

Abstract
UHRF1 is an epigenetic regulator and perform pivotal functions in cell tumorigenesis. We found UHRF1 is increased in breast cancer and patients with high UHRF1 levels have poorer prognoses than those with low UHRF1 levels. However, the underlying mechanisms remain largely unknown. Here, we found overexpression UHRF1 indeed promoted cell proliferation and migration, whereas its downregulation had the opposite functions. In vivo, UHRF1 also accelerated tumor growth. Mechanistically, microarrays were performed in MDA-MB-231 sh-UHRF1 and NC cells and KLF17, with rich CpG islands on its promoter region, finally caused our attention. Then, the expression of UHRF1 and KLF17 was testified negatively correlated in breast cancer cell lines and tissues. Additionally, the inhibition of cell proliferation and migration by UHRF1 depletion can be rescued by KLF17 silencing, suggesting KLF17 is downstream gene of UHRF1. The potential mechanism is that overexpression UHRF1 increased methylation of CpG nucleotides on KLF17 promoter, while UHRF1 silence decreased methylation. Collectively, our results demonstrated that increased UHRF1 can promote breast cancer cell proliferation and migration via silencing of KLF17 expression through CpG island methylation on its promoter.
Keywords: UHRF1, KLF17, proliferation, migration, hypermethylation
Introduction
Ubiquitin-like protein containing PHD and ring finger domain 1 (UHRF1), also known as ICBP90 in humans and Np95 in mice [1], was first isolated by Fujimori A in 1998 and was believed to be associated with cell proliferation due to its strong expression in the testis, spleen, thymus, and lung tissues [2]. As reported, UHRF1 contains multiple domains, including ubiquitin-like domain (UBL), tandem tudor domain (TTD), plant homeodomain (PHD), SET- and RING-associated domain (SRA, where RING domain denotes really interesting new gene domain), all of which are essential for biological functions in cells. The SRA domain was proven to identify hemi-methylated DNA and recruit DNA methyltransferase 1 (DNMT1) to hemi-methylated replication forks to complete DNA replication [3,4], and the RING domain has been shown to be associated with E3 ubiquitin ligase activity [5,6]. Studies have shown that the unmodified N-terminus of histone H3 was recognized by the PHD domain for participation in epigenetic modifications and that TTD domain specifically binds with histone H3 lysine 9 trimethylation (H3K9) for histone deacetylation [7].
In the last 15 years of research on UHRF1, additional detailed functions and mechanisms of UHRF1 were discovered. Previous studies demonstrated that UHRF1 was localized in S-phase nuclei and expressed strongly only in proliferative normal tissues and cells but maintained a constantly high level throughout all stages in cancer cells [8,9], suggesting that UHRF1 is a cell cycle regulator and is essential for cell S-phase entry functions as a checkpoint for G1/S transition. The imbalance of cell cycle processes might induce cell apoptosis. Indeed, other studies showed that downregulation of UHRF1 could induce cell apoptosis [10,11] and might cause a positive feedback loop between UHRF1 and apoptosis because p53/p21Cip1/WAF1-dependent DNA-damage checkpoint signals can lead to reduced UHRF1 expression, which might result in cell apoptosis through DNA-damage checkpoint signals [12]. Furthermore, UHRF1 was revealed to bind a CCAAT box of the topoisomerase II alpha gene promoter and increase its expression in non-proliferating conditions while inhibiting RB expression by combining with the same sites on the RB promoter regions [13,14].
As research has progressed, attention has moved to epigenetics. It was shown that UHRF1 contains a methyl DNA binding domain (SRA) for preferential binding to hemimethylated CG sites and collaborates with DNA methyltransferase protein (DNMT1) throughout S-phase to downregulate genes such as RB, ERa, and VEGF [15-17]. Additionally, recognition of hemi-methylated DNA by the SRA protein UHRF1 was reported as a base-flipping mechanism [18]. Methyltransferases 3A and 3B are also selectively enrolled in nucleosomes and the epigenetic silencing of the viral CMV promoter in embryonic stem cells [19]. UHRF1 also methylates histone H3 and acetylates or deacetylates histones. As shown, UHRF1 co-localizes with histone lysine methytransferase G9a to inhibit p21 promoter activity [20] and also interacts with tat-interactive protein 60 KD (tip60) to decrease the acetylation of its specific binding site H2AK5 [21]. Thus, UHRF1 indeed participates in chromosome stability and chromatin remodeling processes due to the two hallmark functions of UHRF1 methylation and histone H3 deacetylation.
Thus far, several target genes such as RB, ERa, VEGF, p21, and p16 are known to act as tumor suppressors can be downregulated by UHRF1 in primary human cancers. And certain achievements have been reported due to those discoveries, however, the mechanism of UHRF1 in tumorigenesis still remains unclear and requires additional research, especially in breast cancer. In our previous study, we reported that UHRF1 could inhibit MDR1 transcription by directly binding to its promoter and inducing the deacetylation of histones H3 and H4 on the MDR1 promoter [22]. We also found that UHRF1 was responsible for regulating BRCA1 transcription by inducing DNA methylation, histone modifications, and recruitment of transcriptional complexes on the BRCA1 promoter [23].
In our current study, we focus on validating potential downstream genes of UHRF1 by microarray in MDA-MB-231 model cells and finally find KLF17 is downstream gene of UHRF1, by which UHRF1 can promote breast cancer migration and proliferation. This study offers insight into breast cancer progression and suggests that making changes to this mechanism may represent new therapeutic approach to blocking breast cancer development.
Materials and methods
Cell culture
Normal breast cells MCF10A (ATCC) were cultured as previously reported [24]. The human breast cancer cell lines MCF-7, BT549, MDA-MB-468, MDA-MB-231, SK-BR-3 and T47D were obtained from the American Type Culture Collection (ATCC) (Manassas, VA, USA), and human embryonic kidney (HEK) 293T cells were purchased from the cell bank of the Chinese Academy of Sciences (CAS, Shanghai, China). All cells were cultured in completed medium mixed with 10% fetal bovine serum (Thermo, Beijing, China), according to ATCC instructions. 5-Aza-CdR was applied to cells for methylation inhibition, and the cells were exposed continuously at 5 μmol/L for two days (Sigma, St. Louis, MO, USA).
