

Abstract
Gastric cancer (GC) remains the second tumor caused death threat worldwide, and personalized medicine for GC is far from expectation. Finding novel, recurrently mutated genes through next-generation sequencing (NGS) is a powerful and productive approach. However, previous genomic data for GC are based on surgical resected samples while a large proportion of advanced gastric cancer (AGC) patients have already missed the chance for operation. The aim of this study is to assess frequent genomic alteration in AGC via biopsy samples. Here we performed targeted genomic sequencing of 78 AGC patients’ tumor biopsies along with matched lymphocyte samples based on a 118 cancer related gene panel. In total, we observed 301 somatic nonsynonymous genomic alterations in 92 different genes, as well as 37 copy number gain events among 15 different genes (fold change 2-12), and validated the fold changes of ERBB2 copy number gains with IHC and FISH test showed an accuracy of 81.8%. Previously reported driver genes for gastric cancer (TP53, KMT2D, KMT2B, EGFR, PIK3CA, GNAQ, and ARID1A), and several unreported mutations (TGFBR2, RNF213, NF1, NSD1, and LRP2) showed high non-silent mutation prevalence (7.7%-34.6%). When comparing intestinal-type gastric cancer (IGC) with diffuse-type gastric cancer (DGC), TP53 and GNAQ appear to be more frequently mutated in IGC (P=0.028 and P=0.023, respectively), whereas LRP2, BRCA2 and FGFR3 mutations are not observed in IGC, but have 12.8%, 7.7% and 7.7% mutation rates, respectively, in DGC patients. Patients with one or more mutations in adherens junction pathway (CREBBP, EP300, CDH1, CTNNB1, EGFR, MET, TGFBR2 and ERBB2) or TGF-β signaling pathway (CREBBP, EP300, MYST4, KRAS and TGFBR2) showed significantly better overall survival (P=0.007 and P=0.014, respectively), consistent with The Cancer Genome Atlas (TCGA) cohort data. Importantly, 57 (73.1%) patients harbored at least one genomic alteration with potential treatments, making NGS-based drug target screening a viable option for AGC patients. Our study established a comprehensive genomic portrait of AGC, and identified several mutation signatures highly associated with clinical features, survival outcomes, which may be used to design future personalized treatments.
Keywords: Gastric cancer, next-generation sequencing, prognosis, targetable alterations
Introduction
Gastric cancer is a highly heterogeneous disease and one of the most frequent cause of cancer-related mortality worldwide. Almost 1 million new cases of gastric cancer were diagnosed in 2012, half of which occurred in Eastern Asia (mainly in China) [1,2]. Because the early disease is asymptomatic, most gastric cancer patients are diagnosed at advanced stage, when the radical surgery has already missed best timing. While the optimal standard chemotherapy regimen for these patients remains debatable, fluorouracil-containing doublet therapy is the preferred first-line option in eastern Asian countries [3].
So far, a series of NGS studies including TCGA have revealed several genes frequently mutated in gastric cancer, though the findings are sometimes contradictory. Repeatedly mentioned “driver genes” include TP53, CDH1 [4], ARID1A [5], CTNNB1 [6], PIK3CA [7] and RHOA [8,9]. However, these findings are based on gastrostomy specimens, indicating that there’s a chosen bias against those rapidly progress patients, who are disqualified from operations, but potentially suitable candidates for clinical trials. The objective of this study is to establish the feasibility of using endoscopic biopsy of advanced gastric cancer patients for next-generation sequencing, in connection with their first-line standardized chemotherapy results, trying to display a deduced network/core pathway of gastric cancer genomic result to be a complementary of former results. Instead of finding novel driver genes, we employed a targeted genomic sequencing approach based on a 118-cancer genes panel, focusing on linking the known cancer-related driver genes with AGC phenotypes and prognosis.
Methods
Patients and sample collection
A total of 78 fresh-frozen biopsy tumors were collected, histologically confirmed as gastric cancer and none of them had received any previous treatment, along with paired peripheral blood samples used as sequence reference to detect somatic alterations. After biopsy, majority of these patients was treated with first-line fluorouracil plus cisplatin or paclitaxel at the Gastrointestinal Oncology department of Peking University Cancer Hospital from January 2011 to July 2014, and had completed at least two cycles of chemotherapy. All patients gave written informed consents to allow their tissues being used in medical research. Drug administration was carried out according to our previous report [10]. This study was approved by the Ethics Committee of Peking University Cancer Hospital and performed according to the Declaration of Helsinki Principles.
DNA library preparation and target DNA enrichment
Genomic DNA was isolated from fresh-frozen tissues using QIAamp DNA Mini Kit (Lot. 51304, QIAGEN) according to the manufacturer’s instructions and stored at -20°C until use.
