

Abstract
The copper paddle-wheel is the building unit of many metal organic frameworks. Because of the ability of the copper cations to attract polar molecules, copper paddle-wheels are promising for carbon dioxide adsorption and separation. They have therefore been studied extensively, both experimentally and computationally. In this work we investigate the copper–CO2 interaction in HKUST-1 and in two different cluster models of HKUST-1: monocopper Cu(formate)2 and dicopper Cu2(formate)4. We show that density functional theory methods severely underestimate the interaction energy between copper paddle-wheels and CO2, even including corrections for the dispersion forces. In contrast, a multireference wave function followed by perturbation theory to second order using the CASPT2 method correctly describes this interaction. The restricted open-shell Møller–Plesset 2 method (ROS-MP2, equivalent to (2,2) CASPT2) was also found to be adequate in describing the system and used to develop a novel force field. Our parametrization is able to predict the experimental CO2 adsorption isotherms in HKUST-1, and it is shown to be transferable to other copper paddle-wheel systems.
1. Introduction
Metal organic frameworks (MOFs) are a class of three-dimensional nanoporous materials composed of metal nodes connected by organic ligands. The oriented coordination bond between these two components is responsible for the structure of the crystal. The possibility of combining different metals with different ligands provides a large variety of MOF structures. More than 10 000 structures have already been synthesized,1 but this is only a small fraction of the hundreds of thousands of structures that have been predicted computationally.2
MOFs have attracted considerable attention in the past decade for various applications, including gas adsorption and storage,3 gas separation,4 fuel production,5 chemical sensing,6 and catalysis.7
Computational modeling is extensively used to investigate the properties of synthesized materials for a given application and to predict the performance of hypothetical structures. In the case of gas adsorption, the quality of the model directly derives from the accuracy with which one can describe the microscopic interactions between the guest molecules and the framework. Density functional theory (DFT) calculations are routinely used for this purpose.8−10 However, weak interactions, due to dispersion forces arising from electron correlation, are poorly described by standard DFT methods. Corrections need to be introduced for this purpose (see the recent review of Grimme et al.11 and references therein). Alternatively, post-Hartree–Fock methods can be employed to evaluate interaction energies with high accuracy. However, because of the unfavorable scaling with the size of the system, they can hardly be used directly to compute interaction energies in MOFs, whose unit cells typically contains hundreds of atoms.10
This work focuses on the interaction between the carbon dioxide molecule and the copper(II) paddle-wheel, which is a metal organic structure composed of two copper cations connected to four carboxylates anions in a square planar coordination geometry. The smallest example of this structure is the Cu2(formate)4 molecule (Figure 1, left).
Figure 1.
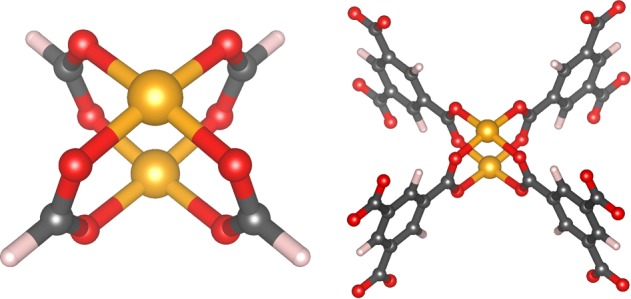
Copper paddle-wheel structure is composed of two coppers atoms bridged though four dicarboxilate anions. Cu2(formate)4 (left) represents the simplest paddle-wheel geometry possible. Dicopper benzyl-1,2,3-trimetylcarboxylate, Cu2(BTC)4 (right), is the building unit of the HKUST-1 framework: each BTC has three caboxylate groups that allow creation of a three-dimensional network.
The copper paddle-wheel is the building unit of many MOFs, including HKUST-1 (Cu3(BTC)2), as shown in Figure 1, right. The structure of HKUST-1 presents three pores (Figure 2, left) and several characteristic adsorption sites for CO2 (Figure 2, right). The biggest pore is characterized by the presence of 12 open metal sites (OMSs), i.e., unsaturated copper cations which are obtained after solvent removal and which are able to attract polar molecules through electrostatic interaction.
Figure 2.

Three different pores in HKUST-1 (left): big pore (blue), medium pore with open metal sites (green), and small pore (yellow). Characteristic sites of adsorption for CO2 (right): open metal site (blue), small pore window (green), small pore center (yellow), and large pore corner (purple).
HKUST-1 is one of the earlier reported MOFs.12 It is among the best performers for natural gas storage,13 and it has also attracted interest for gas separation14−16 and heterogeneous catalysis.17−19 Because of its popularity, much experimental data is available for this framework. Wu et al.20 conducted in situ neutron diffraction studies for CO2 adsorption in HKUST-1 which show that at low loading of CO2 and low temperature (20 K) the open metal site is the strongest adsorption site because it is the only one to be occupied at a 1:1 CO2:Cu ratio of loading. They were also able to rank the strength of the secondary sites by increasing the amount of CO2 and observing the filling in each site: small pore windows sites and center sites are the second and the third, respectively, and large pore corner sites are the fourth in terms of order of filling and therefore interaction energy strength.
In recent work, Grajciar et al.21 showed that DFT dispersion-corrected methods, e.g., Grimme’s pairwise correction for dispersions (D222 and D323) and van der Waals density functionals (vdW-DF24 and vdW-DF225), underestimate the strength of the open metal site and are not able to reproduce the experimental adsorption data obtained by Wu et al.
The van der Waals density functional methods, in particular, were used previously by our group to compute the CO2 binding energy in MOF-74 for different metals8,26−28 and to parametrize the associated force field.29,30 A good agreement with experiment was always observed, giving rise to the question of why the same ab initio methods are not able to model correctly the CO2 interaction with the open metal site in a copper paddle-wheel framework. This underestimation of the interactions in HKUST-1 motivated Grajciar et al. to employ a DFT-coupled clusters corrected (DFT/CC) method31 to study this system and obtain a tailormade correction for the CO2 interaction with HKUST-1. In DFT/CC, the error associated with the PBE density functional is corrected by a term dependent on the pairwise distance between the CO2 atoms and the atoms of the framework. This term was estimated from the difference between the DFT and the CCSD(T)-computed one-dimensional potential energy curves of CO2 interacting with some other reference molecules, i.e., H2, benzene, CO2, and Cu(formate)2.