Patients and samples
Human breast cancer tissues were obtained with written informed consent from Shanghai Cancer Center at Fudan University. Ethics approval was obtained from the Committee of Ethical Research at Shanghai Cancer Center. Approximately 120 specimens (Table 1) of pathologically and normally diagnosed biopsy specimens (3 cm away from breast cancer tissues) were collected between 2009 and 2012 from patients with breast cancer, including 20 with normal tissues. We also collected clinical information from each patient, namely, age, tumor differentiation, and lymph node metastasis. All lesions were diagnosed according to the diagnostic criteria defined by the Evidence-Based Cancer Guidelines (NCCN, 2011).
Table 1.
Clinical-pathological variables and the expression of UHRF1 in the studied cases
| Variables | Number of patients (%) | UHRF1 expression | P value | |
|---|---|---|---|---|
|
| ||||
| Low N (%)a | High N (%)a | |||
| Total | 118 | 57 (45.6) | 61 (54.4) | |
| Age | 0.469 | |||
| ≤50 | 48 (40.7) | 25 (21.2) | 23 (19.5) | |
| >50 | 70 (59.3) | 32 (27.1) | 38 (32.2) | |
| Menopausal status | 0.752 | |||
| Premenopausal | 50 (42.4) | 25 (21.2) | 25 (21.2) | |
| Postmenopausal | 68 (57.6) | 32 (27.1) | 36 (30.5) | |
| Tumor size | 0.105 | |||
| ≤2 cm | 44 (37.3) | 17 (14.4) | 27 (22.9) | |
| >2 cm | 74 (62.7) | 40 (33.9) | 34 (28.8) | |
| Lymph node status | 0.029* | |||
| Negative | 72 (61.0) | 29 (24.6) | 43 (36.4) | |
| Positive | 46 (39.0) | 28 (23.7) | 18 (15.3) | |
| ER status | 0.100 | |||
| Negative | 57 (48.3) | 32 (27.1) | 25 (21.2) | |
| Positive | 61 (51.7) | 25 (21.2) | 36 (30.5) | |
| PR status | 0.854 | |||
| Negative | 59 (50.0) | 29 (24.6) | 30 (25.4) | |
| Positive | 59 (50.0) | 28 (23.7) | 31 (26.3) | |
| HER2 status | 0.233 | |||
| Negative | 81 (71.7) | 43 (38.1) | 38 (33.6) | |
| Positive | 32 (28.3) | 13 (11.5) | 19 (16.8) | |
| Unkown | 5 (4.2) | |||
| Ki67 | 0.442 | |||
| ≤14 | 35 (29.7) | 15 (12.7) | 20 (16.9) | |
| >14 | 83 (70.3) | 42 (35.6) | 41 (34.7) | |
| Grade | 0.184 | |||
| 1 | 6 (5.1) | 5 (4.2) | 1 (0.8) | |
| 2 | 88 (74.6) | 42 (35.6) | 46 (39.0) | |
| 3 | 24 (20.3) | 10 (8.5) | 14 (11.9) | |
| KLF17 level | 0.022* | |||
| Low | 68 (57.5) | 39 (33.1) | 29 (24.5) | |
| High | 50 (42.4) | 18 (15.3) | 32 (27.1) | |
The increased 1.5 fold UHRF1 expression was used as the cutoff.
Indicates P value <0.05.
RNA preparation and reverse transcription of cDNA
Total RNA was extracted from breast cancer cells treated with sh-RNA or overexpression vectors using TRIzol (Life Technologies, Rockville, MD, USA). First-strand cDNA was synthesized from 1 μg total RNA with Primer Mix and RT Enzyme Mix (Toyobo, Japan), according to the manufacturer’s instructions.
Quantitative real-time PCR
Real-time PCR was performed in a 20 μL reaction, which included 3 μL cDNA templates, 5 µM each of forward and reverse primer, and 10 μL SYBR buffer (Takara, Tokyo, Japan). The working conditions were 95°C for 5 min, followed by 95°C for 40 cycles and finally 60°C for 30 s. The primer pairs were used as follows: UHRF1 forward primer: ACACTTGGCTAGTCGTTAATGC, reverse primer: TATGGCCGTCCTCCATCTGT; KLF17 forward primer: AGGGGATGGTGCGATAGATT, reverse primer: GCCTCACCCTCACCTAACAAA; GAPDH forward primer: CCACTAGGCGCTCACTGTT, reverse primer: TGGAATTTGCCATGGGTGGA.
Methylation-specific PCR (MSP)
Genomic DNA was extracted from fresh breast cancer samples and cells as mentioned above using a Genomic DNA Purification Kit (Qiagen, Hilden, Germany). The extracted DNA (1 µg) underwent bisulfite conversion using a sodium bisulfite procedure with the EZ DNA Methylation-Gold Kit (Qiagen, Valencia, Canada), according to the manufacturer’s protocol. The MSP methylation primers were designed by Methyprimer as follows: methylation primers: TTTAGGTTGGAGTGTAATGGC, ATTAACCAAACGTAATAACGCGTA, unmethylation primers: GTTGTTTAGGTTGGAGTGTAATGGT, AATTAACCAAACATAATAACACATA. The PCR products were separated on 2% agarose gels and visualized with ethidium bromide staining.
Bisulfite sequencing
Each of the DNA samples was amplified by PCR using bisulfite primers: TTTGTTGTTTAGGTTGGAGTGTAAT, AATCACTTAAAATCAAAAATTTAAAACC.
The PCR products were cloned into pMD-18T (TaKaRa, Japan) according to the manufacturer’s instructions, and ten positive clones were sequenced. The data were analyzed using the QUMA analyzer software [25].
