

Abstract
Osteosarcoma (OS) is the most common primary bone cancer in adolescents and children. Long noncoding RNAs (lncRNAs) contain > 200 nucleotides and do not have protein-coding ability. Liver-expressed LXR-induced sequence (LeXis) is a newly identified functional lncRNA. However, its expression pattern, biological function, and molecular mechanism in OS progression are unclear. The present study is the first to show that LeXis expression was upregulated in OS tissues. Increased LeXis expression was significantly correlated with high tumor stage, large tumor size, and poor prognosis. Our findings highlight the oncogenic activity of lncRNA LeXis in OS growth. Results of functional assays showed that LeXis promoted OS growth both in vitro and in vivo. Mechanistic investigation showed that LeXis directly interacted with miR-199a and suppressed its expression. Moreover, LeXis increased CTNNB1 expression by functioning as a ceRNA of CTNNB1 against miR-199a. These findings may have important implications for developing novel therapeutic strategies for OS.
Keywords: LeXis, miR-199a, CTNNB1, proliferation, ceRNA
Introduction
Osteosarcoma (OS) is the most common primary bone cancer in adolescents and children [1,2]. Despite recent advancements in surgical technology and chemotherapy for OS, the prognosis of patients with OS remains unfavorable. The 5-year survival rate of patients with OS having distant metastases is approximately 30% [3]. Therefore, there is an urgent need to understand molecular mechanisms underlying OS progression. Only 2% of encoded transcripts are translated into proteins, while majority of transcripts function as noncoding RNAs (ncRNAs). MicroRNAs (miRNAs) play a crucial in various biological processes such as cell proliferation, apoptosis, migration, and invasion [4,5]. miRNAs regulate gene expression by binding to the 3’ untranslated region (3’ UTR) of target mRNA, leading to its degradation or translational suppression [6]. Several studies have determined the functional roles of miRNAs in OS development. miR-199a directly targets and suppresses the expression of receptor tyrosine kinase AXL to inhibits OS progression [7]. Long noncoding RNAs (lncRNAs) are another class of ncRNAs containing > 200 nucleotides and lacking protein-coding ability. Dysregulation of lncRNAs is closely associated with human diseases, including cancers. Emerging evidences indicate that lncRNAs play important roles in gene expression through multiple mechanisms such as transcriptional regulation, epigenetic modification, and post-translational protein modification [8,9]. LncRNAs can act as oncogenes or tumor suppressor genes in OS. LncRNA MALAT1 is upregulated in OS tissues and predicts the poor prognosis of patients with OS. Moreover, MALAT1 promotes the growth and inhibits the apoptosis of OS cells. In addition, MALAT1 interacts with miR-142 and miR-129 to repress the degradation of high-mobility group protein B1 [10]. LIN00161 functions as a tumor suppressor in OS. LIN00161 overexpression enhances cisplatin-induced apoptosis and reverses cisplatin resistance in OS cells by sponging miR-645 and activating IFIT2 [11]. However, few lncRNAs have been functionally characterized in OS to date.
Liver-expressed liver X receptor (LXR)-induced sequence (LeXis) is a newly identified functional lncRNA whose expression increases robustly in response to the consumption of western diet or activation of pharmacologic LXRs. LeXis regulates the expression of genes involved in cholesterol biosynthesis and affects cholesterol level in the liver and plasma by interacting with ribonucleoprotein RALY [12]. However, the expression pattern, biological function, and molecular mechanism of LeXis in OS progression remain unclear. In the present study, we found that LeXis expression was significantly upregulated in OS tissues and that LeXis promoted OS growth by interacting with and suppressing miR-199a. Thus, our results suggest that LeXis is a promising therapeutic target for OS treatment.
Materials and methods
Cell culture and tissue samples
A normal osteoblast cell line (Nhost) and five different OS cell lines (KHOS, 143b, LM7, U2OS, and MG-63) were obtained from Cell Bank of Chinese Academy of Sciences. These cells were cultured in DMEM medium containing 10% fetal bovine serum (FBS, Gibco), 100 mg/ml penicillin and 100 U/ml streptomycin at 37°C. The tumor tissues were obtained from Cangzhou Central Hospital. The use of human tissues was approved by the Medical Ethics Committee of Cangzhou Central Hospital. All of the OS patients were provided written informed consent.
Overexpression or knockdown of LeXis
For knockdown assay, two shRNAs against LeXis were designed. The target sequences of LeXis were provided as follow: sh1: 5’-GTGCAAACAACACTCCAGT-3’, sh2: 5’-CCAACAGGTGCAAACAGAA-3’. The target sequence of CTNNB1 was provided as follow: shCTNNB1: 5’-TGGAGACCTGAGAACCAAT-3’. Scramble shRNA was taken as control. shRNAs against target genes were cloned into shRNA expressing lentiviral vector pLKO.1. Production of lentiviral particles was performed according to the standard protocols. Cells were transfected with lentiviral constructs for 24 hours with 2 μg/ml polybrene (Sigma). After 48 hours, stable cells were selected by using 2 μg/ml puromycin for 7 days.
For overexpression, full-length human LeXis cDNA was cloned into lentiviral expressing vector pLV-puro. Production of lentiviral particles was performed according to the standard protocols. Cells were transfected with lentiviral constructs expressing empty vector or LeXis for 24 hours with 2 μg/ml polybrene (Sigma). After 48 hours, stable cells were selected by using 2 μg/ml puromycin for 7 days.