It is known that a copper–copper magnetic interaction is present in HKUST-1,32 and consequently, the correlation between the electrons of the two coppers can affect the interaction with the CO2. Because of this we investigated the legitimacy of transferring the DFT error for CO2 interaction from the monocopper system Cu(formate)2 to the dicopper paddle-wheel structure (and to the HKUST-1 framework) by using multireference wave function methods. These methods are critical for accurately modeling systems with a relevant magnetic coupling such as the Cu paddle-wheel.33−35 Accordingly, we explored in this work the adequacy of different quantum methods for describing the electronic structure of the system and the interaction between the metal cation and carbon dioxide.
Furthermore, we used our insights to develop a classical force field that is able to accurately describe the Cu paddle-wheel interaction with CO2 and model the adsorption in MOFs containing this building unit. It was estimated36 that among 4764 three-dimensional MOF structures from the Cambridge Structural Database37 (as refined in the CoRE MOF database),38 4.2% of them contains the Cu paddle-wheel and another 3.5% contains the paddle-wheel motif formed by other cations. Cu paddle-wheels are a recurrent building unit among the different MOFs, and with a reliable and transferable force field it would be possible to also screen these frameworks and identify their performance for CO2 adsorption.
2. Computational Methods
The periodic calculations were performed using the Perdew–Burke–Ernzerhof GGA method PBEsol39 to optimize the framework and the second version of van der Waals dispersion-corrected density functional vdW-DF225 to compute the interactions. The plane wave Quantum Espresso 5.4 package40 was employed. We adopted the projector augmented wave (PAW) method41,42 with a cutoff energy of 60 Ry for the wave function and 300 Ry for the electron density. Due to the dimension of the unit cell of HKUST-1 a Γ-point sampling of the Brillouin zone integration was used with a smearing occupation of 0.02 Ry.
For the cluster calculations, geometry optimizations were performed using the unrestricted M06-L/cc-pVDZ43 level of theory, and subsequent single-point energy difference calculations were performed using restricted open-shell MP244 (ROS-MP2) and unrestricted M06-L and M06.43 The Gaussian 09 package45 was employed. We tested the convergence of the basis set using cc-pVDZ, AUG-cc-pVDZ, cc-pVTZ, and AUG-cc-pVTZ.46−49 A spin multiplicity of three was used to model the magnetic state of the copper paddle-wheel clusters. To account for the error in computing the interaction due to the basis set superposition, the counterpoise method by Boys and Bernardi was employed.50 For the ROS-MP2 calculations the frozen orbitals are the 1s for C and O and 1s, 2s, 2p, 3s, and 3p for Cu.
Multireference calculations were performed on the cluster models using the complete active space self-consistent field method (CASSCF)51 followed by second-order perturbation theory (CASPT2)52 using Molcas 8.2.53 All CASSCF/CASPT2 calculations were performed without symmetry. Relativistic basis sets of atomic natural orbital type (ANO-RCC)54 were employed for all atoms. To explore basis set convergence, three different basis sets of increasing size were tested. The first one, BS1, is of double-ζ quality plus polarization; the second one, BS2, is of triple-ζ quality plus polarization on Cu, O, and C atoms and double-ζ quality plus polarization on H atoms; the third one, BS3, is of quadruple-ζ quality plus polarization for Cu and CO2, triple-ζ quality plus polarization on the remaining C and O atoms, and double-ζ quality plus polarization on H atoms. Scalar relativistic effects were included using the Douglas–Kroll–Hess Hamiltonian.55 The computational cost arising from the two-electron integrals was drastically reduced by employing the Cholesky decomposition technique.56 The decomposition threshold was chosen to be 10–4, as this should correspond to an accuracy in total energies of the order of mHartree or higher. In the CASPT2 calculations, in order to prevent possible intruder states, an imaginary shift of 0.1 au was added to the zero-order Hamiltonian. The default IPEA shift of 0.25 au was used. The default choices of the program were employed for freezing orbitals, resulting in the 1s orbitals of C and O being frozen, along with the 1s, 2s, 2p, and 3s orbitals of Cu.
For the cluster models the interaction energy between the framework and the CO2 molecule was computed as the difference between the energy of the supersystem, the framework plus CO2, and the energies of the two isolated components, namely, CO2 and the framework.
The Raspa 2.0 package57 was employed for the force field calculations. In all simulations TraPPE58 Lennard–Jones parameters and charges were used to model CO2–CO2 interactions, while different sets of parameters were used to model the framework–CO2 interaction, as discussed within the results. The details of the simulations are provided in the Supporting Information.
3. Results and Discussion
3.1. Comparison of Simulated and Experimental Isotherms in HKUST-1
The CO2 isotherms computed with the standard force field, i.e., UFF,59 DREIDING,60 and TraPPE,58 are found to be in strong disagreement with the experimental data in the range of pressure from 0 to 1 bar. Figure 3 shows the simulated isotherms computed using the Grand Canonical Monte Carlo (GCMC) technique with different sets of parameters for the dispersion forces and the corresponding experimental isotherms.
Figure 3.
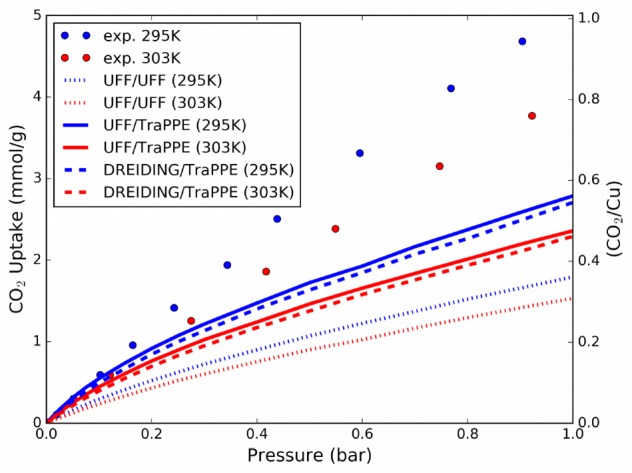
Comparison of experimental (29514 and 303 K31) and simulated adsorption isotherms. TraPPE58 Lennard–Jones parameters and charges are used for CO2–CO2 interactions. To compute the dispersion forces acting between CO2 guest molecules and the crystal, three commonly used approaches are compared. First, we used Lennard–Jones parameters from UFF59 (Lorentz–Berthelot mixing rules). Then we used UFF/TraPPE and DREIDING/TraPPE parameters60 (notation FFframework/FFadsorbate). The point charges for the framework atoms are extracted from a PBEsol DFT calculation using the REPEAT scheme;61 in the Supporting Information we report the charges’ values and compare them with the values obtained by using Bader’s method.62 The framework is assumed to be rigid in all simulations.