Construction of stable UHRF1 cell lines
Lentiviral and retroviral production 293T cells were used in lentiviral production. Lentiviral vectors expressing shRNAs against human KLF17 from the shRNA library were purchased from Genechem (Shanghai, China). Cells were transfected with lentiviral vectors following standard procedures, and viral supernatant was used to infect MDA-MB-231, ZR-75-30 and MCF-7 cells. The cells were treated with puromycin (2 μg/ml) for 48 h.
Cell migration assay
A cell suspension of 1×105 cells in 0.2 ml DMEM medium without FBS was seeded into each well of the upper trans-well chamber, and 500 µL 10% FBS was added to the lower chamber (Corning Costar Corp, America). After incubation for 9 h, the chambers were fixed and stained with 2% crystal violet for 25 min. The number of cells penetrating across the membrane was counted under a microscope in 7 random visual fields.
Cell proliferation assay
In brief, control (NC) and transfected cells were seeded at a density of 1×104 cells/well in a 96-well plate. The proliferation assay was performed every 24 h using CCK8 reagent (Dojindo, Japan). The supernatant was measured using a microplate reader at a wavelength of 450 nm. Each experiment was performed in three parallel wells and was repeated three times.
Mouse experiments
Four- to six-week-old athymic nude mice were injected orthotopically with 5.0×106 cells into the mammary fat pads. After 7 weeks of implantation, the tumors were harvested, weighed and fixed in formalin for further investigation.
Statistical analysis
The means ± the standard deviation (S.D.) were calculated and presented for each data point. Statistical analyses were performed using a paired Student’s t test. For all experiments, P values <0.05 were considered statistically significant.
Results
UHRF1 expression is significantly increased in breast cancer and associated with poor prognosis
To verify UHRF1 expression in breast cancer, we first tested its expression in 20 paired breast cancer and juxtacancerous tissues using real time-PCR assay. The results showed that expression of UHRF1 in breast cancer (BC) tissues was significantly elevated compared with normal or adjacent tissues (Figure 1A). Similar results were also obtained from the Oncomine database according to Curtis breast statistics (Figure 1B). We also observed that the level of UHRF1 was upregulated in several breast cancer cell lines, including MCF-7 and SK-BR-3 etc, compared with normal breast epithelial cells MCF-10A (Figure 1C). Given that the clinical value lay in whether increased UHRF1 expression could impact patient prognosis, we interrogated the Kaplan-Meier plotter database and found that patients with higher UHRF1 levels have shorter disease-free survival times than those with lower UHRF1 expression (Figure 1D).
Figure 1.
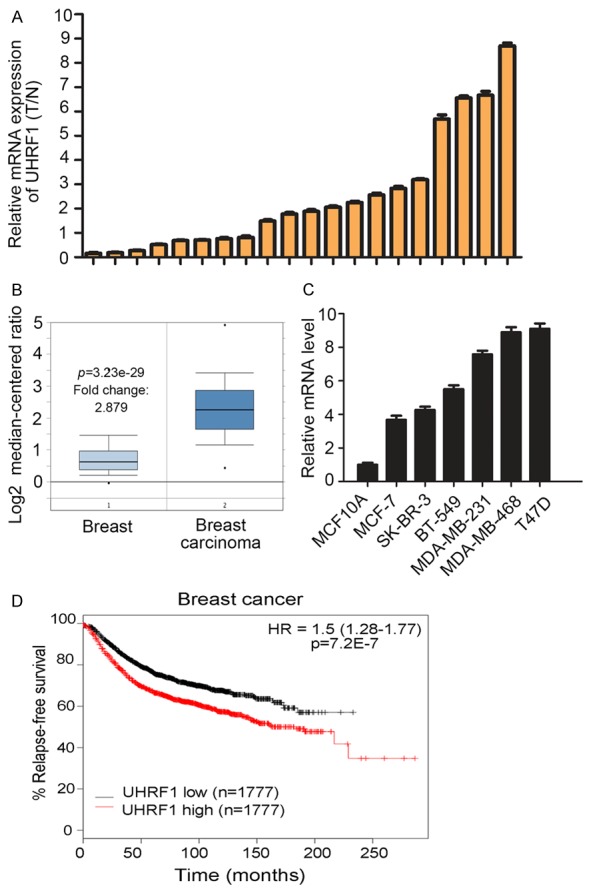
UHRF1 expression is increased in breast cancer and associated with poor prognosis. A. Quantitative analysis of UHRF1 expression level in breast cancer tissues and adjacent normal tissues (n = 20, breast cancer vs. adjacent). B. UHRF1 expression in breast cancer according to Oncomine analysis. C. UHRF1 expression in normal human breast epithelial cells and breast cancer cells. D. Prognosis of patients in two groups based on UHRF1 expression was analyzed by Kaplan-Meier.
UHRF1 expression affects breast cancer cells growth and migration
Stable overexpression and knockdown cell lines were constructed, and the transfected efficiency was validated by western blotting, as presented in Figure 2A and 2B. The cell proliferation was tested by CCK8 assay, and the results showed that overexpression of UHRF1 can significantly promote cell growth, whereas knockdown of UHRF1 expression impedes cell proliferation (Figure 2C). In addition, cells with upregulated UHRF1 have stronger migration ability than vector (PCDH) cells, and the depletion of UHRF1 presents an opposite result (Figure 2D). In vivo, MDA-MB-231 cells transfected with UHRF1 or PCDH were subcutaneously inoculated into nude mice, and the results showed that UHRF1 could accelerate tumor growth (Figure 2E).
Figure 2.
