
| >5 | 14 | 24 | |
| BCLC stage | 0.018* | ||
| A | 17 | 8 | |
| B+C+D | 13 | 22 | |
| Relapse | 0.584 | ||
| Yes | 19 | 21 | |
| No | 11 | 9 | |
| Metastasis | 0.718 | ||
| Yes | 4 | 5 | |
| No | 26 | 25 | |
Abbreviations: AFP, alpha-fetoprotein; BCLC, Barcelona Clinic Liver Cancer; CCAT2, colon cancer-associated transcript 2; HCC, hepatocellular carcinoma.
The values were statistically significant.
The median expression level was used as cutoff.
CCAT2 promotes cell proliferation and tumor growth in vitro and in vivo
We analyzed the biological function of CCAT2 in regulation of cell proliferation using cell biology assays. We analyzed the expression level of CCAT2 in five HCC cell lines (SMMC-7721, PLC/PRF/5, Huh7, SK-Hep-1, and Hep3B) and detected highest level of CCAT2 expression in SKHep-1 cells. SMMC-7721 cells, on the other hand, showed the lowest CCAT2 expression (Figure 2A). To study the effect of CCAT2 on the proliferation of HCC cells, we established SMMC-7721 cells stably overexpressing CCAT2 and SK-Hep-1 cells with stable knockdown of CCAT2 (Figure 2B). As the target site one (sh1) is known to be the most effective site, we chose sh1 for further study. We performed CCK-8 assay to detect the effect of CCAT2 on cell proliferation and found that CCAT2 overexpression significantly increased cell proliferation in SKHep-1 cells. On the contrary, CCAT2 downregulation suppressed cell proliferation in SMMC-7721 cells (Figure 2C and 2D). We performed the colony formation assay to further confirm the role of CCAT2 in cell proliferation. Cells overexpressing CCAT2 showed higher colony numbers as compared to the control cells, while knockdown of CCAT2 showed the opposite effect (Figure 2E and 2F). The growth-enhancing effect of CCAT2 was confirmed with in vivo tumor growth assay. Xenograft tumors grown from cells overexpressing CCAT2 displayed larger mean volumes and formed more rapidly as compared to tumors derived from control cells (Figure 2G). Tumors grown from CCAT2-knockdown cells were smaller than those from control cells (Figure 2H). These results suggest that CCAT2 may promote HCC cell proliferation both in vitro and in vivo.
Figure 2.
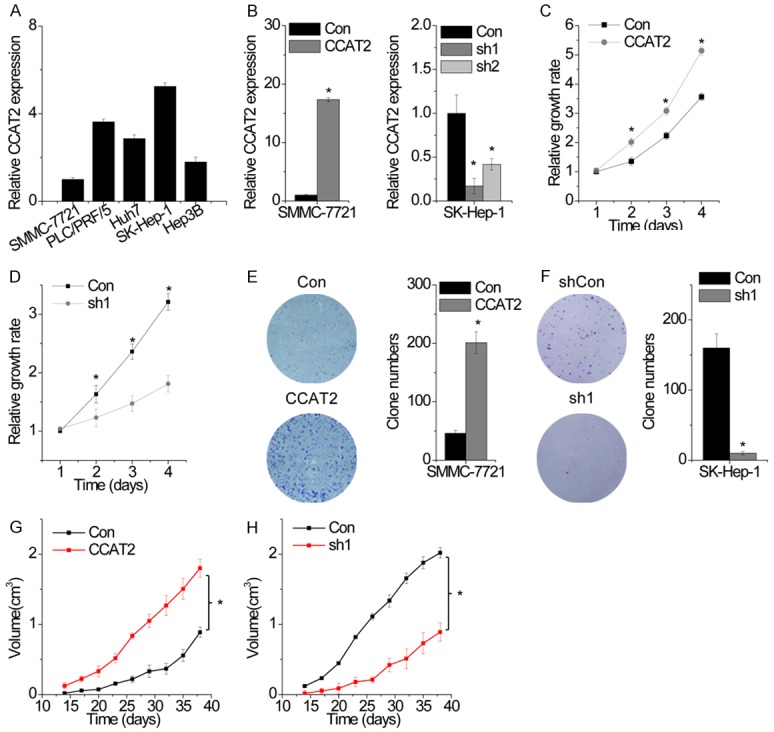
CCAT2 promotes cell proliferation and tumor growth in vitro and in vivo. A. The relative expression of CCAT2 in five HCC cell lines was determined by RT-PCR. The expression of CCAT2 in SMMC-7721 cells was used as a control. B. The relative expression of CCAT2 in control and CCAT2-overexpressing or CCAT2-silenced cells was detected by RT-PCR. C. Cell growth rates were determined with the CCK-8 assay. CCAT2 overexpression in SMMC-7721 cells significantly enhanced cell proliferation as compared to that in control cells. D. CCAT2 depletion inhibited the proliferation of SK-Hep-1 cells. E. Colony formation assay of control and CCAT2-overexpressing SMMC-7721 cells. Representative graphs are shown. F. Colony formation assay of control and CCAT2-silenced SK-Hep-1 cells. Representative graphs are shown. G. Effects of CCAT2 overexpression on tumor growth in vivo. Tumor growth curves are shown. H. Effects of CCAT2 knockdown on tumor growth in vivo. Tumor growth curves are shown. Data are shown as mean ± SD. *P<0.05.