All simulations underestimate the uptake of CO2, which means that the force field underestimates the adsorbate–host interactions. The force field interaction energies for specific sites are compared to those obtained by DFT calculations in Table 1. The binding energy for each site, corresponding to the optimized position of a CO2 molecule in the open metal site, in the small pore window site, and in the small pore center site, are reported.
Table 1. Interaction Energy (kJ/mol) between CO2 and HKUST-1 for Different Adsorption Sitesa.
| method | open metal | window | center |
|---|---|---|---|
| FF (UFF/UFF) | –19.3 | –25.7 | –26.3 |
| FF (UFF/TraPPE) | –19.0 | –27.5 | –29.0 |
| FF (DREIDING/TraPPE) | –19.4 | –27.2 | –28.5 |
| DFT (vdW-DF2) | –22.1 | –30.2 | –26.3 |
| DFT (PBEsol) | –12.1 | –6.7 | –0.8 |
| DFT/CC (Grajciar et al.31) | –28.2 | –23.1 | –23.2 |
The open metal site in the apical position of the copper paddle-wheel, the window, and the center of small octahedral pores. Force field and periodic DFT calculations are compared. Results obtained with PBEsol show the evident inadequacy of pure DFT methods to model noncovalent interactions.
From neutron diffraction in situ experiments by Wu et al. we know that OMSs are the first filled sites; then windows and cage sites get populated by CO2. This observation proves that OMSs have the strongest binding energy. Despite the fact that UFF/UFF, UFF/TraPPE, and DREIDING/TraPPE force fields are giving similar results to the vdW-DF2 method and this could in principle validate the force fields, we clearly see from Table 1 that in all four of these cases the OMS is predicted to be the weakest site. As a consequence, these standard methods erroneously predict that the OMS is poorly occupied, as its interaction energies with CO2 are ∼4kbT and ∼60kbT weaker than other sites at 303 and 20K, respectively. Standard force fields are known to incorrectly model the strong interaction of adsorbate molecules with OMSs in MOFs,63 but vdW-DF2 is also showing the same problem in the case of copper paddle-wheel, while it was found to model accurately the open metal site interaction with CO2 for other MOFs.26,29,30
There are different assumptions in these calculations that may not hold for this system; therefore, the interaction energy between carbon dioxide and the copper atom in HKUST-1 was also computed using other approaches. We considered introduction of the Hubbard correction64 to model the d orbitals of copper, because it was shown to influence the CO2 interaction with the OMS in MOF-74.26,65 The value of U = 3.8 eV, which can reproduce the experimental oxidation energy of copper,66 was used. Also, different versions of the van der Waals density functional were compared to the vdw-DF2 method, i.e., vdW-DF67 and revised-vdW-DF2.68 In all cases the geometry of CO2 was optimized keeping the framework rigid, as obtained from the PBEsol calculation. The results are reported in Table 2. No significant deviations in the interaction energy were found, the only slightly increased value being obtained with vdW-DF, which is known to systematically overestimate dispersion interactions.69
Table 2. CO2 Open Metal Site Interaction Energies in HKUST-1 Computed with Different Dispersion-Corrected DFT Methods.
| method | open metal site interaction |
|---|---|
| vdW-DF | –24.9 kJ/mol |
| vdW-DF2 | –22.1 kJ/mol |
| vdW-DF2+U | –21.4 kJ/mol |
| vdW-DF2-rev | –20.2 kJ/mol |
Finally, the rigid framework assumption was neglected, performing a full optimization of the framework’s atoms with the adsorbed molecule in the OMS, using vdW-DF2. No significant deviation in the binding energy was found: −1.6 kJ/mol of difference from the rigid calculation. Moreover, we noticed an exaggerated distortion of the copper paddle-wheel structure which has not been reported experimentally, suggesting the inadequacy of the vdW-DF2 method to optimize the crystal geometry. The rigidity of the adsorbent was therefore assumed as reasonable.
3.2. Interactions Computed in the Cluster Models
To understand why the vdW-DF2 method underestimates the CO2–Cu interaction in HKUST-1, we analyzed two smaller representative clusters, Cu(formate)2 and Cu2(formate)4. The interaction energy with carbon dioxide was scanned at different distances by keeping the CO2 molecule perpendicular to the CuO4 plane, as shown in Figure 4.
Figure 4.

Path representation of linear scans of CO2 interacting with Cu(formate)2 (left) and Cu2(formate)4 (right). Dotted line, along which the CO2 molecule is displaced, is perpendicular to the CuO4 plane.
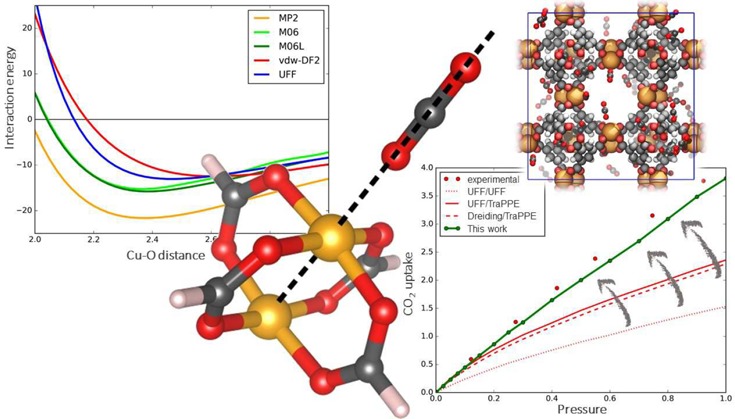
This configuration, referred here as “linear”, was chosen to decrease the number of degrees of freedom for the CO2 position to just one, i.e., the copper–oxygen distance in the axial direction. This configuration also minimizes all pairwise contributions of the interaction but the copper–oxygen one, which is the one vdW-DF2 is failing to model properly. Within HKUST-1, the optimal linear configuration corresponds to a distance of 2.65 Å and a binding energy of −13.4 kJ/mol, computed using vdW-DF2. Figures 5 and 6 show the interaction energy of CO2 as a function of the Cu–O distance computed with different methods in Cu(formate)2 and Cu2(formate)4, respectively.