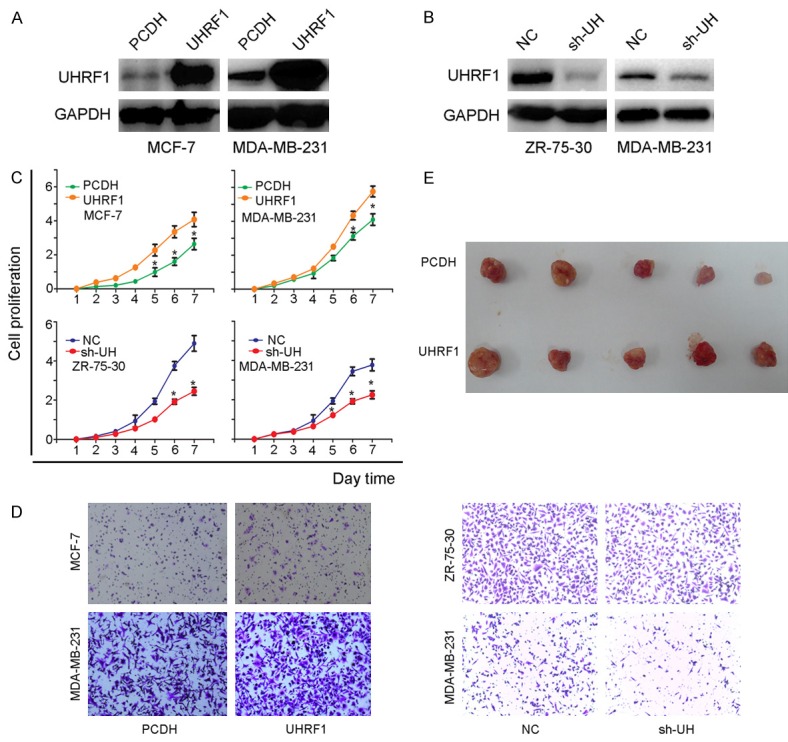
UHRF1 is associated with breast cancer progression. A and B. Stable cell lines were constructed and validated by western blot (overexpression: MCF-7, MDA-MB-231; knockdown: ZR-75-30, MDA-MB-231). C. Proliferation of stable cell lines. D. Migration of stable cell lines. E. Role of UHRF1 in tumor growth in vivo.
KLF17 is a candidate downstream gene of UHRF1
To investigate the potential mechanism of UHRF1 associated with breast cancer, gene microarray was employed to MDA-MB-231 cell models. Based on database analysis from the microarray, approximately 3000 genes changed when the absolute cut-off value was set to 2-fold (Figure 3A). However, when combined with a PubMed search, 38 candidate genes were obtained by analysis from the microarray databases and oncogenes or TSGs search on PubMed. The candidate genes were firstly testified in MDA-MB-231 cells transfected with PCDH or UHRF1 (Figure 3B). Then 23 genes were chose to further confirm in MDA-MB-231 NC and sh-UHRF1 cells. The results showed that only 4 genes were downregulated in UHRF1 overexpression cells and upregulated in UHRF1 downregulation cells. Among them, Krüppel-like factor 17 (KLF17) with rich CpG islands on its promoter was most remarkably one and cause our attention (Figure 3C).
Figure 3.
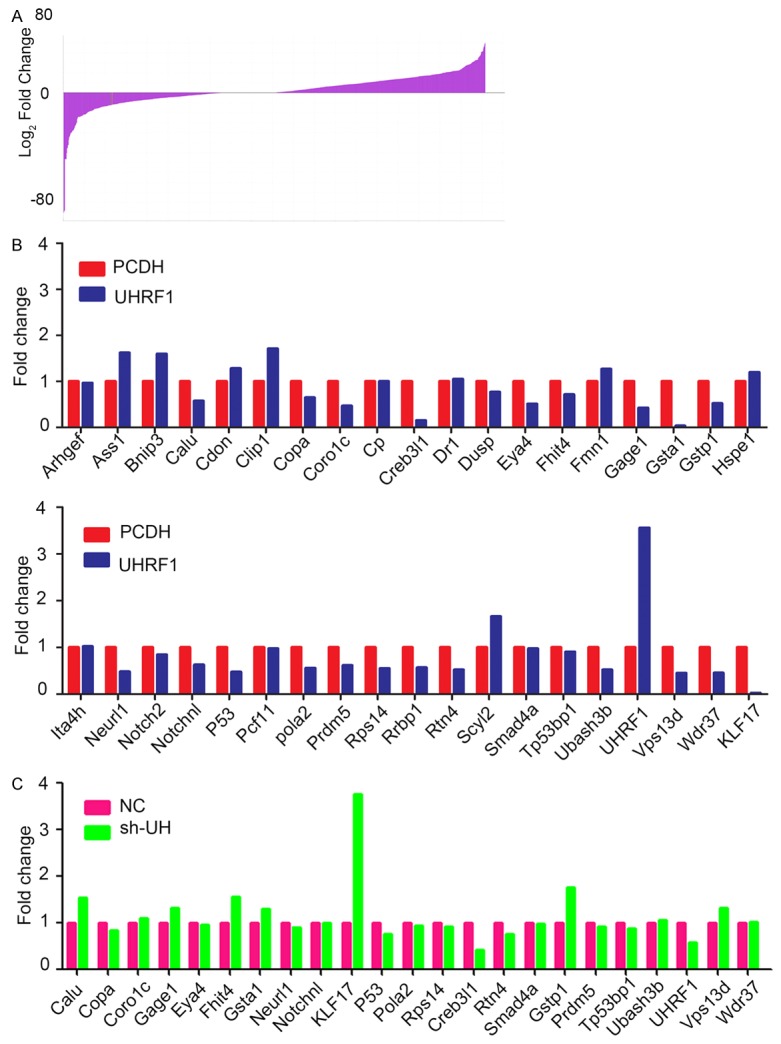
Candidate downstream genes of UHRF1 were screened by microarray. A. Fold changes of the total genes from microarray data (sh-UHRF1 vs. NC). B. Candidate genes firstly testified in UHRF1 overexpression cells by real-time PCR. C. The pickup genes further verified in UHRF1 knockdown cells by real-time PCR.