CCAT2 inhibits cell apoptosis and induces cell cycle progression
As CCAT2 positively regulates cell proliferation, we speculated CCAT2 to be critical for cell apoptosis and cell cycle. We tested this hypothesis by detecting apoptosis by fluorescence-activated cell sorting (FACS) analysis in HCC cells stained for annexin V and 7-aminoactinomycin D (7-AAD). We observed that CCAT2 knockdown in SK-Hep-1 cells resulted in a significantly higher percentage of annexin V-positive cells as compared to the cells expressing a scrambled shRNA (Figure 3A). On the contrary, CCAT2 overexpression inhibited cell apoptosis in SMMC-7721 cells (Figure 3B).
Figure 3.
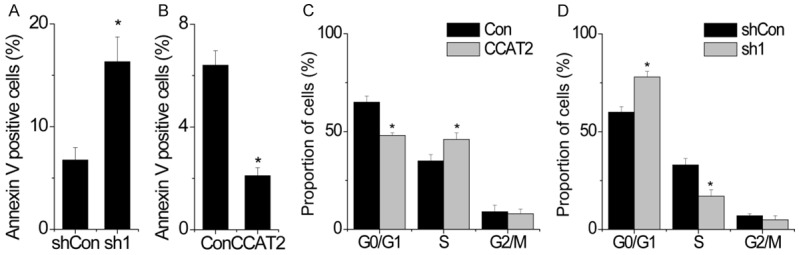
CCAT2 inhibits cell apoptosis and induces cell cycle progression. A. CCAT2-silenced cells were stained with a combination of annexin V and 7-AAD and analyzed by FACS. Cells positive for annexin V staining were counted as apoptotic cells and the percentage of apoptotic cells is reported. B. Cells with CCAT2 overexpression were stained with a combination of annexin V and 7-AAD and analyzed by FACS. Cells positive for annexin V staining were counted as apoptotic cells and the percentage of apoptotic cells is reported. C. FACS analysis showing significant increases or decreases in SMCC-7221 cells overexpressing CCAT2 at S- or G-1 phase, respectively. D. FACS analysis showing significant decreases or increases in CCAT2-silenced SK-Hep-1 cells at S- or G-1 phase, respectively. Data are shown as mean ± SD. *P<0.05.
We analyzed differences in cell cycle distributions following CCAT2 upregulation or downregulation by FACS. CCAT2 overexpression drove cell cycle progression beyond the G1/S transition in SMMC-7721 cells. On the contrary, CCAT2-silenced SK-Hep-1 cells showed a significant arrest in cell cycle at G1/S phase (Figure 3C and 3D).
CCAT2 increases FOXM1 expression
To explore the mechanism by which CCAT2 regulates proliferation in HCC cells, we performed transcriptome microarray assay and identified the target genes regulated by CCAT2 (Figure 4A). Among the targets of CCAT2, FOXM1 is of particular interest because of the remarkable change in its expression upon CCAT2 knockdown and its significant contribution to tumorigenesis and HCC progression. We found that CCAT2 upregulation markedly enhanced both FOXM1 mRNA and protein level, while CCAT2 knockdown significantly decreased FOXM1 expression (Figure 4B and 4C). To determine whether CCAT2 promotes proliferation in a FOXM1-dependent manner, rescue experiments were performed. Overexpression of CCAT2 promotes proliferation of HCC cells, which was rescued by FOXM1 inhibition (Figure 4D and 4E). In contrast, FOXM1 upregulation abolished the inhibition of cell proliferation induced by CCAT2 knockdown (Figure 4F and 4G).
Figure 4.