Figure 5.
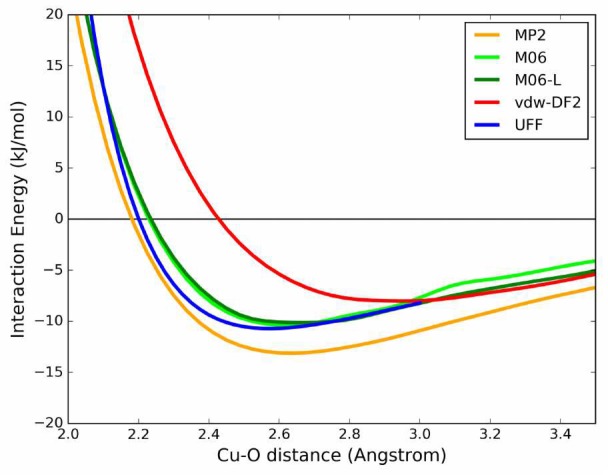
Interaction energy profile for the CO2–Cu(formate)2 linear scan: interaction energy is plotted as a function of the distance between the copper atom and the CO2 molecule’s oxygen.
Figure 6.
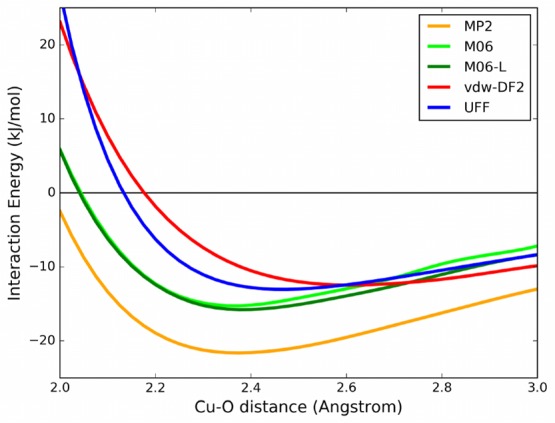
Interaction energy profile for the CO2–Cu2(formate)4 linear scan: interaction energy is plotted as a function of the distance between the CO2 molecule’s oxygen and the closest copper.
Inspection of the energy profiles reported in Figure 5 for Cu(formate)2 shows that the CO2–copper binding energies differ within 4 kJ/mol among the various methods, ranging between −8.0 (vdW-DF2) and −13.1 kJ/mol (MP2). The minimum energy distance for vdW-DF2 is longer than with the other methods, 2.9 Å instead of 2.5–2.6 Å. The M06 and M06-L functionals produce similar energy profiles. Hence, inclusion of the semilocal contribution with Hartree–Fock exchange present in M06 has a minor effect. It is also interesting to note the overall good agreement with the UFF force field. The attraction computed by the force field is mainly due to the Coulombic (REPEAT-TraPPE) interaction, with only a small influence of dispersion forces: the electrostatic contributions represent 96% of the interaction at the optimal distance of 2.5 Å.
In the Cu2(formate)4 case, vdW-DF2, M06, and M06-L underestimate the interaction energy compared with ROS-MP2 by 9.1, 6.4, and 5.8 kJ/mol, respectively. Moreover, if compared to the monocopper system, the ROS-MP2 calculation leads to a binding energy which is 8.5 kJ/mol more stable in this dicopper system.
In a second series of calculations we optimized the position of the CO2 molecule, keeping the Cu2(formate)4 cluster rigid. The CO2 molecule creates an angle with the copper–copper line from 109° (vdW-DF2) to 116° (M06-L) due to both the interaction of the lone pair of CO2 oxygen with the copper and the partially positive CO2 carbon with partially negative oxygen from the paddle-wheel. This optimized configuration is referred here as the “tilted” position, because of the CO2 inclination with respect to the CuO4 plane. The interaction energies between Cu2(formate)4 and the linear and tilted configurations of CO2 computed with different methods are reported in Table 3.
Table 3. Energy of Interaction (kJ/mol) between Cu2(formate)4 and CO2 in Linear and Tilted Conformationa.
| method | linear CO2 interaction energy (kJ/mol) | Cu–O distance (Å) | tilted CO2 interaction energy (kJ/mol) | Cu–O distance (Å) | Cu–O–O angle (deg) |
|---|---|---|---|---|---|
| FF(UFF/UFF) | –13.0 | 2.5 | –14.3 | 2.5 | 127.4° |
| ROS-MP2/cc-pVTZ | –18.2 (−24.5) | 2.4 | –22.9 (−31.3) | (M06-L opt) | (M06-L opt) |
| ROS-MP2/ANO-RCC(BS2) | –20.4 (−38.0) | 2.4 | –24.8 (−43.3) | (M06-L opt) | (M06-L opt) |
| ROS-MP2/aug-cc-pVTZ | –21.6 (−27.1) | 2.4 | –27.2 (−33.1) | (M06-L opt) | (M06-L opt) |
| M06/aug-cc-pVTZ | –15.2 (−17.7) | 2.4 | –21.9 (−25.3) | 2.4 | 114.5° |
| M06-L/aug-cc-pVTZ | –15.8 (−18.6) | 2.4 | –23.3 (−26.0) | 2.4 | 115.9° |
| vdW-DF2/cutoff = 60 Ry | –12.5 | 2.6 | –18.4 | 2.6 | 109.9° |
For all calculations that employ Gaussian basis functions, the energies obtained without counterpoise correction are reported in parentheses. ROS-MP2 calculations without augmented basis function are included to show the variability due to their exclusion in computing interactions.70 ROS-MP2/ANO-RCC calculations are also compared with CASPT2 results in section 3.3: for consistency we used the same basis set as BS2, with triple-ζ quality plus polarization on Cu, O, and C atoms and double-ζ quality plus polarization on H atoms.