The 118 cancers related genes in this panel for captured and targeted next-generation sequencing are listed in Table 1. The exon regions of all 118 genes were specifically enriched using oligonucleotide probes (MyGenostics, Baltimore, MD, USA) as described previously [11,12]. In brief, 1 μg DNA library was used per capture. It was mixed with Buffer BL and GenCap probe (MyGenostics, MD, USA), heated at 95°C for 7 min and 65°C for 2 min, and added preheated Buffer HY (MyGenostics, MD, USA) to keep at 65°C for 22 h for hybridization. Then transferred to the tube with prewashed MyOne beads (Life Technology) and rotated for 1 h. The beads were then washed several times and the bound DNA was then eluted with eluting buffer. The eluted DNA was amplified for 15 cycles using the following program: 98°C for 30 s (1 cycle); 98°C for 25 s, 65°C for 30 s, 72°C for 30 s (15 cycles); 72°C for 5 min (1 cycle). The PCR product was purified using SPRI beads (Beckman Coulter, Inc.) in accordance with manufacturer’s protocol. The enrichment libraries were sequenced on an Illumina HiSeq 2000 sequencer for paired read 100 bp. Short read mapping and alignment were performed using BWA software (Burrows Wheeler Aligner). SNPs and indels were detected using the SOAPsnp software and GATK Indel Genotyper (http://www.broadinstitute.org/gsa/wiki/index.php/; The Genome Analysis Toolkit), respectively. All reference sequences were based on the NCBI37/hg19 assembly of the human genome.
Table 1.
118 cancer-related genes list
| ABL1 | AKAP9 | AKT1 | ALK | APC | ARID1A | ARID2 | ASXL1 |
| ATM | ATRX | BAP1 | BRAF | BRCA1 | BRCA2 | CBL | CDC73 |
| CDH1 | CDK12 | CDKN2A | CEBPA | CIC | CREBBP | CSF1R | CTNNA1 |
| CTNNB1 | CYLD | CYP2D6 | DAXX | DNMT3A | EGFR | EP300 | ERBB2 |
| FAM123B | FBXW7 | FGFR2 | FGFR3 | FLT3 | FOXL2 | FUBP1 | GATA1 |
| GATA3 | GNA11 | GNAQ | GNAS | GRIN2A | HNF1A | HRAS | IDH1 |
| IDH2 | IKZF1 | ITK | JAK2 | JAK3 | KDM5C | KDM6A | KDR |
| KIT | KRAS | LRP2 | MAP2K1 | MAP2K2 | MAP2K4 | MED12 | MEN1 |
| MET | MLH1 | MLL2 | MLL4 | MPL | MSH2 | MSH6 | MYD88 |
| MYH11 | MYST4 | NCOA2 | NF1 | NF2 | NOTCH1 | NOTCH2 | NPM1 |
| NRAS | NSD1 | NTRK1 | PAX5 | PDGFRA | PHOX2B | PIK3CA | PIK3R1 |
| POLR3A | PPP2R1A | PRKAR1A | PTCH1 | PTEN | PTPN11 | PTPRC | RB1 |
| RET | RNF213 | RNF43 | ROS1 | RUNX1 | SETD2 | SF3B1 | SMAD2 |
| SMAD4 | SMARCA4 | SMARCB1 | SMO | SOCS1 | STK11 | TET2 | TGFBR2 |
| TNFAIP3 | TOP1 | TP53 | UTX | VHL | WT1 |
Fluorescence in situ hybridization (FISH) and immunohistochemistry (IHC) for HER2
To assess the accuracy of the gene copy number variation found by our method, HER2 IHC and FISH test were performed as previously described [13]. Briefly, paraffin-embedded blocks of gastric tumors were cut into 4-µm sections. The sample sections together with known positive and negative control sections were baked at 65°C for at least 1 hour, then deparaffinaged and rehydrated. The primary antibody used for IHC was an antibody to HER2 (4B5, Ventana Medical Systems). Antigen epitopes were retrieved by heating at 100°C for 60 min with EDTA, pH 8.5 (Ventana Benchmark CC1 standard program). HER2 IHC was scored based on the ToGA trial screening criteria [13]. HER2 amplification was determined by FISH assays using HER2 FISH pharmDX™ (Dako Denmark A/S) according to the manufacturer’s instruction. HER2 (red) and centromeric probe 17 (CEP17, green) signals per nuclei were counted. A HER2-CEP17 ratio of ≥ 2 was defined as positive for HER2 amplification; chromosome 17 polysomy was defined as ≥ 3 CEP17 signals per cell on average.
Statistical methods
Overall survival in relation to mutation status was evaluated by the Kaplan-Meier survival curve and the log-rank test. The Pearson chi-square test was used for analyzing correlations between cluster membership and clinical-pathological variables (SPSS 11.5.0 for Windows, SPSS Inc, Chicago, IL, USA). A P value of less than 0.05 was taken as statistically significant. Pathway enrichment analysis was performed by online DAVID analysis (version 6.7) [14].
Results
Sequencing results
Of the 78 advanced gastric cancer patients (stage III and IV), 85.9% of them are males. The median age is 59 years old (28-79), with 14.1% ≤ 45 and 47.4% > 60. According to Lauren classification, 33.3% are intestinal-type gastric cancer (IGC), 50% are diffuse-type gastric cancer (DGC), and 16.7% are mixed-type gastric cancer (MGC). For original tumor location, 48.7% are at upper 1/3 of stomach (cardia, GE junction and fundus), 32.1% are at middle 1/3 (gastric body), and 19.2% are at lower 1/3 (antrum origin tumors). Up to May 2015, all patients had been evaluated for clinical response. As the first line chemotherapy, 55 received fluorouracil plus cisplatin and 23 were treated with fluorouracil plus paclitaxel. Thirty-three of them received stable disease (SD), 33 received partial response (PR) and 12 received progressive disease (PD). In the end of the follow-up, 60 patients had progressed in disease, and 49 patients had died.