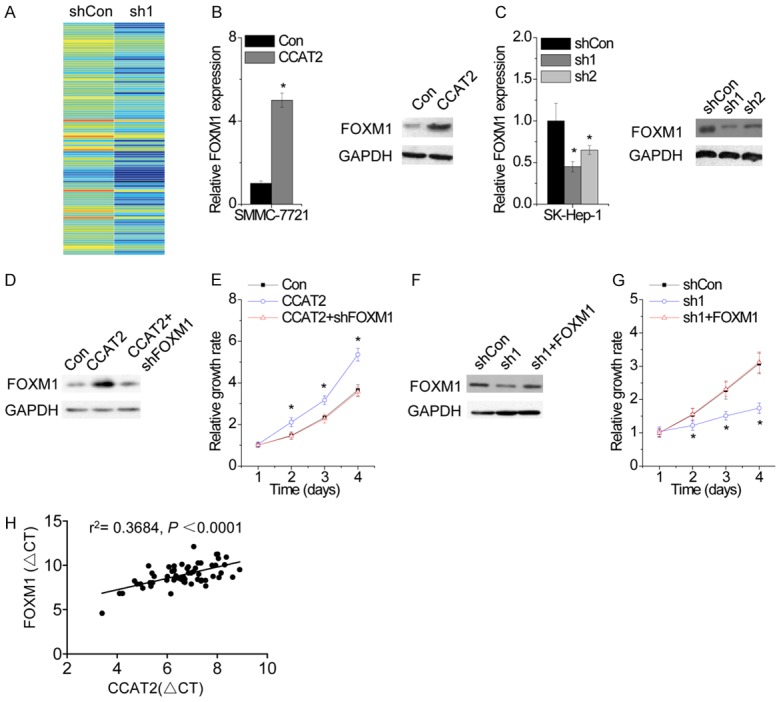
CCAT2 increases FOXM1 expression. A. The mRNA expression profile was detected by microarray in control and CCAT2-silenced SK-Hep-1 cells. B. The effect of CCAT2 overexpression on FOXM1 expression was determined by RT-PCR (left) and western blot (right) analysis. C. The effect of CCAT2 knockdown on FOXM1 expression was determined by RT-PCR (left) and western blot (right) analysis. D. The SMMC-7721 cells expressing CCAT2 was transfected with FOXM1 shRNA. E. The cellular proliferation of SMMC-7721 cells expressing CCAT2 with and without FOXM1 shRNA. F. The SK-Hep-1 cells expressing CCAT2 shRNA was transfected with FOXM1. G. The cellular proliferation of SK-Hep-1 cells expressing CCAT2 shRNA with and without FOXO1 overexpression. H. The correlation between FOXM1 and CCAT2 expression in 60 HCC tissue samples. Data are shown as mean ± SD. *P<0.05.
We confirmed the pathological correlation between CCAT2 and FOXM1 in HCC samples by detecting CCAT2 and FOXM1 expression in the same set of 60 HCC tissues. We found a positive correlation between CCAT2 and FOXM1 in HCC tissues (r2=0.3684, P<0.0001; Figure 4H). Together these results demonstrate that CCAT2 exerts its function via regulation of FOXM1 expression.
CCAT2 functions as a competitive endogenous RNA (ceRNA) of FOXM1 through competitive interaction with miR-34a
We explored the mechanism by which CCAT2 regulates FOXM1 expression. We first performed cellular fractionation to analyze the subcellular localization of CCAT2 and found CCAT2 to be mainly localized in the cytoplasm (Figure 5A), suggestive of its potential role in posttranscriptional regulation. The interaction between lncRNAs and miRNAs provides an additional layer of control in gene regulation [13]. Using Targetscan and Microinspector software, we found a set of miRNAs that may potentially bind to CCAT2. Of these miRNA candidates, miR-34a was thought to directly bind to CCAT2. miR-34a has been reported to exert tumor-suppressive function through negative regulation of FOXM1 expression in HCC [14]. Dual luciferase assay showed a significant decrease in luciferase activity following cotransfection of miR-34a and wild-type CCAT2 expression vector but not mutant CCAT2 with mutation in miR-34a-binding site (CCAT2-mut) (Figure 5B). To further confirm the direct interaction between CCAT2 and miR-34a, we performed an RIP assay to pull-down endogenous miRNAs associated with CCAT2, followed by RT-PCR detection (Figure 5C). We found that the amount of CCAT2 was significantly higher in SMCC-7721 cells with miR-34a as compared to those carrying the empty vector (MS2), negative control IgG, and CCAT2-mut. To investigate whether CCAT2 was regulated by miR-34a in an AGO2-dependent manner, we performed anti-AGO2 RIP using SK-Hep-1 cells with upregulated miR-35a expression. The endogenous CCAT2 pull-down by AGO2 was specifically enriched in cells overexpressing miR-34a (Figure 5D), indicating that miR-34a is a CCAT2-targeting microRNAs. We further clarified the regulatory relationship between CCAT2 and miR-34a. Overexpression of CCAT2 significantly inhibited miR-34a expression (Figure 5E), whereas overexpression of miR-34a failed to affect CCAT2 expression (Figure 5F). These data indicate that miR-34a directly binds to CCAT2, but fails to induce CCAT2 degradation.