On the basis of quantum calculations, the tilted conformation binding energy is ca. 5.5–7.5 kJ/mol larger than the linear conformation binding energy. The force field model, based on pairwise interactions, underestimates this difference at only 1.3 kJ/mol.
Finally, we tested the possible additive effect on the CO2 binding energy by adding a second CO2 molecule bonded symmetrically on the other copper of Cu2(formate)4. The binding energies computed with ROS-MP2 for this system do not show any significant deviation (−21.0 and −26.9 kJ/mol for the linear and tilted conformations, respectively), and therefore, any additive effect can reasonably be neglected.
To summarize, vdW-DF2 underestimates the CO2–Cu2(formate)4 binding energy by 9.1 and 8.8 kJ/mol, respectively, for the linear and tilted configurations, if compared to the ROS-MP2/aug-cc-pVTZ calculations. Considering the ROS-MP2 results, we are now able to improve our model for HKUST-1 and similar copper paddle-wheel MOFs.
3.3. Multireference Calculations
To have more insight into the interaction between CO2 and Cu2(formate)4, we performed wave function-based multireference complete active space calculations, followed by second-order perturbation theory.
A variety of active spaces were explored, including an active space with 2 electrons in 2 orbitals (2,2) and one with 10 electrons in 10 orbitals (10,10). The (2,2) CASSCF calculation is equivalent to a restricted open-shell (ROS)-HF calculation, while the (2,2) CASPT2 calculation is equivalent to the ROS-MP2 calculation. Notice that a singlet CASSCF (2,2) active space indeed corresponds to a multireference calculation in the sense that it generates a wave function that is the combination of two configuration state functions (or Slater determinants). Both the singlet and the triplet lowest spin states were explored. In all cases the singlet state is the ground state and lies 3 kJ/mol lower than the triplet state. This result is in good agreement with the experimental values obtained for MOF-11: 3.4 and 5.3 kJ/mol, respectively, for the water bound and the anhydrous structure.71 It is also in good agreement with the 3.2 kJ/mol value Maurice et al. calculated with DDCI3 on a similar system, copper acetate monohydrate.72
In the following we will discuss the energetics and electronic structure configurations of the singlet. However, as discussed above and also in the literature,33−35 it is reasonable to expect that the open-shell singlet and the triplet potential energy surfaces have a parallel shape. The singlet state is a linear combination of two electronic configurations with 50% weight each (See Table S1). The first configuration corresponds to orbital MO1 doubly occupied (MO12, Figure 7a) and the second to orbital MO2 doubly occupied (MO2, Figure 7b). In the (2,2) calculations these orbitals are the only ones included in the active space. They have an average occupation number of about 1 each (because each of them has only a 50% probability of being doubly occupied). In the (10,10) calculation, the other orbitals included in the active space are π and π* orbitals on the O and C atoms of the paddles. They have occupations of 2 and 0, respectively, within each pair. Additional details regarding the active space orbitals, including visual plots of the (10,10) orbitals, are presented in the Supporting Information.
Figure 7.
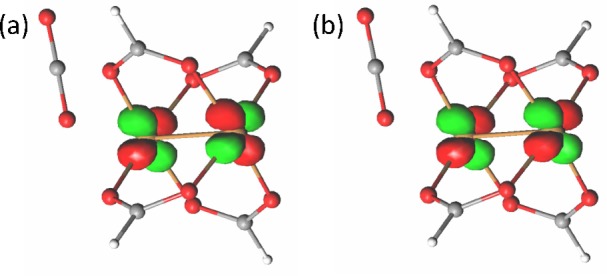
Two molecular orbitals MO1 (a) and MO2 (b), in the tilted dicopper system at equilibrium, with their occupation number in parentheses. In the linear system they look similar. Their occupation number is 1. They correspond to an overall configuration of 0.51 MO12 + 0.49 MO2.
The binding energies are reported in Table 4. In the dicopper system, the binding energy of CO2 to Cu is significantly larger than in the monocopper case, as already discussed in section 3.2. This behavior can be explained by inspection of the electronic configuration of the Cu2 system. The two Cu atoms are close enough to have electronic communication, and the overall wave function is a superposition of two electronic configurations. A multiconfigurational method is therefore needed to correctly describe this system in the singlet ground state. The monocopper system, on the other hand, has a single configuration, which is reasonably well described by MP2. The triplet state of the Cu2 system is also single configurational.
Table 4. CASPT2 Interaction Energies (kJ/mol) between Cu2(formate)4 and CO2 in Linear and Tilted Conformations for Different Active Spaces and Different Basis Sets for the Singlet Ground Statea.
| configuration | active space | BS1 | BS2 | BS3 |
|---|---|---|---|---|
| linear | (2,2) | –15.0 (−43.2) | –18.7 (−33.5) | –20.2 (−31.7) |
| linear | (10,10) | –14.8 (−46.6) | –18.6 (−36.8) | –20.1 (−35.0) |
| tilted | (2,2) | –17.7 (−49.3) | –23.5 (−40.0) | –25.8 (−39.6) |
| tilted | (10,10) | –15.8 (−51.1) | –21.8 (−41.7) | –23.9 (−41.1) |
The distance between CO2 and copper is 2.4 Å for both the linear and the tilted conformations. Values include counterpoise correction. Values without counterpoise correction are in parentheses.
The interaction energies for the singlet and triplet states are very similar (within 1 kJ/mol), and only the singlet energies are reported in Table 4. Our results show that an active space of (2,2) followed by PT2, equivalent to ROS-MP2, is sufficient to describe the binding of this system, as the binding energy does not change by more than 2 kJ/mol when increasing the active space to (10,10).