Western blot
Western blotting was performed according to the standard protocol. In briefly, cell lysis were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane. The PVDF membrane was incubated with 1:1000 anti-CTNNB1 (Cell signaling) and 1:10000 anti-GAPDH (Cell signaling) overnight. Then the membrane was incubated with 1:10000 anti-mouse IgG-HRP conjugate (Jackson) or anti-rabbit IgG-HRP conjugate (Jackson) secondary antibodies. Blots were detected by ECL system (Millipore) and analyzed on an imager (GE Healthcare).
Colony formation
2 × 103 cells were seeded in 6-well plates, and then cultured for 7 days. Cells were fixed by 4% paraformaldehyde and stained with 0.5% crystal violet.
Apoptosis assay
The apoptosis was detected by using Apoptosis Detection Kit (KeyGEN, Nanjing, China) according to the manufacturer’s instructions. Cells were stained with fluorescein isothiocyanate-conjugated Annexin V and 7-Aminoactinomycin D (7-AAD). Cells were then analyzed with a FACScan flow cytometer, and the data were analyzed by Kaluza software.
Cell cycle assay
The cell cycle was analyzed using an in situ cell proliferation kit FLUOS (Roche) according to the manufacturer’s instruction. Briefly, cells were labeled with BrdU for 40 min before trypsinization. Cells were fixed by adding 70% ice-cold ethanol. Fixed cells were incubated with 4 M HCl at room temperature for 30 min then washed with PBS. The cells were then incubated with anti-BrdU-FLUOS antibody at room temperature for 45 min. Then cell suspensions were incubated with 7-AAD for 5 min and immediately analyzed with a FACScan flow cytometer. The data were analyzed by Kaluza software.
CCK-8 assay
Cell proliferation was determined by the CCK-8 (Dojindo) according to the manufacturer’s instructions. Cell proliferation rate was detected by measuring the absorbance at 450 nm with the Microplate Reader (Bio-Rad).
RNA extraction and quantitative real-time PCR (qPCR)
Total RNAs from cells or tissues were exacted by using TRIzol (Invitrogen) reagent according to the manufacturer’s instructions. First-strand cDNA was synthesized by using the PrimeScript™ RT reagent Kit (Takara) according to the manufacturer’s instructions. qPCR was performed using FastStart Universal SYBR Green Master (Roche) in the ABI StepOne-Plus System. GAPDH was taken as internal reference. Data were normalized to control group. Primers sequences were provided as follows: LeXis-F: 5’-CAGGCAGCTATGTCCCTTATG-3’, LeXis-R: 5’-GTAGAGGTTCACCCTTCCAATC-3’; CTNNB1-F: 5’-CTTCACCTGACAGATCCAAGTC-3’, CTNNB1-R: 5’-CCTTCCATCCCTTCCTGTTTAG-3’. Comparative quantification was determined using the 2-ΔΔCt method.
Dual-luciferase assays
The LeXis and LeXis-mut (mutant in miR-199a binding site) was cloned into pmirGLO plasmid, respectively. The pmirGLO-LeXis or pmirGLO-LeXis-mut was cotransfected with miR-199a mimics or miRNA negative control (miR-NC) into cells by Turbofect reagent (Thermo). Luciferase activity was determined by the Dual-luciferase Reporter Assay System (Promega). The luciferase activity was normalized to Renilla luciferase activity.
RNA immunoprecipitation (RIP)
Cells were co-transfected with pLV-MS2 or pLV-LeXis-MS2 or pLV-LeXis-mut-MS2 and pMS2-GFP (Addgene). After 48 hours of transfection, cells were subjected to a RIP assay by using 5 μg GFP antibody (Abcam) or negative control IgG using RNA Immunoprecipitation Kit (Millipore) according to the manufacturer’s instructions.
For anti-AGO2 RIP, cells were transfected with miR-NC or miR-199a mimics. After 48 hours of transfection, cells were used to perform anti-AGO2 RIP assay (Abcam) using 5 μg anti-AGO2 antibody (Millipore) as described above.
Immunohistochemistry
In the immunocytochemical assay, the slides were rehydrated and immersed in 3% hydrogen peroxide solution for 15 min; pretreated by microwave for 25 min in 0.01 mol/L citrate buffer at 95°C; and cooled for 60 min at room temperature. In between each incubation step, the sections were washed with PBS. The slides were blocked by 10% normal goat serum for 30 min at 37°C, washed, and then incubated overnight at 4°C with diluted Ki-67 antibody. After washing with PBS, the slides were visualized using GTVisionTMIII Detection System/Mo&Rb (GeneTech, GK500710) following the manufacturer’s instructions.
Statistical analysis
Data were analyzed using the SPSS 19.0 software. P value less than 0.05 was considered statistically significant.