Figure 5.
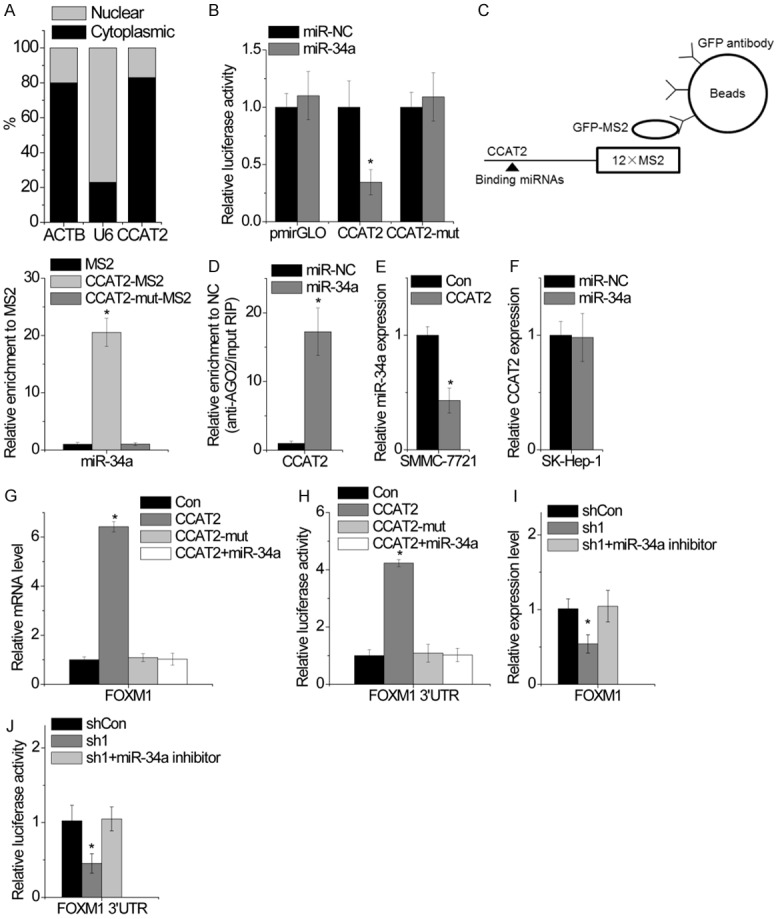
CCAT2 functions as a ceRNA of FOXM1 through its competitive interaction with miR-34a. A. The cellular location of CCAT2 in SMMC-7721 cells. B. Luciferase activity in SMMC-7721 cells cotransfected with miR-34a and empty luciferase reporter or one containing CCAT2 or CCAT2-mut. Data are presented as the relative ratio of firefly luciferase activity to Renilla luciferase activity. C. MS2-RIP followed by microRNA RT-PCR to detect microRNAs endogenously associated with CCAT2. D. Anti-AGO2 RIP was performed in SMMC-7721 cells transiently overexpressing miR-34a, followed by RT-PCR to detect CCAT2 associated with AGO2. E. The effect of CCAT2 overexpression on miR-34a expression was detected by RT-PCR. F. The effect of miR-34a overexpression on CCAT2 expression was detected by RT-PCR. G. The mRNA level of FOXM1 expression in SMCC-7721 cells overexpressing CCAT2 or CCAT2-mut cotransfected with miR-34a was analyzed by qPCR. H. The relative luciferase activity of FOXM1 3’UTR in SMCC-7721 cells overexpressing CCAT2 or CCAT2-mut cotransfected with miR-34a was analyzed by qPCR. I. The mRNA level of FOXM1 expression in CCAT2-silenced SK-Hep-1 cells cotransfected with miR-34a inhibitor was analyzed by qPCR. J. The relative luciferase activity of FOXM1 3’UTR in CCAT2-silenced SK-Hep-1 cells cotransfected with miR-34a inhibitor was analyzed. Data are shown as mean ± SD. *P<0.05.