We sequenced 1,051,300 bp target region bases for each sample, achieving an average of 400× coverage for each target. In total, we identified 22,532 mutations and 293 indels, including 258 high-accuracy non-silent somatic point mutations in 87 different genes, and 43 somatic indels in 23 different genes after the following multistep filtering process: 1) tumor sample mutations were filtered with peripheral blood mutations; 2) mutations were filtered with normal person databases (1000 genome database version 2012/ESP6500 database/Inhouse normal person database); 3) variant allele fraction (VAF) > 0.05, and all synonymous mutations were deleted; 4) continuous two or more point mutations of same gene were considered as noise. Sixty-five of the 78 patients were detected with somatic mutations in the 118 genes; the median and mean mutant genes per sample were 3 and 3.5 (range, 0-25) respectively. The G>A and C>T transition mutations were dominated, which is consistent with previous studies [6]. We also found 37 copy number gain events among 15 different genes (fold change 2-12), including 11 patients with ERBB2 overexpression (fold change 2-12), 5 patients with MET overexpression (fold change 2-6), 4 patients with CDK12 overexpression (fold change 2-9), and 3 patients with FGFR2 overexpression (fold change 3-6).
Of the 92 mutated genes, 12 were reported as frequently mutated in GC. They are TP53 (34.6%), KMT2D (15.4%), KMT2B (11.5%), PIK3CA (9.0%), EGFR (9.0%), GNAQ (9.0%), ARID1A (7.7%), EP300 (6.4%), NOTCH1 (6.4%), BRCA2 (5.1%), CTNNB1 (3.8%), and CDH1 (3.8%). We also uncovered 5 previously unreported genes. They are TGFBR2 (15.4%), NF1 (9.0%), RNF213 (9.0%), NSD1 (7.7%), and LRP2 (7.7%). TGFBR2, located in 3p24.1, is a member of TGFB receptor superfamily that forms a heterodimeric complex with TGFBR1. This complex binds TGF-β and regulates the transcription of a subset of cell proliferation related genes. Ten of the 12 patients detected with TGFBR2 mutations harbored E125fs deletion or insertion, which was confirmed as somatic in chronic myelomonocytic leukemia [15], but was first reported in GC cohort. Identification of mutations in NF1 (located in 17q11.2) has been challenging due to its large size, lack of mutational hot spots, and the presence of pseudogenes [16]. NF1 negatively regulate RAS and plays a role in the control of cell growth, with the mutation rate 10% in TCGA GC cohort [7]. RNF213, located in 17q25.3, is involved in angiogenesis via the non-canonical Wnt signaling pathway inhibition and promoting vessel regression [17], highly mutated in desmoplastic melanoma (25%) [18] and 11.1% patients mutated in TGCA GC cohort. NSD1 (located in 5q35.3) is a histone methyltransferase which enhances androgen receptor transactivation, frequently mutated in head and neck squamous cell carcinoma (11.8%) [19], and 7.3% patients mutated in TCGA GC cohort.
Gene alterations and clinicopathological factors
When comparing mutations between intestinal type gastric cancer (IGC) and diffuse type gastric cancer (DGC), the total number of mutations between IGC and DGC is not significantly different (mean values, 3.5 [standard deviation (SD), 3.2] and 3.7 [SD, 5.0], respectively; P=0.83). TP53 non-silent mutation was more common in IGC (46.2% in IGC and 25.53% in DGC, P=0.028, Figure 1). Similar trend was observed in GNAQ as well (13.9% in IGC and 2.6% in DGC, P=0.023). Several cell cycle related genes, including TP53, were previously reported frequently altered in IGC [20]. TP53 mutations have been detected even in early lesions such as intestinal metaplasia [21]. While a total of 5 DGC patients have LRP2 mutations (K1805o, T2557A, P4538Q, G4097S, and R2175Q), 3 DGC patients have BRCA2 mutations (E3175o, K1489N, and K2496X), and 3 DGC patients have FGFR3 mutations (E320X, Q286R, and R401H), none IGC patients harbored these mutations. Only three patients have CDH1 mutations, with two DGC (D254N and T61I) and one mixed-type gastric cancer (I250S). Two DGC patients have APC mutations (S277G and A2760S), which is consistent with a previous report [22]. The frequencies of mutated spots are compared with TCGA GC cohort (Figure 2).
Figure 1.

Summary of the somatic mutations and Indels of all 78 patients. Tumors are divided into three groups: intestinal-type gastric cancer, diffuse-type gastric cancer and mixed-type gastric cancer and arranged from left to right by the number of non-silent mutations per sample in each group, shown in the top track. Clinical features are shown in the middle panel, primary tumor location are labeled as Up (upper 1/3 of stomach), Mid (middle 1/3 of stomach) and Low (lower 1/3 of stomach). Bottom panel shows the summary of somatic mutation of 38 significant mutated genes. Right bar chart shows the prevalence of each mutation category in each gene. (WT: wild type; FS indel: frameshift indel; AA del: amino acid deletion; IF indel: in-frame indel; OE: overexpression; NA: not available).
Figure 2.

Comparing hot spots between PUCH and TCGA cohorts of selected genes. Lollipop plots showing the type and location of mutations in TP53, GNAQ, LRP2, FGFR3, BRCA2 and TGFBR2. We compared our study (bottom plot; PUCH) with TCGA 286 gastric cancer patients (top plot; TCGA), percentage means mutation rate in each cohort.