Results
LeXis expression increases in OS tissues
First, we performed qPCR to detect LeXis expression level in OS tissues and corresponding noncancerous tissues obtained from 60 patients with OS. LeXis expression was higher in OS tissues than in the corresponding noncancerous tissues (Figure 1A). Next, we examined LeXis expression in five OS cell lines, namely, KHOS, 143b, LM7, U2OS, and MG-63, and a normal osteoblast cell line Nhost and found that LeXis expression was higher in OS cells than in Nhost cells (Figure 1B). Of the OS cell lines analyzed, MG-63 showed the highest and U2OS showed the lowest LeXis expression. To determine the clinical significance of LeXis expression, we analyzed the association between LeXis expression and clinicopathological features of patients with OS. We found that increased LeXis expression was significantly associated with high clinical stage and large tumor size (Table 1). Results of survival analysis showed that LeXis expression was associated with the prognosis of patients with OS, with increased LeXis expression being associated with reduced survival rate of patients with OS (Figure 1C). These data suggest that LeXis functions as an oncogene to promote OS progression and development.
Figure 1.
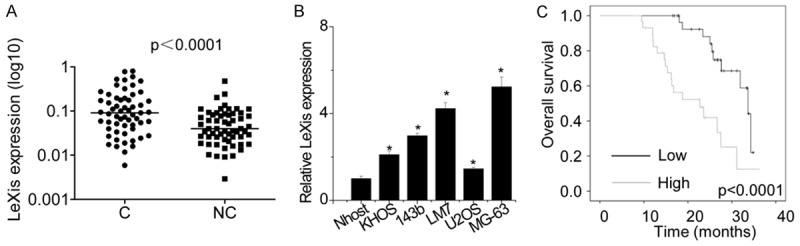
The LeXis expression is increased in OS. A. Expression of lncRNA LeXis was measured in 60 pairs of OS cancerous tissues (C) and adjacent noncancerous bone tissues (NC) using qPCR. B. Expression of lncRNA LeXis was detected in five OS cell lines (KHOS, 143b, LM7, U2OS and MG-63) and normal osteoblast cells (NHOst) using qPCR (*P < 0.05). C. Kaplan-Meier survival curve and log-rank test were used to evaluate the association of LeXis expression with overall survival rate. Patients were segregated into LeXis-high group and LeXis-low according to the median of LeXis expression in OS tissues.
Table 1.
Correlation between LeXis expression and clinicopathological features in OS patients
| Variables | LeXis expression levels | P | |
|---|---|---|---|
|
| |||
| Low | High | ||
| Gender | |||
| Male | 15 | 16 | 0.796 |
| Female | 15 | 14 | |
| Age | |||
| > 20 | 13 | 12 | 0.793 |
| ≤ 20 | 17 | 18 | |
| Location | |||
| Femur/Tibia | 19 | 17 | 0.598 |
| Elsewhere | 11 | 13 | |
| Tumor size (cm) | |||
| ≤ 5 | 17 | 8 | 0.018 |
| > 5 | 13 | 22 | |
| Clinical stage | |||
| I+IIA | 18 | 7 | 0.004 |
| IIB/III | 12 | 23 | |
| Distant Metastasis | |||
| Yes | 23 | 20 | 0.39 |
| No | 7 | 10 | |
P value was acquired by Pearson chi-square test. The median expression level was used as the cutoff.
LeXis enhances the proliferation and inhibits the apoptosis of OS cells in vitro
The positive correlation between LeXis expression and tumor size observed in the present study suggested that LeXis affected OS growth. To clarify this, we constructed stable LeXis-knockout MG-63 cells by using two different shRNA-expressing lentiviruses. Results of qPCR indicated that transfection of MG-63 cells with the lentiviruses expressing sh1 and sh2 effectively suppressed LeXis expression. Moreover, results of qPCR showed that sh1 was the most effective shRNA for suppressing LeXis expression (Figure 2A); hence, it was used for performing subsequent assays. Results of CCK-8 assay showed that the proliferation of LeXis-knockdown MG-63 cells was significantly lower than that of control cells (Figure 2B). Similarly, we found that LeXis-knockdown MG-63 cells formed less number of clones than control cells (Figure 2C). We also constructed stable LeXis-overexpressing U2OS cells (Figure 2D) and found that LeXis overexpression significantly promoted cell proliferation and clone formation (Figure 2E and 2F).
Figure 2.