Basis set effects were explored for the CASPT2 calculations. Table 4 shows that on going from BS1 to BS2 the uncorrected binding energy decreases by about 10 kJ/mol, while it remains almost unchanged on going from BS2 to BS3. The counterpoise-corrected binding energies change by 3–6 kJ/mol going from BS1 to BS2 while again undergoing little change when going from BS2 to BS3. The CASPT2 results with the (2,2) active space reported in Table 4 should be compared to the ROS-MP2/ANO-RCC (BS2) results reported in Table 3. The only difference between these two sets of results is that those in Table 3 are obtained for the triplet, while those in Table 4 are obtained for the open-shell singlet and with unfrozen 3p orbitals for Cu. The two sets of values including counterpoise corrections differ by less than 2 kJ/mol, and more generally the most accurate CASPT2/BS3 energies agrees well with the ROS-MP2/aug-cc-pVTZ values, especially in the linear conformation (difference of 1.5 kJ/mol).
3.4. Correction of the Force Field
In order to model properly the interaction of carbon dioxide with the open metal site in a classical force field, we needed to correct the potential energy curve based on our first-principle calculations. The most representative path for different CO2–Cu distances is the one where the energy is mainly influenced by the interaction with the cation rather than the interaction with other atoms of the cluster (or framework). Hence, we fitted the linear CO2–Cu2(formate)4 curve obtained with the ROS-MP2/aug-cc-pVTZ method to obtain the new parameters for the force field. Only the Cu–O van der Waals potential was tuned while keeping the standard UFF parameters for all other atoms pairs and REPEAT (PBEsol derived) point charges to model electrostatic interactions. For the Cu–O interaction, a Buckingham potential was adopted to correctly represent the repulsion at short distance, and an r–8 attractive term was added to account for the stabilization observed in the ROS-MP2 calculations. The details about the fitting and the coefficient for the Cu–O potential are reported in the Supporting Information. The optimal CO2 interaction with Cu2(formate)4, which corresponds to the tilted conformation, computed with the fitted force field parameters has a value of −23.2 kJ/mol. This result is consistent with the UFF difference between the linear and the tilted configurations of −1.3 kJ/mol. We notice that by applying this relatively simple but effective correction, obtained without modifying the pairwise interaction with other atoms and without introducing a specific contribution based on the Cu–CO2 angle, the minimum interaction energy obtained for Cu2(formate)4 is in fair agreement with the ROS-MP2 result of −27.2 kJ/mol.
Finally, we replicated the GCMC simulations in HKUST-1 using our UFF parameters with the corrected Cu–O potential. The comparison with experimental data is reported in Figure 8. The simulations are still slightly underestimating the measured uptake, and this is reflecting the previously mentioned underestimation of ca. 4 kJ/mol for the interaction energy in the optimal tilted configuration. However, the assumptions made for the force field are sufficient to obtain a good representation of the uptake around ambient temperature and a significant improvement with respect to the standard force fields.
Figure 8.
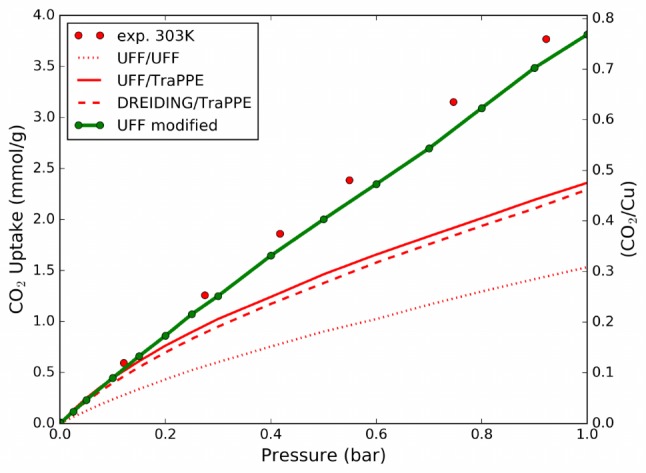
Comparison between the experimental31 and the simulated isotherms for CO2 inside HKUST-1 at 303 K. Modified UFF force field is obtained by fitting the Cu–O potential on ROS-MP2 calculations.
The minimum energy of interaction computed with our new force field in the three main adsorption sites of HKUST-1, i.e., open metal, small pore window, and small pore center sites, are now ranked correctly, −27.3, −26.8, and −26.8 kJ/mol, respectively, and the OMS stability is not underestimated any longer compared to the in situ experimental results.
As a starting point for our correction, we used UFF/UFF mixed parameters instead of UFF/TraPPE or DREIDING/TraPPE, because from the simulated isotherm (Figure 8) we can observe that these last force fields are already predicting the experimental uptake at very low pressure (below 0.1 bar), even if the open metal site interaction is strongly underestimated. This is an artifact due to a fortuitous error cancellation with the overestimation of the interaction in other sites, i.e., the small pores centers (see Table 1), which are already saturated at 0.82 mmol/g, as clearly shown by the deviation of the simulated isotherm from the experimental one. Therefore, employing the conventionally used UFF/UFF, UFF/TraPPE, or DREIDING/TraPPE mixed parameters to describe the guest–host interaction in an analysis of the site occupancy would lead to wrong conclusions, i.e., that in HKUST-1 the open metal sites are very poorly occupied at low uptake.73 Ulterior comparisons with experimental data is provided in the Supporting Information: CO2 uptake at higher pressure and different temperatures74 and the heat of desorption as a function of the uptake.31,75,76
3.5. Investigation of the “Double” Open Metal Site Interaction in Cu–TDPAT
To further test the reliability of our force field, we investigated another interesting copper paddle-wheel metal organic framework, Cu–TDPAT, first synthesized by Li et al.77 The crystalline structure is characterized by the presence of strong adsorption sites for CO2, where both oxygens of the guest molecule are attracted to two different copper cations (Figure 9), leading to an interaction energy which is roughly double with respect to the conventional single open metal site of copper paddle-wheel.
Figure 9.
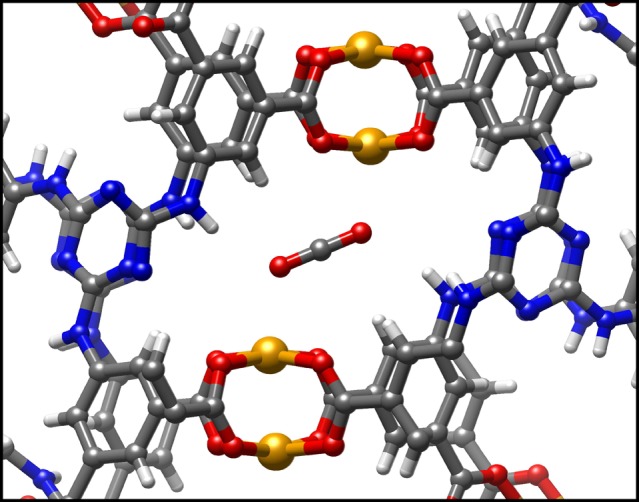
CO2 molecule adsorbed in the double open metal site of Cu–TDPAT.