UHRF1 promotes cancer cell progression by down-regulating KLF17 expression
To further investigate the relationship between UHRF1 and KLF17, PCR or real-time PCR was performed, and the results showed that KLF17 genes were increased in sh-UHRF1 cells vs. NC cells (Figure 4A). In contrast, the overexpression of UHRF1 in MCF-7 cells showed a decreased expression of KLF17 compared with vector (PCDH) cells (Figure 4B). In addition to the stable cell models, we examined this relationship in several breast cancer cell lines and 20 paired tissues. Similar findings of this inverse correlation were also found in the cell lines and tissues (Figure 4C and 4D). In addition, the UHRF1 mRNA levels of 118 breast cancer samples were detected by real-time PCR and were divided into low- and high-expression groups after setting the cut-off to 1.5-fold. The correlation between UHRF1 and KLF17 expression and clinicopathological characteristics in breast cancer is presented in Table 1. The results showed that UHRF1 displays correlations with lymph node status and KLF17 expression. The above results indicated that UHRF1 has a negative correlation with KLF17. Then we performed rescue experiments to inspect whether UHRF1 promotes breast cancer progression through downstream gene KLF17 using three stable cell lines with NC, sh-UHRF1 and sh-UHRF1+sh-KLF17 groups (Figure 4E). We found that the decreased proliferation and migration by sh-UHRF1 can be partially rescued by KLF17 silencing, indicating that KLF17 might be a potent downstream gene of UHRF1 (Figure 4F and 4G).
Figure 4.
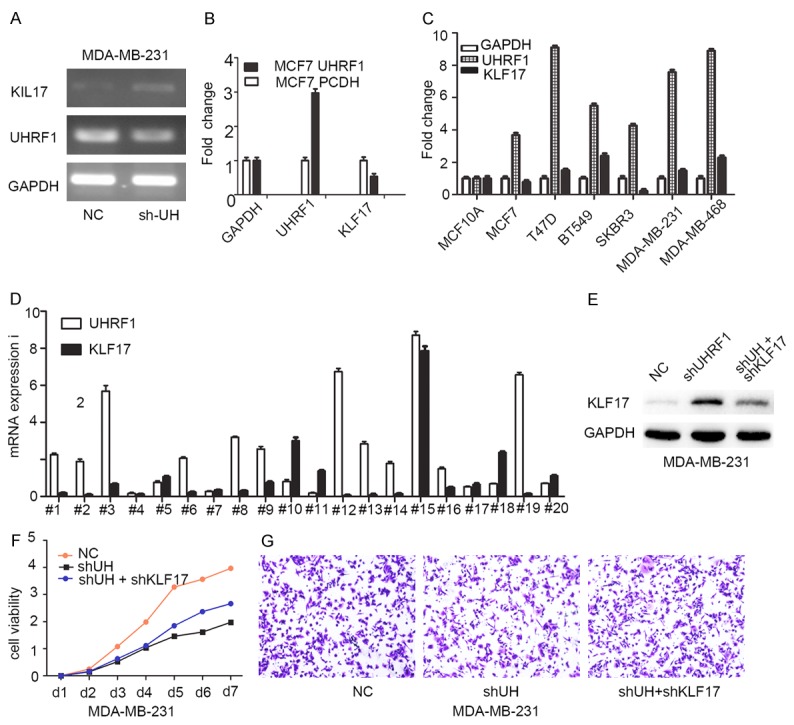
UHRF has an inverse relationship with KLF17 expression. A. Increased KLF17 expression in sh-UHRF1 cells. B. Downregulation of KLF17 in overexpression cells. C. Inverse relationship between UHRF1 and KLF17 levels in several breast cancer cell lines. D. Inverse relationship between UHRF1 and KLF17 level in 20 paired breast cancer tissues. E. Constructed stable cell lines for rescue experiments. F. Proliferation inhibited by sh-UHRF1 can be rescued by sh-KLF17. G. Rescued migration experiments in three stable cell lines.
KLF17 gene promoter is methylated by UHRF1
UHRF1 is acknowledged as an epigenetic factor of DNA methylation, and a CpG island also exists on the KLF17 promoter (Figure 5A). Thus, we questioned whether KLF17 downregulation by UHRF1 is associated with DNA methylation. MDA-MB-231 and MCF-7 cells were treated with 5-aza-dC, an inhibitor of methylation, and we observed that KLF17 expression was augmented in the 5-aza-dC group compared with the vehicle group (Figure 5B), suggesting that KLF17 expression can be regulated by methylation. Bisulfite sequencing PCR (BSP) was performed to detect CpG island methylation status, and the results revealed that the overexpression of UHRF1 increases the methylation of CpG islands and reduces the expression of KLF17, whereas depletion of UHRF1 decreases the methylation of CpG nucleotides with elevated expression of KLF17 (Figure 5C). Further, the promoter methylation statuses were also detected by MSP in 8 breast cancer samples, and the majority of them were methylated, suggesting that KLF17 downregulation by UHRF1 may be caused by DNA methylation (Figure 5D).
Figure 5.