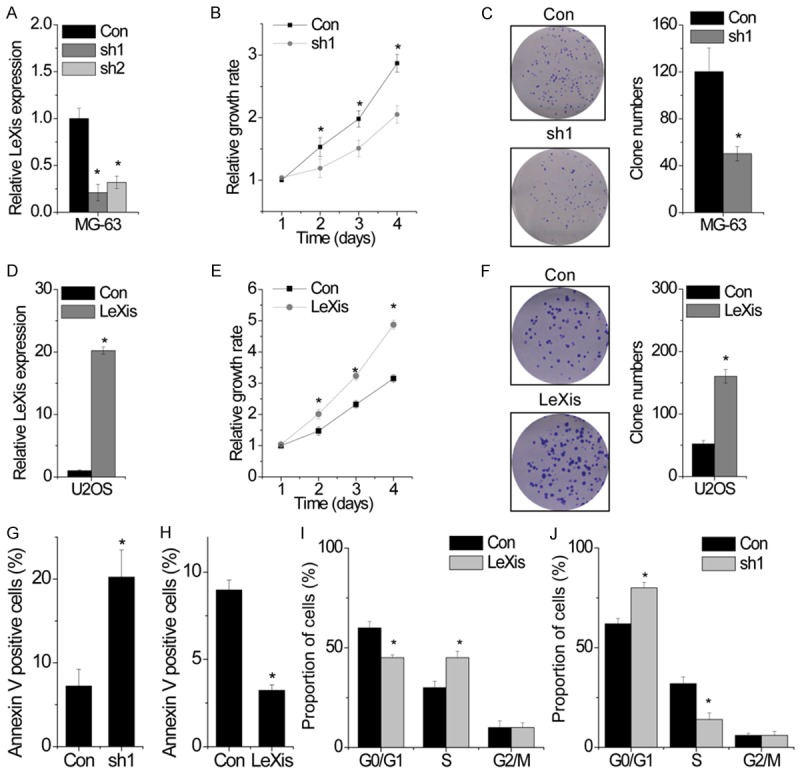
LeXis enhances OS cell proliferation and inhibits apoptosis in vitro. (A) The relative expression of LeXis in control and LeXis silenced MG-63 cells was detected by qPCR. (B) The cell growth rates were determined by performing CCK-8 assay. Knockdown LeXis in MG-63 cells significantly suppressed cell proliferation, relative to control cells. (C) Colony formation assay of control and LeXis silenced MG-63 cells. Representative graphs are shown. (D) The relative expression of LeXis in control and LeXis overexpressed U2OS cells was detected by qPCR. (E) The cell growth rates were determined by performing CCK-8 assay. Overexpression of LeXis in U2OS cells significantly suppressed cell proliferation, relative to control cells. (F) Colony formation assay of control and LeXis overexpressed U2OS cells. Representative graphs are shown. (G, H) Cells with LeXis knockdown (G) or overexpression (H) were stained with a combination of annexin V and 7-AAD and analyzed by FACS. Cells positive for annexin V staining were counted as apoptotic cells, and the percentage of apoptotic cells is shown. (I, J) FACS analysis showing significant decreases or increases of cells in G0/G1 or S phase, respectively, in U2OS cells with LeXis overexpression (I). In contrast, cells in S phase population were significantly decreased when LeXis was silenced in MG-63 cells (J). Data are shown as mean ± SD; *P < 0.05 (Student’s t test).
Next, we performed flow cytometry analysis to analyze the effect of LeXis on cell cycle and apoptosis and to determine the mechanism through which LeXis regulated cell proliferation. We found that the percentage of Annexin V-positive cells was significantly higher among LeXis-knockdown MG-63 cells than among control cells (Figure 2G). In contrast, protective effects were observed in LeXis-overexpressing U2OS cells (Figure 2H). However, LeXis knockdown and overexpression did not affect cell cycle distribution (Figure 2I and 2J). Together, these results indicate that LeXis promotes the proliferation and inhibits the apoptosis of OS cells.
LeXis promotes OS growth in vivo
Based on the above findings that LeXis promoted cell proliferation, migration, and invasion of OS cells, we investigated the effects of LeXis on cancer growth in vivo. The mean volumes of xenograft tumors generated from LeXis-knockout MG-63 cells were lower than those of tumors generated from control cells (Figure 3A). In contrast, tumors generated from LeXis-overexpressing U2OS cells were larger than those generated from control cells (Figure 3B). Results of immunohistochemical staining showed reduced Ki67 expression in xenograft tumors generated from LeXis-knockdown cells (Figure 3C) and increased Ki67 expression in xenograft tumors generated from LeXis-overexpressing cells (Figure 3D). These results highlight the important role of LeXis in OS growth.
Figure 3.
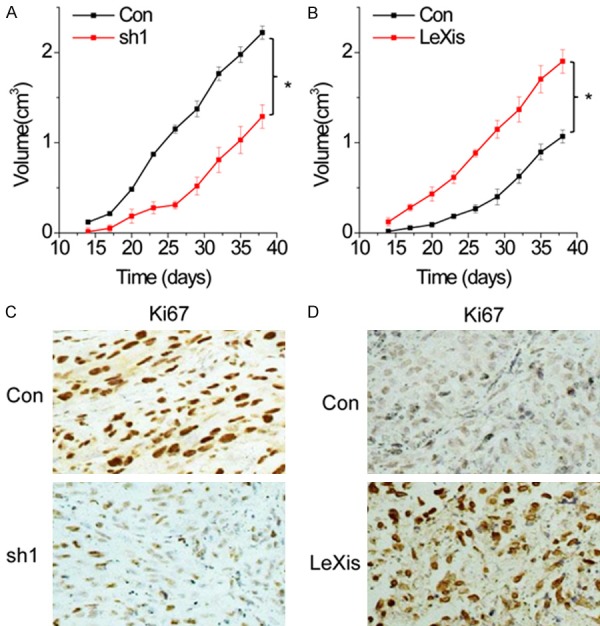
LeXis promotes OS growth in vivo. A. Effects of LeXis knockdown on tumor growth in vivo. The tumor growth curves were shown. B. Effects of LeXis overexpression on tumor growth in vivo. The tumor growth curves were shown. C, D. Immunohistochemical staining of Ki67 in xenograft tumor tissues. The degree of tumor proliferation was determined by Ki67 staining. Data are shown as mean ± SD; *P < 0.05 (Student’s t test).