We suspected CCAT2 to regulate FOXM1 expression through competitive interaction with miR-34a. We confirmed this hypothesis by transient transfection of wild-type CCAT2- or CCAT2-mut-overexpressing SMMC-7721 cells with miR-34a. Ectopic expression of wild-type CCAT2, but not CCAT2-mut, significantly upregulated FOXM1 expression. Upregulation of miR-34a abolished the increased FOXM1 expression induced by CCAT2 (Figure 5G). To confirm these results, we examined whether CCAT2 increased FOXM1 expression through the regulation of FOXM1 3’UTR. The luciferase reporter vector containing FOXM1 3’UTR was transfected into SMMC-7721 cells overexpressing wild-type or mutant CCAT2 with or without miR-34a. Overexpression of wild-type CCAT2, but not CCAT2-mut, increased the luciferase activity of FOXM1 3’UTR. The effect of CCAT2 on the luciferase activity of FOXM1 3’UTR was abolished by miR-34a (Figure 5H). On the contrary, miR-34a inhibition overcame the suppression of FOXM1 by CCAT2 knockdown (Figure 5I). Moreover, miR-34a silencing rescued the inhibition of the luciferase activity of FOXM1 3’UTR mediated by CCAT2 knockdown (Figure 5J). Taken together, CCAT2 functions as a ceRNA of FOXM1 and competes with FOXM1 mRNA for the common miR-34a.
FOXM1 activates CCAT2 expression
The positive correlation between CCAT2 and FOXM1 expression drew us to suspect the regulatory role of FOXM1 in CCAT2 transcription. We found that FOXM1 overexpression significantly increased CCAT2 expression (Figure 6A), whereas FOXM1 shRNA dramatically suppressed CCAT2 level (Figure 6B). To determine whether FOXM1 regulates CCAT2 transcription, CCAT2 promoter luciferase constructs (-1500/0) were cotransfected with FOXM1. The luciferase reporter assay revealed transactivation of CCAT2 promoter activity by FOXM1 (Figure 6C), which was inhibited upon FOXM1 silencing (Figure 6D). Furthermore, ChIP assay confirmed the direct binding of FOXM1 to the promoter of CCAT2 in HCC cells (Figure 6E). Taken together, these results demonstrate that CCAT2 is a direct target gene of FOXM1 and there exists a positive feedback regulation between CCAT2 and FOXM1.
Figure 6.
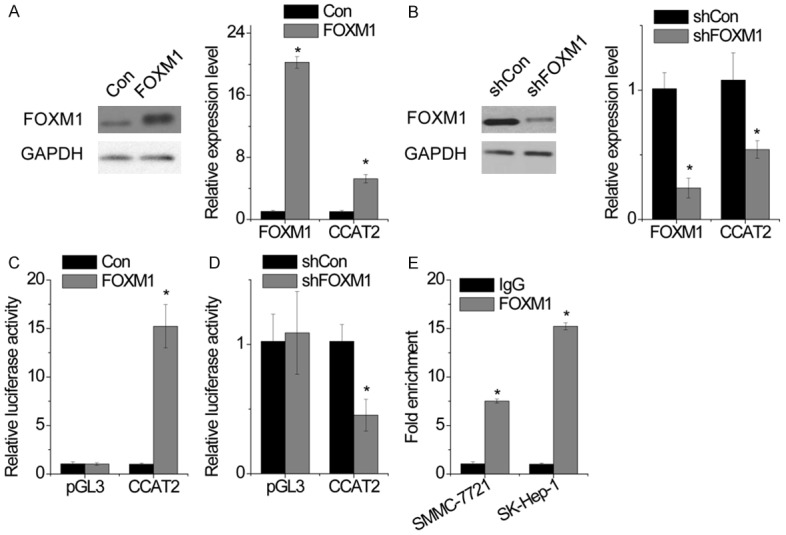
FOXM1 activates CCAT2 expression. A. SMMC-7721 cells were transfected with empty vector or FOXM1 and CCAT2 expression was determined by RT-PCR. B. SK-Hep-1 cells were transfected with control or FOXM1 shRNA and CCAT2 expression was determined by RT-PCR. C. Luciferase activity in SMMC-7721 cells cotransfected with FOXM1 and empty luciferase reporter or one containing CCAT2 promoter. D. Luciferase activity in SK-Hep-1 cells cotransfected with FOXM1 shRNA and empty luciferase reporter or one containing CCAT2 promoter. E. The binding level of FOXM1 at CCAT2 promoter was detected by ChIP assay, followed by RT-PCR. Data are shown as mean ± SD. *P<0.05.