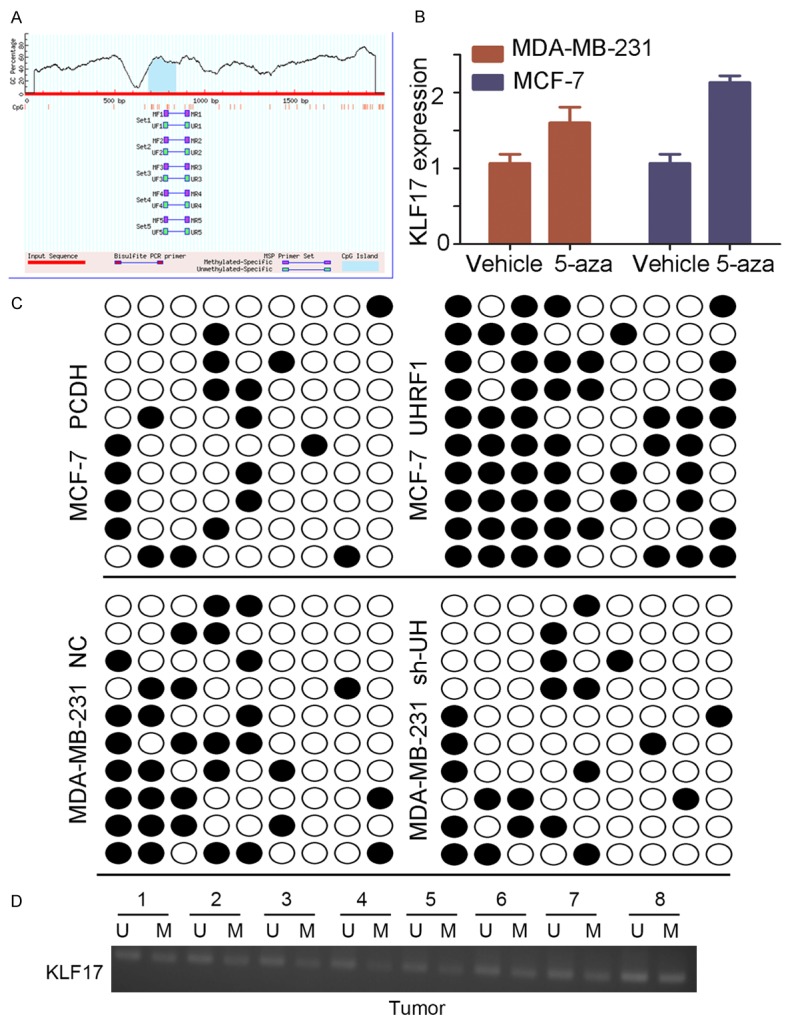
UHRF1 reduces the expression of Klf17 expression by increasing methylation of CpG nucleotides. A. Predicted CpG islands in KLF17 promoter. B. KLF17 expression in MDA-MB-231 and MCF-7 cells before and after 5-aza-CdR treatment. C. CpG island methylation status of KLF17 gene in stable cell lines. QUMA analysis revealed that KLF17 methylation frequency is markedly increased in UHRF1 overexpression cells and decreased in sh-UHRF1 cells. Ten individual clones are shown per cell line. CpG dinucleotides are represented as dark squares for methylated cytosines and as open squares for unmethylated cytosines. D. CpG island methylation status of KLF17 gene in breast cancer tissues analyzed by MSP.
Discussion
Breast cancer is recognized as a worldwide disease with high incidence and mortality [26]. Although great achievements have been made in new anti-cancer agents such as small molecular inhibitor, monoclonal antibody target drug, chemotherapy regimens etc., many obstacles still remain in breast cancer treatment [27,28]. Therefore, it is essential to explore the crucial genes and underlying mechanisms implicated in breast cancer.
A large body of evidence has suggested that UHRF1 plays a vital role in numerous carcinogenesis and cancer progression processes [29,30], and it has also been documented that UHRF1 is associated with breast cancer growth and metastasis, drug resistance and radiotherapy [31-33]. However, less is known as to how UHRF1 influences cancer cell progression. Therefore, in our study, we aimed to test the role of UHRF1 in breast cancer and focus on identifying a potential mechanism. The results show that UHRF1 indeed promotes cell proliferation and migration, and the mechanism is associated with downregulation of KLF17 level through CpG island methylation on its promoter.
Specifically, we found that the exogenous level of UHRF1 is elevated in breast cancer tissues and cell lines, increased UHRF1 promotes cell proliferation and migration, and patients with high UHRF1 expression are likely to have poor prognoses, similar to the results of previous studies [31,34,35]. Over the past years of studies on UHRF1, the majority have demonstrated that UHRF1 is an epigenetic modification factor associated with DNA methylation and histone deacetylation or methylation [36]. Based on previous studies, UHRF1 not only acts as an epigenetic factor but also as a transcription factor because it can bind to CCAAT sites to regulate expression of several genes, including RB, VEGF, ERα, etc [15-17]. Therefore, we used microarray techniques to examine potential downstream genes that might be useful as anti-cancer targets.
In previous studies, UHRF1 has been shown to be overexpressed and to coordinate tumor suppressor gene (TSG) silencing in several cancers due to its promoter hypermethylation. The investigated cases include kiss1 in bladder cancer, SOCS3 and 3OST2 in endometrial carcinoma [37], and MEG3 in hepatocellular carcinoma [38], suggesting that the role of UHRF1 in maintaining cancer cell progression was associated with TSGs promoter methylation. Thus, we applied MDA-MB-231 cell models in microarray analysis and obtained 4 candidate genes. The subsequent real-time PCR analysis validated that KLF17 is the most remarkable alteration one which also has CpG islands on its promoter.
Krüppel-like factor 17 (KLF17), belong to a family factor, has been identified as playing a negative role in the breast cancer EMT program [39] that a process in which epithelial cells obtain mesenchymal features and is closely associated with tumor metastasis. Several regulation factors for this process have been identified [40]. To further validate whether methylation is caused by decline of KLF17 in UHRF1 stable cell lines, MSP and BSP were performed, and the KLF17 promoter was indeed associated with methylation, which was also evidenced by treatment with the methylation inhibitor 5-aza-2’-deoxycytidine (5-aza-CdR) in cancer cell lines. However, we did not show how the methylation occurred, and this topic requires further investigation.
In conclusion, we found that UHRF1 is an oncogene in breast cancer that can promote breast cancer proliferation and migration. Using gene microarray, KLF17 was identified as a downstream gene of UHRF1 with CpG islands in its promoter. Our results demonstrate that increased UHRF1 can promote breast cancer cell proliferation and migration by epigenetic silencing of KLF17 expression. These results may offer insight into the breast cancer progression process and suggest that making changes to this mechanism may represent a new therapeutic approach to blocking breast cancer development.
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (81472669 and 81272923).
Disclosure of conflict of interest
None.