LeXis directly interacts with miR-199a
We next investigated the mechanism through which LeXis promoted OS growth. Results of fractionation assays showed that LeXis was localized in both the nucleus and cytoplasm of MG-63 cells (Figure 4A), suggesting that it performed different functions. Recent studies have suggested that many RNA transcripts act as competing endogenous RNAs (ceRNAs) by competitively binding to miRNAs. We predicted potential miRNAs that interacted with LeXis by using StarBase and TargetScan prediction algorithms. Bioinformatics analysis showed that LeXis contained binding sites for miR-199a. Direct interaction between miR-199a and LeXis was validated by performing RIP assay [13] to pull down endogenous miRNAs that interacted with LeXis. The LeXis RIP in MG-63 cells was significantly enriched for miR-199a compared to the empty vector (MS2), IgG and LeXis-mut with mutation in miR-199a binding sites (Figure 4B). In addition, we constructed luciferase reporter vectors containing wild-type LeXis or LeXis-mut. We found that transfection of miR-199a reduced the luciferase activity of the reporter vector expressing wild-type LeXis but not of an empty vector or the reporter vector expressing LeXis-mut (Figure 4C). miRNAs degrade target mRNAs in an AGO2-dependent manner [14]. To investigate whether miR-199a regulated LeXis in AGO2-dependent manner, we performed anti-AGO2 RIP assay by using miR-199a-overexpressing U2OS cells. Pulldown of endogenous LeXis by using AGO2 showed high LeXis level in miR-199a-overexpressing U2OS cells (Figure 4D), suggesting that miR-199a targeted LeXis. Ectopically expressed LeXis but not LeXis-mut reduced miR-199a expression (Figure 4E). miR-199a overexpression did not alter LeXis expression (Figure 4F), indicating that miR-199a could bind to LeXis but did not induce its degradation. Next, we examined miR-199a expression in OS tissues and corresponding noncancerous tissues obtained from 60 patients with OS. We found that miR-199a expression was significantly lower in OS tissues than in corresponding noncancerous tissues (Figure 4G). Correlation analysis showed a negative correlation between LeXis and miR-199a expression in OS tissues (r2 = 0.6626, P < 0.0001; Figure 4H). These results suggest that LeXis associates with miR-199a and functions as a ceRNA.
Figure 4.
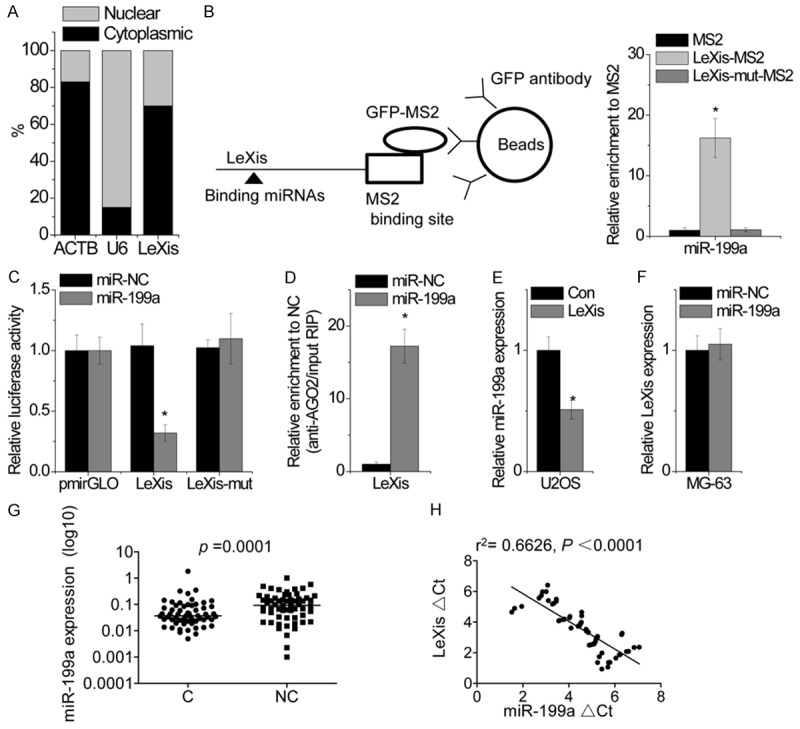
LeXis directly interacts with miR-199a. A. Representative analysis of LeXis distribution by cellular fractionation of MG-63 cells. ACTB mRNA and U6 RNA served as controls for cytoplasmic and nuclear RNAs, respectively. B. MS2-RIP followed by microRNA qPCR to detect microRNAs endogenously associated with LeXis. C. Luciferase activity in MG-63 cells cotransfected with miR-199a and luciferase reporters containing nothing, LeXis or mutant transcript. Data are presented as the relative ratio of firefly luciferase activity to renilla luciferase activity. D. Anti-AGO2 RIP was performed in MG-63 cells transiently overexpressing miR-199a, followed by qPCR to detect LeXis associated with AGO2. E. The effect of LeXis overexpression on miR-199a expression was detected by qPCR in U2OS cells. F. The effcct of miR-199a overexpression on LeXis expression was determined by qPCR in MG-63 cells. G. Expression of miR-199a was measured in 60 pairs of OS cancerous tissues (C) and adjacent noncancerous bone tissues (NC) using qPCR. H. The correlation between LeXis and miR-199a expression in OS tissues. Data are shown as mean ± SD; *P < 0.05 (Student’s t test).