Notably, ARID1A mutation was more frequent in younger patients (less than 45 years old), compared to patients between 45 and 60 and elder patients (older than 60) (36.4% vs. 0% vs. 5.4%, P<0.001). Additionally, TGFBR2 mutation was more frequent in lower 1/3 stomach (5.3% vs. 12.0% vs. 40.0%, P=0.004) than in upper and middle 1/3 stomach. A similar trend was also found in JAK2 (2.6% vs. 0% vs. 20.0%, P=0.013) and MET mutation (0% vs. 0% vs. 13.3%, P=0.013).
Pathway enrichment of mutations
We mapped high frequency mutations into different pathways, and found that several pathways were significantly enriched (Table 2). Cell movement related pathways (including adherens junction, focal adhesion and regulation of actin cytoskeleton), ERBB signaling, MAPK signaling and JAK-STAT pathway are the top enriched altered pathways. To find correlation between enriched pathways and prognosis, we analyzed association of mutated pathways with survival data. We found that patients with one or more mutations in adherens junction pathway (CREBBP, EP300, CDH1, CTNNB1, EGFR, MET, TGFBR2 and ERBB2) showed significantly better overall survival (P=0.007). This better prognostic significance was successfully verified in TCGA GC dataset with disease-free survival time (P=0.014). A similar trend was also found in TGF-β signaling pathway (CREBBP, EP300, MYST4, KRAS and TGFBR2), in that patients with mutations in one or more of these genes were associated with better overall survival and disease-free survival in PUCH (P=0.021) and TCGA cohort (P=0.032) (Figure 3).
Table 2.
Mutation pathway enrichment
| Category | Term | Count | % | P-value | Benjamini | genes |
|---|---|---|---|---|---|---|
| KEGG | Adherens junction | 8 | 21.6 | 1.30E-07 | 1.30E-06 | CREBBP EP300 CDH1 CTNNB1 EGFR MET TGFBR2 ERBB2 |
| KEGG | Focal adhesion | 7 | 18.9 | 6.40E-04 | 3.40E-03 | CTNNB1 EGFR MET PTEN PIK3CA ERBB2 BRAF |
| KEGG | ErbB signaling pathway | 5 | 13.5 | 1.20E-03 | 5.50E-03 | EGFR PIK3CA KRAS ERBB2 BRAF |
| KEGG | MAPK signaling pathway | 7 | 18.9 | 2.80E-03 | 1.20E-02 | EGFR FGFR3 NF1 TGFBR2 TP53 KRAS BRAF |
| KEGG | Regulation of actin cytoskeleton | 6 | 16.2 | 5.70E-03 | 2.40E-02 | APC EGFR FGFR3 PIK3CA KRAS BRAF |
| AKEGG | Jak-STAT signaling pathway | 5 | 13.5 | 9.60E-03 | 3.30E-02 | CREBBP EP300 JAK2 JAK3 PIK3CA |
| PANTHER | P00059: p53 pathway | 7 | 18.9 | 4.50E-04 | 1.20E-02 | CREBBP EP300 MYST4 PTEN PIK3CA ATM TP53 |
| PANTHER | P00057: Wnt signaling pathway | 9 | 24.3 | 5.60E-03 | 7.10E-02 | ARID1A CREBBP EP300 MYST4 APC CDH1 CTNNB1 GNAQ TP53 |
| PANTHER | P00018: EGF receptor signaling pathway | 6 | 16.2 | 6.50E-03 | 6.70E-02 | EGFR PIK3CA ATM KRAS ERBB2 BRAF |
| PANTHER | P00005: Angiogenesis | 7 | 18.9 | 1.00E-02 | 8.80E-02 | NOTCH1 APC CTNNB1 FGFR3 PIK3CA KRAS BRAF |
| PANTHER | P00031: Inflammation mediated by chemokine and cytokine signaling pathway | 8 | 21.6 | 1.30E-02 | 9.30E-02 | JAK2 JAK3 GNAQ MYH11 PTEN PIK3CA KRAS BRAF |
| PANTHER | P00048: PI3 kinase pathway | 5 | 13.5 | 1.60E-02 | 1.00E-01 | JAK2 GNAQ PTEN PIK3CA KRAS |
| PANTHER | P00052: TGF-beta signaling pathway | 5 | 13.5 | 4.20E-02 | 2.00E-01 | CREBBP EP300 MYST4 TGFBR2 KRAS |
| PANTHER | P00047: PDGF signaling pathway | 5 | 13.5 | 6.90E-02 | 2.90E-01 | JAK2 JAK3 PIK3CA KRAS BRAF |
| BIOCARTA | TGF beta signaling pathway | 5 | 13.5 | 8.60E-05 | 9.90E-03 | CREBBP EP300 APC CDH1 TGFBR2 |
Figure 3.