Abbreviations
- BSP
Bisulfite sequencing PCR
- BC
breast cancer
- UHRF1
Ubiquitin-like containing PHD and Ring Finger domain 1
- DNMT1
DNA methyltransferase1
- HCC
hepatic cell carcinoma
- KLF17
Krüppel-like factor 17
- MSP
Methylation-specific PCR
- NC
negative control
- PHD
Plant Homo domain
- QUMA
Quantification toll for methylation analysis
- Ring
Really Interesting New Gene
- SRA
Set and Ring associated
- TSGs
Tumor suppressor genes
- TTD
Tandem Tudor domain
- Tip60
Tat-interactive protein, 60 kDa
- UBL
Ubiquitin-like
- 5-aza-CdR
5-aza-2’-deoxycytidine
References
- 1.Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 2.Fujimori A, Matsuda Y, Takemoto Y, Hashimoto Y, Kubo E, Araki R, Fukumura R, Mita K, Tatsumi K, Muto M. Cloning and mapping of Np95 gene which encodes a novel nuclear protein associated with cell proliferation. Mamm Genome. 1998;9:1032–1035. doi: 10.1007/s003359900920. [DOI] [PubMed] [Google Scholar]
- 3.Unoki M, Nishidate T, Nakamura Y. ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene. 2004;23:7601–7610. doi: 10.1038/sj.onc.1208053. [DOI] [PubMed] [Google Scholar]
- 4.Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 5.Karagianni P, Amazit L, Qin J, Wong J. ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol Cell Biol. 2008;28:705–717. doi: 10.1128/MCB.01598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Citterio E, Papait R, Nicassio F, Vecchi M, Gomiero P, Mantovani R, Di Fiore PP, Bonapace IM. Np95 is a histone-binding protein endowed with ubiquitin ligase activity. Mol Cell Biol. 2004;24:2526–2535. doi: 10.1128/MCB.24.6.2526-2535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arita K, Isogai S, Oda T, Unoki M, Sugita K, Sekiyama N, Kuwata K, Hamamoto R, Tochio H, Sato M, Ariyoshi M, Shirakawa M. Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc Natl Acad Sci U S A. 2012;109:12950–12955. doi: 10.1073/pnas.1203701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uemura T, Kubo E, Kanari Y, Ikemura T, Tatsumi K, Muto M. Temporal and spatial localization of novel nuclear protein NP95 in mitotic and meiotic cells. Cell Struct Funct. 2000;25:149–159. doi: 10.1247/csf.25.149. [DOI] [PubMed] [Google Scholar]
- 9.Miura M, Watanabe H, Sasaki T, Tatsumi K, Muto M. Dynamic changes in subnuclear NP95 location during the cell cycle and its spatial relationship with DNA replication foci. Exp Cell Res. 2001;263:202–208. doi: 10.1006/excr.2000.5115. [DOI] [PubMed] [Google Scholar]
- 10.Abbady AQ, Bronner C, Trotzier MA, Hopfner R, Bathami K, Muller CD, Jeanblanc M, Mousli M. ICBP90 expression is downregulated in apoptosis-induced Jurkat cells. Ann N Y Acad Sci. 2003;1010:300–303. doi: 10.1196/annals.1299.052. [DOI] [PubMed] [Google Scholar]
- 11.Muto M, Kanari Y, Kubo E, Takabe T, Kurihara T, Fujimori A, Tatsumi K. Targeted disruption of Np95 gene renders murine embryonic stem cells hypersensitive to DNA damaging agents and DNA replication blocks. J Biol Chem. 2002;277:34549–34555. doi: 10.1074/jbc.M205189200. [DOI] [PubMed] [Google Scholar]
- 12.Arima Y, Hirota T, Bronner C, Mousli M, Fujiwara T, Niwa S, Ishikawa H, Saya H. Down-regulation of nuclear protein ICBP90 by p53/p21Cip1/WAF1-dependent DNA-damage checkpoint signals contributes to cell cycle arrest at G1/S transition. Genes Cells. 2004;9:131–142. doi: 10.1111/j.1356-9597.2004.00710.x. [DOI] [PubMed] [Google Scholar]
- 13.Hopfner R, Mousli M, Jeltsch JM, Voulgaris A, Lutz Y, Marin C, Bellocq JP, Oudet P, Bronner C. ICBP90, a novel human CCAAT binding protein, involved in the regulation of topoisomerase IIalpha expression. Cancer Res. 2000;60:121–128. [PubMed] [Google Scholar]
- 14.Jeanblanc M, Mousli M, Hopfner R, Bathami K, Martinet N, Abbady AQ, Siffert JC, Mathieu E, Muller CD, Bronner C. The retinoblastoma gene and its product are targeted by ICBP90: a key mechanism in the G1/S transition during the cell cycle. Oncogene. 2005;24:7337–7345. doi: 10.1038/sj.onc.1208878. [DOI] [PubMed] [Google Scholar]
- 15.Macaluso M, Montanari M, Noto PB, Gregorio V, Bronner C, Giordano A. Epigenetic modulation of estrogen receptor-alpha by pRb family proteins: a novel mechanism in breast cancer. Cancer Res. 2007;67:7731–7737. doi: 10.1158/0008-5472.CAN-07-1476. [DOI] [PubMed] [Google Scholar]
- 16.Achour M, Jacq X, Ronde P, Alhosin M, Charlot C, Chataigneau T, Jeanblanc M, Macaluso M, Giordano A, Hughes AD, Schini-Kerth VB, Bronner C. The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene. 2008;27:2187–2197. doi: 10.1038/sj.onc.1210855. [DOI] [PubMed] [Google Scholar]
- 17.Liu W, Qiao RH, Wang DM, Huang XW, Li B, Wang D. UHRF1 promotes human osteosarcoma cell invasion by downregulating the expression of Ecadherin in an Rb1 dependent manner. Mol Med Rep. 2016;13:315–320. doi: 10.3892/mmr.2015.4515. [DOI] [PubMed] [Google Scholar]