LeXis functions as a ceRNA of CTNNB1
CTNNB1, which encodes β-catenin, is a target gene of miR-199a and plays a critical role in Wnt/β-catenin signaling and tumor growth [15,16]. We hypothesized that LeXis regulated CTNNB1 expression by suppressing miR-199a. We found that LeXis upregulation increased both the mRNA and protein expression of CTNNB1, whereas LeXis knockdown exerted an opposite effect (Figure 5A and 5B). We also found that LeXis expression was positively correlated with CTNNB1 expression in OS tissues (r2 = 0.3376, P < 0.0001; Figure 5C). To determine whether LeXis regulated the stability of CTNNB1 mRNA, we treated different U2OS clones with α-amanitin to block new RNA synthesis and measured the decrease in CTNNB1 mRNA level over a 24-h period. LeXis overexpression increased the half-life of CTNNB1 mRNA (Figure 5D), whereas LeXis depletion decreased the half-life of CTNNB1 mRNA (Figure 5E). To verify whether LeXis regulated cell proliferation in β-catenin-dependent manner, we silenced CTNNB1 expression in LeXis-overexpressing OS cells. We found that CTNNB1 downregulation abolished OS cell proliferation induced by LeXis overexpression (Figure 5F and 5G).
Figure 5.
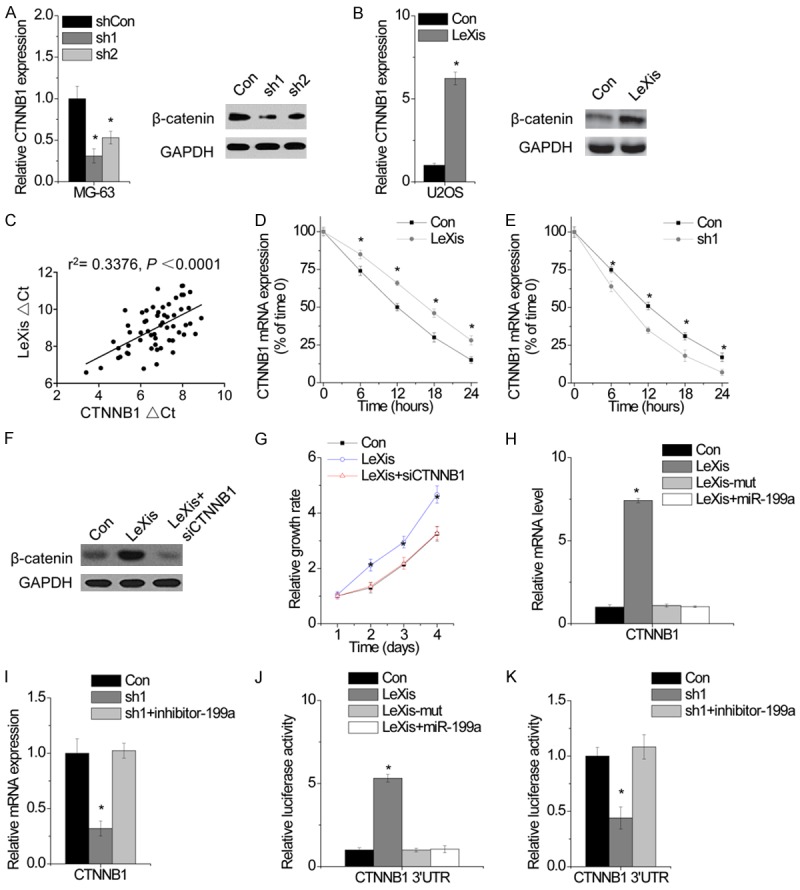
LeXis functions as a ceRNA of β-catenin. A. The mRNA (left) and protein expression of CTNNB1 in MG-63 cells with or without LeXis knockdown was detected by qPCR and western blot, respectively. B. The mRNA (left) and protein expression of CTNNB1 in MG-63 cells with or without LeXis overexpression was detected by qPCR and western blot, respectively. C. The correlation between LeXis and CTNNB1 mRNA expression in 60 OS tissues. D. The stability of CTNNB1 over time was measured by qPCR relative to time 0 after blocking new RNA synthesis with α-amanitin (50 mM) in U2OS cells with LeXis overexpression and normalized to 18S rRNA (a product of RNA polymerase I that is unchanged by α-amanitin). E. The stability of CTNNB1 over time was measured by qPCR relative to time 0 after blocking new RNA synthesis with α-amanitin (50 mM) in MG-63 cells with LeXis knockdown and normalized to 18S rRNA (a product of RNA polymerase I that is unchanged by α-amanitin). F. siRNA against CTNNB1 was tranfected into U2OS cells with LeXis overexpression. G. The cell proliferation of U2OS cells expressing LeXis with and without CTNNB1 siRNA. H. The mRNA level of CTNNB1 expression in LeXis or LeXis-mut overexpressed U2OS cells cotransfected with miR-199a was analyzed by qPCR. I. The mRNA level of CTNNB1 expression in LeXis silenced MG-63 cells cotransfected with miR-199a inhibitor was analyzed by qPCR. J. The relative luciferase activity of CTNNB1 3’UTR in LeXis or LeXis-mut overexpressed U2OS cells cotransfected with miR-199a was analyzed by qPCR. K. The relative luciferase activity of CTNNB1 3’UTR in LeXis silenced MG-63 cells cotransfected with miR-199a inhibitor was analyzed. Data are shown as mean ± SD; *P < 0.05 (Student’s t test).