Reduction in CCAT2 expression by UTMD-mediated siRNA delivery results in inhibition of HCC growth in vivo
Ultrasound-targeted microbubble destruction technology has been recognized as a promising technology for drug and gene delivery in vivo [15]. We evaluated the therapeutic potential of UTMD-mediated siRNA delivery that specifically targets CCAT2 and found that CCAT2 siRNA injection diminished the tumor volume. Moreover, the tumor size in mice treated with CCAT2 siRNA-microbubbles was much smaller than that in mice injected with CCAT2 siRNA (Figure 7). These results suggest that targeting FOXM1 may represent a promising approach for cancer treatment.
Figure 7.
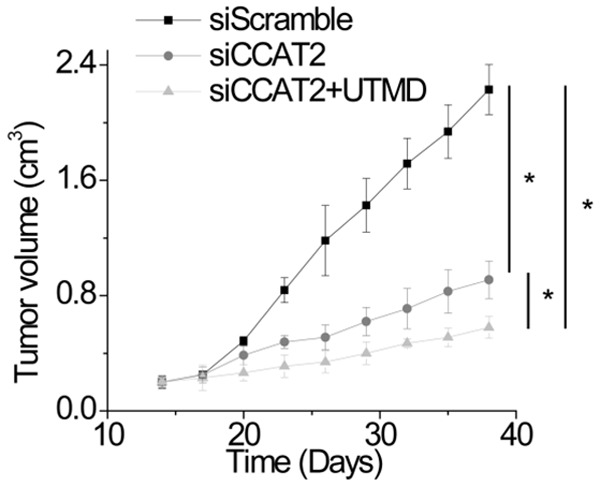
Reduction in CCAT2 expression by UTMD-mediated siRNA delivery results in inhibition of HCC growth in vivo. Once palpable tumors were established and reached 200 mm3, mice were divided into the following treatment groups: G1, the group injected with scramble siRNA; G2, the group injected with CCAT2 siRNA; G3, the group injected with CCAT2 siRNA-microbubbles. The tumor size was measured.
Discussion
In recent years, lncRNAs have gained enormous attention as a novel and critical regulator of gene expression, tumorigenesis, and tumor progress. In the present study, we evaluated differential expression of CCAT2 in HCC tissues and non-tumorous tissues and found a significant increase in CCAT2 expression in HCC tissues. Upregulation of CCAT2 expression in HCC patients predicts unfavorable prognosis. Through gain- and loss-of-function assays, we demonstrated a positive regulatory role of CCAT2 in cell proliferation and tumor growth in vitro and in vivo.
Studies have reported lncRNA to regulate gene expression by different mechanisms, including gene transcription, genomic imprinting, and chromatin modification [5]. We performed transcriptome microarray analysis to reveal the molecular mechanism involved in CCAT2-induced cell proliferation and demonstrated that FOXM1 expression was regulated by CCAT2. Cellular fractionation analysis results showed that CCAT2 is mainly located in the cytoplasm, suggestive of its potential role in post-transcriptional regulation. Recent studies have focused on the interaction between lncRNAs and miRNAs. For instance, lncRNA-ATB upregulated zinc finger E-box-binding homeobox 1 (ZEB1) and ZEB2 by competitive binding to the miR-200 family, thereby inducing epithelial-mesenchymal transition and invasion [16]. LncRNA ZFAS1 activated ZEB1, MMP14, and MMP16 expression through the suppression of miR-150, inducing HCC metastasis [17]. These findings encouraged us to explore the underlying mechanisms involved with miRNAs by which CCAT2 regulates FOXM1 expression. Here, we provided strong evidence that CCAT2 regulates FOXM1 expression through competitive binding to miR-34a. RIP and luciferase reporter assays indicated a directly interaction between CCAT2 and miR-34a. Restoration of miR-34a expression rescued FOXM1 expression and 3’UTR luciferase activity induced by CCAT2 overexpression. In addition, CCAT2 and FOXM1 expression levels were positively correlated in clinical HCC samples, further supporting the hypothesis that FOXM1 is a target of CCAT2.