Because of this reason, Cu–TDPAT is one of the top performing MOFs for both gravimetric and volumetric CO2 uptake at ambient pressure.78 The conventional unit cell of Cu–TDPAT contains 48 copper cations: 24 of them compose 12 double open metal sites, while the remaining 24 atoms compose 24 single open metal sites, with a conformation very similar to the OMS of HKUST-1. Due to the large dimension of the unit cell (960 atoms), the crystal is too big to perform a DFT calculation with an accuracy comparable with our previous calculation on HKUST-1. Consequently, we employed the extended charge equilibration (EQeq) method79 to compute the partial charges of the framework. This method is able to self-consistently compute point charges for MOFs, with results very similar to the charges obtained by fitting the electrostatic potential from a quantum calculation, e.g., REPEAT. HKUST-1 itself was successfully tested in the original paper presenting the EQeq method.79 Compared to the quantum electrostatic potential fitting, this method is drastically faster (a few minutes instead of hours for HKUST-1) and applicable to a unit cell containing a large number of atoms, which is practically forbidden to DFT calculations. The result obtained for the copper paddle-wheel is consistent with our PBEsol calculation in HKUST-1. Using the EQeq method the average point charges for Cu–TDPAT are 0.905 and −0.398 for the copper and the carboxylic oxygen, respectively, versus 0.914 and −0.57 for HKUST-1 computed using REPEAT. With the new set of parameters, we compared the results of the GCMC simulations to experimental data (Figure 10).
Figure 10.

Comparison of experimental77 and simulated adsorption of CO2 in Cu–TDPAT at 298 K using different sets of parameters. Force field developed in this work is reported as “UFF modified”, while UFF/UFF and UFF/TraPPE are the conventionally used standard sets of parameters. In both plots the uptake is converted to CO2 molecules per copper ratio, and the equivalence to the number of double open metal sites (0.25 CO2/Cu) and the number of total open metal sites (0.75 CO2/Cu) is highlighted with a dotted line. Experimental heat of desorption (black dots, right picture) has been computed through the Virial–Langmuir method, while the simulated values (colored lines) are computed from the guest molecules number fluctuation in the GCMC simulation.
This comparison shows good agreement, as for HKUST-1, which gives us some confidence in the transferability of our force field to model CO2 adsorption. Moreover, it becomes more evident how UFF/UFF and UFF/TraPPE parametrizations do not capture the strong interaction between CO2’s oxygen and copper.
4. Conclusions
In this work we have shown that the Cu–Cu interaction in copper paddle-wheel systems is the reason why DFT methods, even when they include dispersion corrections, systematically underestimate the interaction between CO2 and copper paddle-wheel motif. Our calculations confirm the presence of the copper–copper coupling, influencing the attraction of the CO2, and suggest that the monocopper cluster Cu(formate)2 is not a realistic model to describe this interaction.
One thus needs an electronic structure theory that properly describes the Cu–Cu interaction, such as the ROS-MP2 wave function. We show that if this interaction is included in our calculations, the prediction of the binding energies is in better agreement with the experimental data. To justify the choice of ROS-MP2 method, which is equivalent to a (2,2) CASPT2 calculation, we performed a number of multireference calculations over a variety of active spaces, basis sets, and spin states. We concluded that the ROS-MP2 level of theory is good enough to model the Cu2(formate)4–CO2 interaction.
Using the ROS-MP2 results, we reparametrized the UFF pairwise potential to correctly model the interaction of CO2 with the open metal site in HKUST-1, which was severely underestimated by conventional force fields. The results obtained from our new force field agree with experimental isotherms as well as with in situ PXRD studies, which found the open metal site to be the strongest adsorption site for CO2 rather than the small pore center site. The correction proposed in this work acts in proximity of the open metal site, and this local character of the correction terms allows us to transfer the same parameters to other MOFs containing the copper paddle-wheel motif.
To test this transferability, we employed our force field to model the CO2 interaction with the “double” open metal sites of Cu–TDPAT framework, and we showed a significant improvement with respect to conventional UFF parameters in this case as well. The modified set of parameters proposed makes it possible now to accurately describe the adsorption behavior for this class of MOFs. In this study we focused on CO2, but similar effects can be expected for other polar molecules.
Acknowledgments
The research of D.O., D.T., and B.S. was partially supported by the National Center of Competence in Research (NCCR) “Materials’ Revolution: Computational Design and Discovery of Novel Materials (MARVEL)” of the Swiss National Science Foundation (SNSF) and by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 666983, MaGic). Part of the computational work was made possible through a grant from the Swiss National Supercomputing Center (CSCS) under Project no. s611. S.J.G. and L.G. were supported as part of the Inorganometallic Catalyst Design Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award DE-SC0012702.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcc.7b02302.