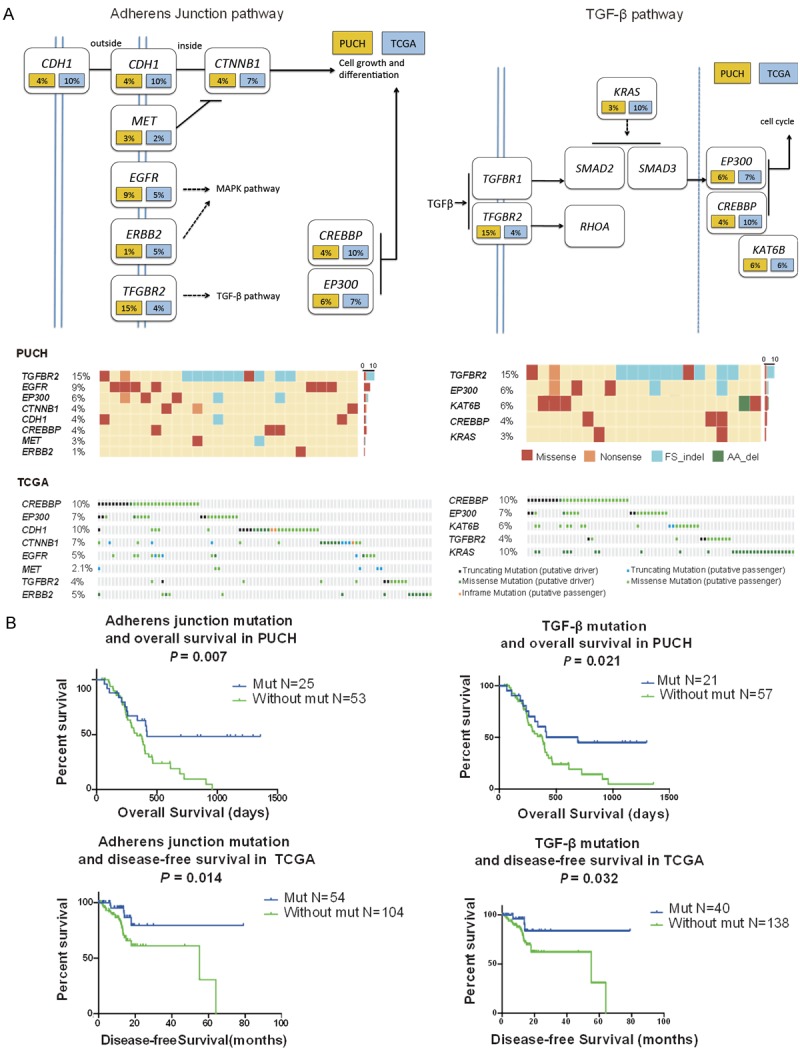
Altered genes in adherens junction and TGF-β pathways (PUCH and TCGA cohort), and association with prognosis. A. Oncomaps and schematics of genetic alterations in adherens junction pathway and TGF-β pathway, the frequencies (%) in PUCH cohort (yellow) and TCGA cohort (blue) are shown. B. Patients with one or more mutations in both pathways showed significantly better overall survival in PUCH cohort, and the same trend in disease-free survival data in TCGA cohort.
Potential druggable alterations
Using online database DGIdb 2.0 [23] for mutation druggability annotation, we found that 73.1% of AGC patients harbored at least one actionable alteration reported so far (Figure 4). These actionable targets included well-studied ERBB2 (n=13, 16.7%), EGFR (n=8, 10.3%), PIK3CA (n=8, 10.3%), MET (n=7, 9.0%), and NOTCH1 (n=5, 6.4%), as well as promising targets CDK12 (n=5, 6.4%) and BRCA2 (n=4, 5.1%), both are potential targets of PARP inhibitors.
Figure 4.

Distribution of potential druggable alterations observed in this study, based on DGIdb.
We also validated the sequencing-based ERBB2 copy number gains with routine clinical IHC and FISH test results (Figure 5), and showed that 9 out of 11 (81.8%) were correctly matched (Table 3).
Figure 5.
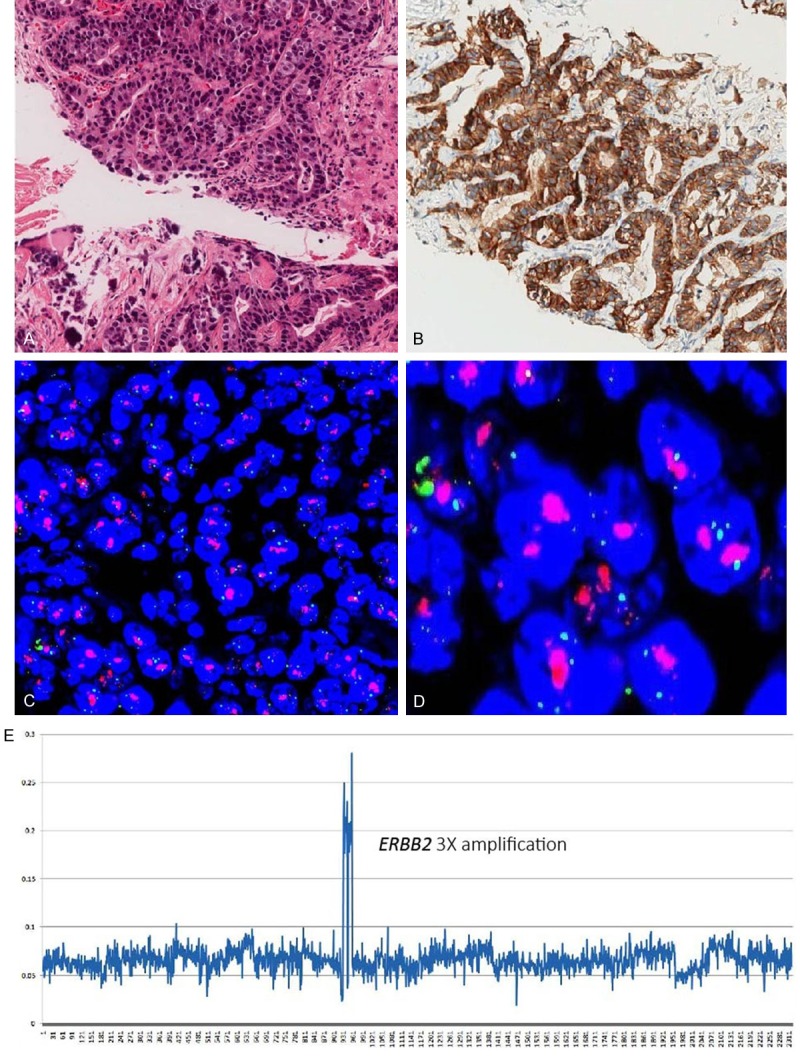
IHC and FISH validation of HER2 expression in Sample S884. A. H&E stain result; B. Strong membrane immunostaining for the HER2 protein, IHC score 3+; C and D. FISH test showed HER2 overexpression; E. NGS method detected HER2 amplification.