The FOXM1 transcription factor is a key factor in cancer initiation and progression. Previous study revealed FOXM1 as a key downstream effector of PI3K-AKT, ATM/p53-E2F, and p38-MAPK-MK2 signaling cascades involved in cancer progression [18-20]. In our present study, for the first time, we revealed that FOXM1 also activates lncRNA CCAT2 expression. Through luciferase reporter and ChIP assays, we demonstrated that FOXM1 directly binds to CCAT2 promoter and activates its transcription. Therefore, CCAT2 and FOXM1 formed a positive feedback regulation crucial for HCC progress.
Several studies have employed UTMD for siRNA or shRNA delivery in vitro and in vivo [21-23]. UTMD stimulates sonoporation and changes in the permeability of the cell membrane, facilitating efficient delivery of siRNA or DNA into cells [24]. In this study, we evaluated the therapeutic potential of UTMD-mediated siRNA delivery that specifically targets CCAT2. We observed that UTMD-mediated siCCAT2 delivery significantly suppressed tumor growth in vivo, indicative of its therapeutic potential for HCC treatment.
In summary, overexpression of CCAT2 in HCC promotes tumor growth in a FOXM1-dependent manner and there exists a positive feedback regulation between FOXM1 and CCAT2 (Figure 8). Thus, CCAT2-FOXM1 may be a novel target for the treatment of HCC.
Figure 8.
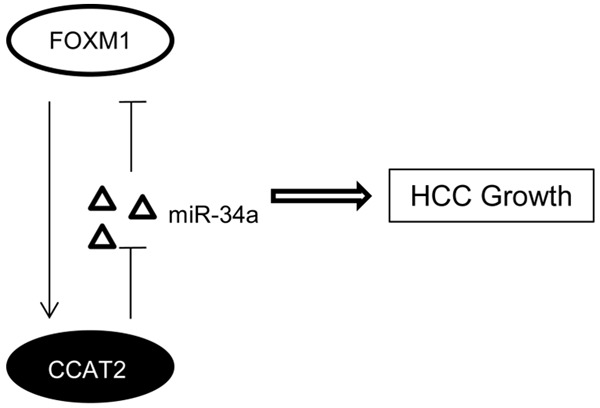
Schematic model of CCAT2 functions during HCC growth. CCAT2 promotes HCC cell growth by competitive binding to miR-34a and upregulation of FOXM1. On the other hand, FOXM1 binds to CCAT2 promoter to activate its transcription.
Acknowledgements
This work was supported by Natural Science Foundation of Liaoning province (No. 20170540397).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Kanda M, Sugimoto H, Kodera Y. Genetic and epigenetic aspects of initiation and progression of hepatocellular carcinoma. World J Gastroenterol. 2015;21:10584–10597. doi: 10.3748/wjg.v21.i37.10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. 2013;10:542–552. doi: 10.1038/nrgastro.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 6.Huang JL, Zheng L, Hu YW, Wang Q. Characteristics of long non-coding RNA and its relation to hepatocellular carcinoma. Carcinogenesis. 2014;35:507–514. doi: 10.1093/carcin/bgt405. [DOI] [PubMed] [Google Scholar]
- 7.Li M, Wang Y, Cheng L, Niu W, Zhao G, Raju JK, Huo J, Wu B, Yin B, Song Y, Bu R. Long noncoding RNAs in renal cell carcinoma: a systematic review and clinical implications. Oncotarget. 2017 doi: 10.18632/oncotarget.17053. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klingenberg M, Matsuda A, Diederichs S, Patel T. Non-coding RNA in hepatocellular carcinoma: mechanisms, biomarkers and therapeutic targets. J Hepatol. 2017 doi: 10.1016/j.jhep.2017.04.009. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 9.Lai MC, Yang Z, Zhou L, Zhu QQ, Xie HY, Zhang F, Wu LM, Chen LM, Zheng SS. Long noncoding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med Oncol. 2012;29:1810–1816. doi: 10.1007/s12032-011-0004-z. [DOI] [PubMed] [Google Scholar]
- 10.Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H, Schroeder R, Trauner M, Zatloukal K. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology. 2007;132:330–342. doi: 10.1053/j.gastro.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 11.Redis RS, Sieuwerts AM, Look MP, Tudoran O, Ivan C, Spizzo R, Zhang X, de Weerd V, Shimizu M, Ling H, Buiga R, Pop V, Irimie A, Fodde R, Bedrosian I, Martens JW, Foekens JA, Berindan-Neagoe I, Calin GA. CCAT2, a novel long non-coding RNA in breast cancer: expression study and clinical correlations. Oncotarget. 2013;4:1748–1762. doi: 10.18632/oncotarget.