To investigate whether LeXis regulated CTNNB1 expression by interacting with miR-199a, we determined CTNNB1 expression in LeXis- and LeXis-mut-overexpressing U2OS cells. Overexpression of LeXis but not of LeXis-mut increased CTNNB1 expression (Figure 5H). We transfected LeXis-overexpressing cells with miR-199a and found that miR-199a overexpression abrogated LeXis overexpression-induced increase in CTNNB1 expression (Figure 5H). In contrast, miR-199a inhibition rescued LeXis knockdown-induced suppression of CTNNB1 expression (Figure 5I). To confirm whether the above results were associated with the regulation of the 3’ UTR of CTNNB1 mRNA, we constructed a luciferase reporter vector containing the 3’ UTR of CTNNB1 mRNA. LeXis overexpression increased the luciferase activity of CTNNB1 3’ UTR-containing luciferase reporter vector, whereas miR-199a overexpression decreased the luciferase activity of CTNNB1 3’ UTR-containing luciferase reporter vector (Figure 5J). Conversely, LeXis knockdown decreased the luciferase activity of CTNNB1 3’ UTR-containing luciferase reporter vector, which was rescued by inhibiting miR-199a (Figure 5K). These results suggest an important role of LeXis in regulating CTNNB1 expression by competitively binding to miR-199a.
Discussion
Emerging evidences have shown that lncRNAs play critical roles in several cancers. LncRNAs regulate gene expression through different mechanisms. For example, lncRNA FAL1 enhances cell proliferation and interacts with and regulates the stability of epigenetic repressor BMI1 to modulate the transcription of target genes, including CDKN1A [17]. LncRNA BRM increases YAP1 expression and promotes the self-renewal of liver cancer stem cells by interacting with BRM [18]. LncRNA DANCR associates with CTNNB1 mRNA to block its degradation mediated by miR-214, miR-320a, and miR-199a [15]. LncRNA ATB upregulates ZEB1 and ZEB2 expression by competitively binding to miR-200 family and induces the metastasis of liver cancer [13]. These findings indicate that lncRNAs perform their functions by interacting with different molecules, including mRNAs, miRNAs, and proteins. However, the functional role of lncRNA LeXis in cancers is unclear to date. The present study is the first to show that LeXis promoted the proliferation and inhibited the apoptosis of OS cells. Increased LeXis expression was correlated with high clinical stage, large tumor size, and poor prognosis. Results of mechanistic investigation showed that LeXis physically interacted with miR-199a, thus suppressing its binding to the 3’ UTR of CTNNB1 mRNA and increasing CTNNB1 expression. We also observed a positive correlation between LeXis and CTNNB1 expression in OS tissues. These results strongly suggest that LeXis functions as a ceRNA of CTNNB1 in OS. Moreover, we found that LeXis was located in both the nucleus and cytoplasm, suggesting that it performed different functions by associating with different molecules.
miR-199a acts as a tumor suppressor in several cancers. miR-199a directly targets oncoproteins such as CCR7, YAP1, and PHLPP1 [19-21] and suppresses tumor growth, metastasis, and chemotherapy resistance. A recent study reported that long noncoding monocytic RNA acts as a ceRNA to sequester miR-199a [22]. However, it is unclear whether other lncRNAs function as ceRNAs of miR-199a. In the present study, we found that LeXis associated with miR-199a and suppressed its expression. In addition, we observed a negative correlation between LeXis and miR-199a expression in OS tissues. Use of StarBase and TargetScan prediction databases suggested that LeXis interacted with other miRNAs. However, further studies are need to determine whether LeXis competes with other mRNAs of common miRNAs to regulate cellular phenotypes.
In conclusion, the present study is the first to show that LeXis expression is upregulated in OS tissues and that increased LeXis expression is correlated with high tumor stage, large tumor size, and poor prognosis. Our findings highlight the oncogenic activity of lncRNA LeXis in OS growth. Results of functional assays indicate that LeXis promotes OS growth both in vitro and in vivo. Furthermore, we found that LeXis positively regulates CTNNB1 expression by functioning as a ceRNA against miR-199a. These findings may have important implications for developing novel therapeutic strategies for OS.
Disclosure of conflict of interest
None.