Table 3.
Sequencing-based, IHC, and FISH results of ERBB2 overexpression
| Sample | IHC | FISH | Fold change* | Sequencing-routine match |
|---|---|---|---|---|
| S906 | 2+ | + | 2 | yes |
| S928 | 3+ | + | 2 | yes |
| S867 | 3+ | + | 3 | yes |
| S876 | 3+ | NA | 3 | yes |
| S884 | 3+ | + | 3 | yes |
| S959 | 3+ | + | 3 | yes |
| S878 | NA | - | 4 | no |
| S954 | 3+ | + | 4 | yes |
| S879 | 3+ | + | 5 | yes |
| S926 | 1+ | - | 9 | no |
| S941 | 3+ | + | 12 | yes |
NA, not available;
sequencing-based copy number change results.
Discussion
An increasing number of NGS studies focusing on gastric cancer has been released in the past decade [6,8,24-28]. The most comprehensive study by TCGA [7] has classified gastric cancer into four distinct molecular subtypes: EBV positive, MSI, CNV and genome stable. Along with other high-quality studies, these investigations provided the molecular portraits of GC. However, all were based on operation-acquired tissues, which could lead to an underneath potential bias that the later stages GC patients were lacking in this landscape. To our knowledge, this is the first study using NGS to evaluate large scale AGC patients using biopsy tumor samples. Systematic comparison of mutation differences between early-stage and advanced stage GCs are not available by now, though publications of other tumor types showed contrasting results of same mutation in different clinical stages, for instance, in early-stage colorectal cancer (CRC) patients, the overall survival between BRAF mutant and wild-type patients differed non-significantly, while in late-stage patients, BRAF mutant patients showed significantly poorer overall survival [29].
Our study identified several previously reported GC-associated genes (eg, TP53, KMT2D, KMT2B, EGFR, PIK3CA, GNAQ, and ARID1A), which confirmed the importance of these genes in GC development. In addition, we found several recurrently mutated genes (TGFBR2, RNF213, NF1, NSD1, LRP2), particular TGFBR2, with a mutation rate of 15.9%. Ten of the 12 mutated patients were detected with deletion or insertion at the same spot E125fs, which is reported for the first time in gastric cancer. Twenty to thirty percent of all CRCs, and 90% of microsatellite unstable (MSI) CRCs have TGFBR2 mutations [30], and several reports have indicated that TGFBR2 mutations may be associated with significantly improved survival in MSI colon cancer patients [31]. TGFBR2 mutation was reported in the ovarian metastatic clone instead of primary site in a diffuse-type gastric cancer patient, and gastric organoid modeling experiment showed its metastasis suppressor activity [32].
Importantly, we found that mutations in adherens junction and TGF-β signaling pathways were associated with better overall survival time, and the prognosis significance of both signatures was successfully confirmed in TCGA cohort. While the adherens junctions and focal adhesions as the top two enriched pathways has been reported previously in a 100 patients’ cohort [25], the relationship with good prognosis is reported for the first time here. TGF-β can act as a tumor suppressor at early stages of tumorigenesis, but enhance tumor progression, invasion and metastasis as a major inducer of epithelial to mesenchymal transition at later stages [33]. Our results may help differentiate GC patients with dissimilar survival outcomes, and determine whether more aggressive chemotherapy or novel targeted therapy could be used on those patients with predicted poor prognosis.
Identification of new therapeutic approached is one of the important goals that NGS can aid clinicians [34]. An increasing number of clinical trials with novel small molecular compounds are launched at different stages of clinical trials, mostly started with advanced stages of patients due to minimal harm. In this AGC cohort, we discovered that more than 70% of patients may potentially benefit from existing small molecule compounds (DGIdb). Among them, 8 patients had oncogenic PIK3CA mutation (E726K, E950o, M811T, M1043V, V850fs, E545K, and W11C). E545K was reported as a hot-spot mutation detected in 24% breast cancer patients [35]. A phase II clinical trial (NCT02451956) using AZD5363 (an AKT inhibitor) and paclitaxel in PIK3CA mutated or amplified patients has started at 2016 in South Korea [36]. Three patients were detected with FGFR2 amplification, which is the target of Dovitinib or AZD4547 in three phase II clinical trials worldwide (NCT01719549, NCT01921673 and NCT01457846).