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai Y, He J, Zhang D. Long noncoding RNA CCAT2 promotes breast tumor growth by regulating the Wnt signaling pathway. Onco Targets Ther. 2015;8:2657–2664. doi: 10.2147/OTT.S90485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang C, Wu D, Gao L, Liu X, Jin Y, Wang D, Wang T, Li X. Competing endogenous RNA networks in human cancer: hypothesis, validation, and perspectives. Oncotarget. 2016;7:13479–13490. doi: 10.18632/oncotarget.7266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X, Chen W, Miao R, Zhou Y, Wang Z, Zhang L, Wan Y, Dong Y, Qu K, Liu C. miR-34a induces cellular senescence via modulation of telomerase activity in human hepatocellular carcinoma by targeting FoxM1/c-Myc pathway. Oncotarget. 2015;6:3988–4004. doi: 10.18632/oncotarget.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen ZY, Yang F, Lin Y, Zhang JS, Qiu RX, Jiang L, Zhou XX, Yu JX. New development and application of ultrasound targeted microbubble destruction in gene therapy and drug delivery. Curr Gene Ther. 2013;13:250–274. doi: 10.2174/15665232113139990003. [DOI] [PubMed] [Google Scholar]
- 16.Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, Wang SB, Wang YZ, Yang Y, Yang N, Zhou WP, Yang GS, Sun SH. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666–681. doi: 10.1016/j.ccr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Li T, Xie J, Shen C, Cheng D, Shi Y, Wu Z, Deng X, Chen H, Shen B, Peng C, Li H, Zhan Q, Zhu Z. Amplification of long noncoding RNA ZFAS1 promotes metastasis in hepatocellular carcinoma. Cancer Res. 2015;75:3181–3191. doi: 10.1158/0008-5472.CAN-14-3721. [DOI] [PubMed] [Google Scholar]
- 18.Grant GD, Brooks L 3rd, Zhang X, Mahoney JM, Martyanov V, Wood TA, Sherlock G, Cheng C, Whitfield ML. Identification of cell cycleregulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol Biol Cell. 2013;24:3634–3650. doi: 10.1091/mbc.E13-05-0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu G, Zhou A, Xue J, Huang C, Zhang X, Kang SH, Chiu WT, Tan C, Xie K, Wang J, Huang S. FoxM1 promotes breast tumorigenesis by activating PDGF-A and forming a positive feedback loop with the PDGF/AKT signaling pathway. Oncotarget. 2015;6:11281–11294. doi: 10.18632/oncotarget.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Behren A, Muhlen S, Acuna Sanhueza GA, Schwager C, Plinkert PK, Huber PE, Abdollahi A, Simon C. Phenotype-assisted transcriptome analysis identifies FOXM1 downstream from Ras-MKK3-p38 to regulate in vitro cellular invasion. Oncogene. 2010;29:1519–1530. doi: 10.1038/onc.2009.436. [DOI] [PubMed] [Google Scholar]
- 21.Park DH, Jung BK, Lee YS, Jang JY, Kim MK, Lee JK, Park H, Seo J, Kim CW. Evaluation of in vivo antitumor effects of ANT2 shRNA delivered using PEI and ultrasound with microbubbles. Gene Ther. 2015;22:325–332. doi: 10.1038/gt.2014.120. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Chang S, Sun J, Zhu S, Pu C, Li Y, Zhu Y, Wang Z, Xu RX. Targeted microbubbles for ultrasound mediated short hairpin RNA plasmid transfection to inhibit survivin gene expression and induce apoptosis of ovarian cancer A2780/DDP cells. Mol Pharm. 2015;12:3137–3145. doi: 10.1021/mp500835z. [DOI] [PubMed] [Google Scholar]
- 23.Wang HH, Song YX, Bai M, Jin LF, Gu JY, Su YJ, Liu L, Jia C, Du LF. Ultrasound targeted microbubble destruction for novel dual targeting of HSP72 and HSC70 in prostate cancer. Asian Pac J Cancer Prev. 2014;15:1285–1290. doi: 10.7314/apjcp.2014.15.3.1285. [DOI] [PubMed] [Google Scholar]
- 24.Yin T, Wang P, Li J, Zheng R, Zheng B, Cheng D, Li R, Lai J, Shuai X. Ultrasound-sensitive siRNA-loaded nanobubbles formed by heteroassembly of polymeric micelles and liposomes and their therapeutic effect in gliomas. Biomaterials. 2013;34:4532–4543. doi: 10.1016/j.biomaterials.2013.02.067. [DOI] [PubMed] [Google Scholar]