References
- 1.Arndt CA, Rose PS, Folpe AL, Laack NN. Common musculoskeletal tumors of childhood and adolescence. Mayo Clin Proc. 2012;87:475–487. doi: 10.1016/j.mayocp.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen R, Wang G, Zheng Y, Hua Y, Cai Z. Long non-coding RNAs in osteosarcoma. Oncotarget. 2017;8:20462–20475. doi: 10.18632/oncotarget.14726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reed DR, Hayashi M, Wagner L, Binitie O, Steppan DA, Brohl AS, Shinohara ET, Bridge JA, Loeb DM, Borinstein SC, Isakoff MS. Treatment pathway of bone sarcoma in children, adolescents, and young adults. Cancer. 2017;123:2206–2218. doi: 10.1002/cncr.30589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leichter AL, Sullivan MJ, Eccles MR, Chatterjee A. MicroRNA expression patterns and signalling pathways in the development and progression of childhood solid tumours. Mol Cancer. 2017;16:15. doi: 10.1186/s12943-017-0584-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kushlinskii NE, Fridman MV, Braga EA. Molecular mechanisms and microRNAs in osteosarcoma pathogenesis. Biochemistry (Mosc) 2016;81:315–328. doi: 10.1134/S0006297916040027. [DOI] [PubMed] [Google Scholar]
- 6.Ram Kumar RM, Boro A, Fuchs B. Involvement and clinical aspects of MicroRNA in osteosarcoma. Int J Mol Sci. 2016:17. doi: 10.3390/ijms17060877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tian R, Xie X, Han J, Luo C, Yong B, Peng H, Shen J, Peng T. miR-199a-3p negatively regulates the progression of osteosarcoma through targeting AXL. Am J Cancer Res. 2014;4:738–750. [PMC free article] [PubMed] [Google Scholar]
- 8.Ma G, Tang M, Wu Y, Xu X, Pan F, Xu R. LncRNAs and miRNAs: potential biomarkers and therapeutic targets for prostate cancer. Am J Transl Res. 2016;8:5141–5150. [PMC free article] [PubMed] [Google Scholar]
- 9.Chang W. Non-coding RNAs and berberine: a new mechanism of its anti-diabetic activities. Eur J Pharmacol. 2017;795:8–12. doi: 10.1016/j.ejphar.2016.11.055. [DOI] [PubMed] [Google Scholar]
- 10.Liu K, Huang J, Ni J, Song D, Ding M, Wang J, Huang X, Li W. MALAT1 promotes osteosarcoma development by regulation of HMGB1 via miR-142-3p and miR-129-5p. Cell Cycle. 2017;16:578–587. doi: 10.1080/15384101.2017.1288324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Zhang L, Zheng X, Zhong W, Tian X, Yin B, Tian K, Zhang W. Long non-coding RNA LINC00161 sensitises osteosarcoma cells to cisplatin-induced apoptosis by regulating the miR-645-IFIT2 axis. Cancer Lett. 2016;382:137–146. doi: 10.1016/j.canlet.2016.08.024. [DOI] [PubMed] [Google Scholar]
- 12.Sallam T, Jones MC, Gilliland T, Zhang L, Wu X, Eskin A, Sandhu J, Casero D, Vallim TQ, Hong C, Katz M, Lee R, Whitelegge J, Tontonoz P. Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature. 2016;534:124–128. doi: 10.1038/nature17674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, Wang SB, Wang YZ, Yang Y, Yang N, Zhou WP, Yang GS, Sun SH. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666–681. doi: 10.1016/j.ccr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 14.Shang R, Zhang F, Xu B, Xi H, Zhang X, Wang W, Wu L. Ribozyme-enhanced single-stranded Ago2-processed interfering RNA triggers efficient gene silencing with fewer off-target effects. Nat Commun. 2015;6:8430. doi: 10.1038/ncomms9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan SX, Wang J, Yang F, Tao QF, Zhang J, Wang LL, Yang Y, Liu H, Wang ZG, Xu QG, Fan J, Liu L, Sun SH, Zhou WP. Long noncoding RNA DANCR increases stemness features of hepatocellular carcinoma by derepression of CTNNB1. Hepatology. 2016;63:499–511. doi: 10.1002/hep.27893. [DOI] [PubMed] [Google Scholar]
- 16.Song X, Xin N, Wang W, Zhao C. Wnt/betacatenin, an oncogenic pathway targeted by H. pylori in gastric carcinogenesis. Oncotarget. 2015;6:35579–35588. doi: 10.18632/oncotarget.5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu X, Feng Y, Zhang D, Zhao SD, Hu Z, Greshock J, Zhang Y, Yang L, Zhong X, Wang LP, Jean S, Li C, Huang Q, Katsaros D, Montone KT, Tanyi JL, Lu Y, Boyd J, Nathanson KL, Li H, Mills GB, Zhang L. A functional genomic approach identifies FAL1 as an oncogenic long noncoding RNA that associates with BMI1 and represses p21 expression in cancer. Cancer Cell. 2014;26:344–357. doi: 10.1016/j.ccr.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu P, Wang Y, Wu J, Huang G, Liu B, Ye B, Du Y, Gao G, Tian Y, He L, Fan Z. LncBRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat Commun. 2016;7:13608. doi: 10.1038/ncomms13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou M, Wang S, Hu L, Liu F, Zhang Q, Zhang D. miR-199a-5p suppresses human bladder cancer cell metastasis by targeting CCR7. BMC Urol. 2016;16:64. doi: 10.1186/s12894-016-0181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren K, Li T, Zhang W, Ren J, Li Z, Wu G. miR-199a-3p inhibits cell proliferation and induces apoptosis by targeting YAP1, suppressing Jagged1-Notch signaling in human hepatocellular carcinoma. J Biomed Sci. 2016;23:79. doi: 10.1186/s12929-016-0295-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mussnich P, Rosa R, Bianco R, Fusco A, D’Angelo D. MiR-199a-5p and miR-375 affect colon cancer cell sensitivity to cetuximab by targeting PHLPP1. Expert Opin Ther Targets. 2015;19:1017–1026. doi: 10.1517/14728222.2015.1057569. [DOI] [PubMed] [Google Scholar]
- 22.Chen MT, Lin HS, Shen C, Ma YN, Wang F, Zhao HL, Yu J, Zhang JW. PU.1-regulated long noncoding RNA lnc-MC controls human monocyte/macrophage differentiation through interaction with MicroRNA 199a-5p. Mol Cell Biol. 2015;35:3212–3224. doi: 10.1128/MCB.00429-15. [DOI] [PMC free article] [PubMed] [Google Scholar]