In conclusion, our study of the genomic and clinical data of 78 advanced stage gastric cancer patients could help provide a more comprehensive genomic portrait of GC. We also identified individuals of AGC with potential targetable genomic alterations. Subgroup with adherens junction mutations or TGF-β pathway mutations predicts better prognosis. These findings could help devise personalized treatment strategies, and NGS of biopsy samples before and after therapy will also be used for prediction and monitoring tumor progressions in the future.
Acknowledgements
This work was supported by Beijing Municipal Administration of Hospital Clinical Medicine Development of Special Funding Support (ZYLX201701), National Natural Science Foundation of China (No. 81472789), and National Basic Research Program of China (No. 2014CBA02002).
Disclosure of conflict of interest
None.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Wadhwa R, Song S, Lee JS, Yao Y, Wei Q, Ajani JA. Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol. 2013;10:643–655. doi: 10.1038/nrclinonc.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen L, Shan YS, Hu HM, Price TJ, Sirohi B, Yeh KH, Yang YH, Sano T, Yang HK, Zhang X, Park SR, Fujii M, Kang YK, Chen LT. Management of gastric cancer in Asia: resource-stratified guidelines. Lancet Oncol. 2013;14:e535–547. doi: 10.1016/S1470-2045(13)70436-4. [DOI] [PubMed] [Google Scholar]
- 4.Nadauld LD, Ford JM. Molecular profiling of gastric cancer: toward personalized cancer medicine. J. Clin. Oncol. 2013;31:838–839. doi: 10.1200/JCO.2012.47.1714. [DOI] [PubMed] [Google Scholar]
- 5.Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, Lee SP, Ho SL, Chan AK, Cheng GH, Roberts PC, Rejto PA, Gibson NW, Pocalyko DJ, Mao M, Xu J, Leung SY. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–1223. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 6.Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, Lim KH, Ong CK, Huang D, Chin SY, Tan IB, Ng CC, Yu W, Wu Y, Lee M, Wu J, Poh D, Wan WK, Rha SY, So J, Salto-Tellez M, Yeoh KG, Wong WK, Zhu YJ, Futreal PA, Pang B, Ruan Y, Hillmer AM, Bertrand D, Nagarajan N, Rozen S, Teh BT, Tan P. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, Yamamoto S, Tatsuno K, Katoh H, Watanabe Y, Ichimura T, Ushiku T, Funahashi S, Tateishi K, Wada I, Shimizu N, Nomura S, Koike K, Seto Y, Fukayama M, Aburatani H, Ishikawa S. Recurrent gain-offunction mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet. 2014;46:583–587. doi: 10.1038/ng.2984. [DOI] [PubMed] [Google Scholar]
- 9.Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, Tsui WY, Ho SL, Chan AK, Man JL, Foglizzo V, Ng MK, Ching YP, Cheng GH, Xie T, Fernandez J, Li VS, Clevers H, Rejto PA, Mao M, Leung SY. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–582. doi: 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 10.Gao J, Lu M, Yu JW, Li YY, Shen L. Thymidine Phosphorylase/beta-tubulin III expressions predict the response in Chinese advanced gastric cancer patients receiving first-line capecitabine plus paclitaxel. BMC Cancer. 2011;11:177. doi: 10.1186/1471-2407-11-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, Wolfgang CL, Klein AP, Diaz LA Jr, Allen PJ, Schmidt CM, Kinzler KW, Papadopoulos N, Hruban RH, Vogelstein B. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66. doi: 10.1126/scitranslmed.3002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, Lehle M, Ruschoff J, Kang YK ToGA Trial Investigators. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 14.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 15.Mason CC, Khorashad JS, Tantravahi SK, Kelley TW, Zabriskie MS, Yan D, Pomicter AD, Reynolds KR, Eiring AM, Kronenberg Z, Sherman RL, Tyner JW, Dalley BK, Dao KH, Yandell M, Druker BJ, Gotlib J, O’Hare T, Deininger MW. Age-related mutations and chronic myelomonocytic leukemia. Leukemia. 2016;30:906–913. doi: 10.1038/leu.2015.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest. 2017;97:146–157. doi: 10.1038/labinvest.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scholz B, Korn C, Wojtarowicz J, Mogler C, Augustin I, Boutros M, Niehrs C, Augustin HG. Endothelial RSPO3 controls vascular stability and pruning through non-canonical WNT/Ca(2+)/NFAT signaling. Dev Cell. 2016;36:79–93. doi: 10.1016/j.devcel.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 18.Shain AH, Garrido M, Botton T, Talevich E, Yeh I, Sanborn JZ, Chung J, Wang NJ, Kakavand H, Mann GJ, Thompson JF, Wiesner T, Roy R, Olshen AB, Gagnon A, Gray JW, Huh N, Hur JS, Busam KJ, Scolyer RA, Cho RJ, Murali R, Bastian BC. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat Genet. 2015;47:1194–1199. doi: 10.1038/ng.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grabsch HI, Tan P. Gastric cancer pathology and underlying molecular mechanisms. Dig Surg. 2013;30:150–158. doi: 10.1159/000350876. [DOI] [PubMed] [Google Scholar]