- 18.Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 19.Meilinger D, Fellinger K, Bultmann S, Rothbauer U, Bonapace IM, Klinkert WE, Spada F, Leonhardt H. Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep. 2009;10:1259–1264. doi: 10.1038/embor.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim JK, Esteve PO, Jacobsen SE, Pradhan S. UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic Acids Res. 2009;37:493–505. doi: 10.1093/nar/gkn961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Achour M, Fuhrmann G, Alhosin M, Rondé P, Chataigneau T, Mousli M, Schini-Kerth VB, Bronner C. UHRF1 recruits the histone acetyltransferase Tip60 and controls its expression and activity. Biochem Biophys Res Commun. 2009;390:523–528. doi: 10.1016/j.bbrc.2009.09.131. [DOI] [PubMed] [Google Scholar]
- 22.Jin W, Liu Y, Xu SG, Yin WJ, Li JJ, Yang JM, Shao ZM. UHRF1 inhibits MDR1 gene transcription and sensitizes breast cancer cells to anticancer drugs. Breast Cancer Res Treat. 2010;124:39–48. doi: 10.1007/s10549-009-0683-8. [DOI] [PubMed] [Google Scholar]
- 23.Jin W, Chen L, Chen Y, Xu SG, Di GH, Yin WJ, Wu J, Shao ZM. UHRF1 is associated with epigenetic silencing of BRCA1 in sporadic breast cancer. Breast Cancer Res Treat. 2010;123:359–373. doi: 10.1007/s10549-009-0652-2. [DOI] [PubMed] [Google Scholar]
- 24.Jiang HL, Sun HF, Gao SP, Li LD, Hu X, Wu J, Jin W. Loss of RAB1B promotes triple-negative breast cancer metastasis by activating TGFbeta/SMAD signaling. Oncotarget. 2015;6:16352–16365. doi: 10.18632/oncotarget.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008;36:W170–W175. doi: 10.1093/nar/gkn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 27.Kordbacheh T, Law WY, Smith IE. Sanctuary site leptomeningeal metastases in HER-2 positive breast cancer: a review in the era of trastuzumab. Breast. 2016;26:54–58. doi: 10.1016/j.breast.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Akin S, Babacan T, Sarici F, Altundag K. A novel targeted therapy in breast cancer: cyclin dependent kinase inhibitors. J BUON. 2014;19:42–46. [PubMed] [Google Scholar]
- 29.Wan X, Yang S, Huang W, Wu D, Chen H, Wu M, Li J, Li T, Li Y. UHRF1 overexpression is involved in cell proliferation and biochemical recurrence in prostate cancer after radical prostatectomy. J Exp Clin Cancer Res. 2016;35:34. doi: 10.1186/s13046-016-0308-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou L, Shang Y, Jin Z, Zhang W, Lv C, Zhao X, Liu Y, Li N, Liang J. UHRF1 promotes proliferation of gastric cancer via mediating tumor suppressor gene hypermethylation. Cancer Biol Ther. 2015;16:1241–1251. doi: 10.1080/15384047.2015.1056411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li XL, Xu JH, Nie JH, Fan SJ. Exogenous expression of UHRF1 promotes proliferation and metastasis of breast cancer cells. Oncol Rep. 2012;28:375–383. doi: 10.3892/or.2012.1792. [DOI] [PubMed] [Google Scholar]
- 32.Fang L, Shanqu L, Ping G, Ting H, Xi W, Ke D, Min L, Junxia W, Huizhong Z. Gene therapy with RNAi targeting UHRF1 driven by tumor-specific promoter inhibits tumor growth and enhances the sensitivity of chemotherapeutic drug in breast cancer in vitro and in vivo. Cancer Chemother Pharmacol. 2012;69:1079–1087. doi: 10.1007/s00280-011-1801-y. [DOI] [PubMed] [Google Scholar]
- 33.Li X, Meng Q, Rosen EM, Fan S. UHRF1 confers radioresistance to human breast cancer cells. Int J Radiat Biol. 2011;87:263–273. doi: 10.3109/09553002.2011.530335. [DOI] [PubMed] [Google Scholar]
- 34.Zhu M, Xu Y, Ge M, Gui Z, Yan F. Regulation of UHRF1 by microRNA-9 modulates colorectal cancer cell proliferation and apoptosis. Cancer Sci. 2015;106:833–839. doi: 10.1111/cas.12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang D, Xue H, Yu Y, Lv F, You W, Zhang B. Elevated expression of UHRF1 predicts unfavorable prognosis for patients with hepatocellular carcinoma. Int J Clin Exp Pathol. 2015;8:9416–9421. [PMC free article] [PubMed] [Google Scholar]
- 36.Rothbart SB, Dickson BM, Ong MS, Krajewski K, Houliston S, Kireev DB, Arrowsmith CH, Strahl BD. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev. 2013;27:1288–1298. doi: 10.1101/gad.220467.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H, Zhang C, Sheng Y, Yao S, Liu Z, Zhang C, Zhang T. Frequent SOCS3 and 3OST2 promoter methylation and their epigenetic regulation in endometrial carcinoma. Am J Cancer Res. 2015;5:180–190. [PMC free article] [PubMed] [Google Scholar]
- 38.Zhuo H, Tang J, Lin Z, Jiang R, Zhang X, Ji J, Wang P, Sun B. The aberrant expression of MEG3 regulated by UHRF1 predicts the prognosis of hepatocellular carcinoma. Mol Carcinog. 2016;55:209–219. doi: 10.1002/mc.22270. [DOI] [PubMed] [Google Scholar]
- 39.Gumireddy K, Li A, Gimotty PA, Klein-Szanto AJ, Showe LC, Katsaros D, Coukos G, Zhang L, Huang Q. KLF17 is a negative regulator of epithelial-mesenchymal transition and metastasis in breast cancer. Nat Cell Biol. 2009;11:1297–1304. doi: 10.1038/ncb1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang HL, Sun HF, Gao SP, Li LD, Huang S, Hu X, Liu S, Wu J, Shao ZM, Jin W. SSBP1 Suppresses TGFbeta-driven epithelial-to-mesenchymal transition and metastasis in triple-negative breast cancer by regulating mitochondrial retrograde signaling. Cancer Res. 2016;76:952–964. doi: 10.1158/0008-5472.CAN-15-1630. [DOI] [PubMed] [Google Scholar]