- 21.Ochiai A, Yamauchi Y, Hirohashi S. p53 mutations in the non-neoplastic mucosa of the human stomach showing intestinal metaplasia. Int J Cancer. 1996;69:28–33. doi: 10.1002/(SICI)1097-0215(19960220)69:1<28::AID-IJC6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 22.Tahara E. Genetic alterations in human gastrointestinal cancers. The application to molecular diagnosis. Cancer. 1995;75:1410–1417. doi: 10.1002/1097-0142(19950315)75:6+<1410::aid-cncr2820751504>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 23.Wagner AH, Coffman AC, Ainscough BJ, Spies NC, Skidmore ZL, Campbell KM, Krysiak K, Pan D, McMichael JF, Eldred JM, Walker JR, Wilson RK, Mardis ER, Griffith M, Griffith OL. DGIdb 2.0: mining clinically relevant druggene interactions. Nucleic Acids Res. 2016;44:D1036–1044. doi: 10.1093/nar/gkv1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, Liu J, Yue YG, Wang J, Yu K, Ye XS, Do IG, Liu S, Gong L, Fu J, Jin JG, Choi MG, Sohn TS, Lee JH, Bae JM, Kim ST, Park SH, Sohn I, Jung SH, Tan P, Chen R, Hardwick J, Kang WK, Ayers M, Hongyue D, Reinhard C, Loboda A, Kim S, Aggarwal A. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449–456. doi: 10.1038/nm.3850. [DOI] [PubMed] [Google Scholar]
- 25.Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, Tsui WY, Ho SL, Chan AK, Man JL, Foglizzo V, Ng MK, Chan AS, Ching YP, Cheng GH, Xie T, Fernandez J, Li VS, Clevers H, Rejto PA, Mao M, Leung SY. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–582. doi: 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 26.Ushiku T, Ishikawa S, Kakiuchi M, Tanaka A, Katoh H, Aburatani H, Lauwers GY, Fukayama M. RHOA mutation in diffuse-type gastric cancer: a comparative clinicopathology analysis of 87 cases. Gastric Cancer. 2016;19:403–411. doi: 10.1007/s10120-015-0493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei Z, Tan IB, Das K, Deng N, Zouridis H, Pattison S, Chua C, Feng Z, Guan YK, Ooi CH, Ivanova T, Zhang S, Lee M, Wu J, Ngo A, Manesh S, Tan E, Teh BT, So JB, Goh LK, Boussioutas A, Lim TK, Flotow H, Tan P, Rozen SG. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology. 2013;145:554–565. doi: 10.1053/j.gastro.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 28.Shah MA, Khanin R, Tang L, Janjigian YY, Klimstra DS, Gerdes H, Kelsen DP. Molecular classification of gastric cancer: a new paradigm. Clin Cancer Res. 2011;17:2693–2701. doi: 10.1158/1078-0432.CCR-10-2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen KH, Lin YL, Liau JY, Tsai JH, Tseng LH, Lin LI, Liang JT, Lin BR, Hung JS, Chang YL, Yeh KH, Cheng AL. BRAF mutation may have different prognostic implications in early- and latestage colorectal cancer. Med Oncol. 2016;33:39. doi: 10.1007/s12032-016-0756-6. [DOI] [PubMed] [Google Scholar]
- 30.Grady WM. Polymerase slippage restoration of frameshifted TGFBR2 in colorectal cancer: a Novel Paradigm. Gastroenterology. 2015;148:1276–1279. doi: 10.1053/j.gastro.2015.04.023. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe T, Wu TT, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB 3rd, Hamilton SR. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344:1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nadauld LD, Garcia S, Natsoulis G, Bell JM, Miotke L, Hopmans ES, Xu H, Pai RK, Palm C, Regan JF, Chen H, Flaherty P, Ootani A, Zhang NR, Ford JM, Kuo CJ, Ji HP. Metastatic tumor evolution and organoid modeling implicate TGFBR2 as a cancer driver in diffuse gastric cancer. Genome Biol. 2014;15:428. doi: 10.1186/s13059-014-0428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts K, Bhatia K, Stanton P, Lord R. Proteomic analysis of selected prognostic factors of breast cancer. Proteomics. 2004;4:784–792. doi: 10.1002/pmic.200300633. [DOI] [PubMed] [Google Scholar]
- 34.Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome Med. 2015;7:80. doi: 10.1186/s13073-015-0203-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramirez-Ardila DE, Helmijr JC, Look MP, Lurkin I, Ruigrok-Ritstier K, van Laere S, Dirix L, Sweep FC, Span PN, Linn SC, Foekens JA, Sleijfer S, Berns EM, Jansen MP. Hotspot mutations in PIK3CA associate with first-line treatment outcome for aromatase inhibitors but not for tamoxifen. Breast Cancer Res Treat. 2013;139:39–49. doi: 10.1007/s10549-013-2529-7. [DOI] [PubMed] [Google Scholar]
- 36.Maron SB, Catenacci DV. Novel targeted therapies for esophagogastric cancer. Surg Oncol Clin N Am. 2017;26:293–312. doi: 10.1016/j.soc.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]