Supplementary results for the multireference calculations; details of the classical simulations, coefficients of the force field, and comparison between the ROS-MP2 and the fitted Cu–O potential; atomic coordinates for the cluster models (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Furukawa H.; Cordova K. E.; O’Keeffe M.; Yaghi O. M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444. 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]
- Wilmer C. E.; Leaf M.; Lee C. Y.; Farha O. K.; Hauser B. G.; Hupp J. T.; Snurr R. Q. Large-scale screening of hypothetical metal-organic frameworks. Nat. Chem. 2012, 4, 83–89. 10.1038/nchem.1192. [DOI] [PubMed] [Google Scholar]
- Morris R. E.; Wheatley P. S. Gas storage in nanoporous materials. Angew. Chem., Int. Ed. 2008, 47, 4966–4981. 10.1002/anie.200703934. [DOI] [PubMed] [Google Scholar]
- Li J.-R.; Kuppler R. J.; Zhou H.-C. Selective gas adsorption and separation in metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. 10.1039/b802426j. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Lin W. Metal-organic frameworks for artificial photosynthesis and photocatalysis. Chem. Soc. Rev. 2014, 43, 5982–5993. 10.1039/C4CS00103F. [DOI] [PubMed] [Google Scholar]
- Kreno L. E.; Leong K.; Farha O. K.; Allendorf M.; Van Duyne R. P.; Hupp J. T. Metal-organic framework materials as chemical sensors. Chem. Rev. 2012, 112, 1105–1125. 10.1021/cr200324t. [DOI] [PubMed] [Google Scholar]
- Liu J.; Chen L.; Cui H.; Zhang J.; Zhang L.; Su C.-Y. Applications of metal-organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. 10.1039/C4CS00094C. [DOI] [PubMed] [Google Scholar]
- Poloni R.; Lee K.; Berger R. F.; Smit B.; Neaton J. B. Understanding trends in CO2 adsorption in metal-organic frameworks with open-metal sites. J. Phys. Chem. Lett. 2014, 5, 861–865. 10.1021/jz500202x. [DOI] [PubMed] [Google Scholar]
- Evans J. D.; Fraux G.; Gaillac R.; Kohen D.; Trousselet F.; Vanson J.-M.; Coudert F.-X. Computational chemistry methods for nanoporous materials. Chem. Mater. 2017, 29, 199–212. 10.1021/acs.chemmater.6b02994. [DOI] [Google Scholar]
- Odoh S. O.; Cramer C. J.; Truhlar D. G.; Gagliardi L. Quantum-chemical characterization of the properties and reactivities of metal-organic frameworks. Chem. Rev. 2015, 115, 6051–6111. 10.1021/cr500551h. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Hansen A.; Brandenburg J. G.; Bannwarth C. Dispersion-corrected mean-field electronic structure methods. Chem. Rev. 2016, 116, 5105–5154. 10.1021/acs.chemrev.5b00533. [DOI] [PubMed] [Google Scholar]
- Chui S. S.-Y.; Lo S. M.-F.; Charmant J. P.; Orpen A. G.; Williams I. D. A chemically functionalizable nanoporous material [Cu3 (TMA) 2 (H2O) 3] n. Science 1999, 283, 1148–1150. 10.1126/science.283.5405.1148. [DOI] [PubMed] [Google Scholar]
- Mason J. A.; Veenstra M.; Long J. R. Evaluating metal-organic frameworks for natural gas storage. Chem. Sci. 2014, 5, 32–51. 10.1039/C3SC52633J. [DOI] [Google Scholar]
- Min Wang Q.; Shen D.; Bülow M.; Ling Lau M.; Deng S.; Fitch F. R.; Lemcoff N. O.; Semanscin J. Metallo-organic molecular sieve for gas separation and purification. Microporous Mesoporous Mater. 2002, 55, 217–230. 10.1016/S1387-1811(02)00405-5. [DOI] [Google Scholar]
- Hamon L.; Jolimaître E.; Pirngruber G. D. CO2 and CH4 separation by adsorption using Cu-BTC metal- organic framework. Ind. Eng. Chem. Res. 2010, 49, 7497–7503. 10.1021/ie902008g. [DOI] [Google Scholar]
- Ge L.; Zhou W.; Rudolph V.; Zhu Z. Mixed matrix membranes incorporated with size-reduced Cu-BTC for improved gas separation. J. Mater. Chem. A 2013, 1, 6350–6358. 10.1039/c3ta11131h. [DOI] [Google Scholar]
- Wee L. H.; Janssens N.; Bajpe S. R.; Kirschhock C. E.; Martens J. A. Heteropolyacid encapsulated in Cu 3 (BTC) 2 nanocrystals: an effective esterification catalyst. Catal. Today 2011, 171, 275–280. 10.1016/j.cattod.2011.03.017. [DOI] [Google Scholar]
- Pérez-Mayoral E.; Čejka J. [Cu3 (BTC) 2]: a metal-organic framework catalyst for the Friedländer reaction. ChemCatChem 2011, 3, 157–159. 10.1002/cctc.201000201. [DOI] [Google Scholar]
- Ye J.-Y.; Liu C.-J. Cu 3 (BTC) 2: CO oxidation over MOF based catalysts. Chem. Commun. 2011, 47, 2167–2169. 10.1039/c0cc04944a. [DOI] [PubMed] [Google Scholar]
- Wu H.; Simmons J. M.; Srinivas G.; Zhou W.; Yildirim T. Adsorption sites and binding nature of CO2 in prototypical metal- organic frameworks: A combined neutron diffraction and first-principles study. J. Phys. Chem. Lett. 2010, 1, 1946–1951. 10.1021/jz100558r. [DOI] [Google Scholar]
- Grajciar L.; Nachtigall P.; Bludský O.; Rubeš M. Accurate ab initio description of adsorption on coordinatively unsaturated Cu2+ and Fe3+ sites in MOFs. J. Chem. Theory Comput. 2015, 11, 230–238. 10.1021/ct500711z. [DOI] [PubMed] [Google Scholar]
- Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Dion M.; Rydberg H.; Schröder E.; Langreth D. C.; Lundqvist B. I. van der Waals density functional for general geometries. Phys. Rev. Lett. 2004, 92, 246401. 10.1103/PhysRevLett.92.246401. [DOI] [PubMed] [Google Scholar]
- Lee K.; Murray É. D.; Kong L.; Lundqvist B. I.; Langreth D. C. Higher-accuracy van der Waals density functional. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 82, 081101. 10.1103/PhysRevB.82.081101. [DOI] [Google Scholar]
- Lee K.; Howe J. D.; Lin L.-C.; Smit B.; Neaton J. B. Small-molecule adsorption in open-site metal-organic frameworks: a systematic density functional theory study for rational design. Chem. Mater. 2015, 27, 668–678. 10.1021/cm502760q. [DOI] [Google Scholar]
- Poloni R.; Smit B.; Neaton J. B. CO2 capture by metal-organic frameworks with van der Waals density functionals. J. Phys. Chem. A 2012, 116, 4957–4964. 10.1021/jp302190v. [DOI] [PubMed] [Google Scholar]
- Poloni R.; Smit B.; Neaton J. B. Ligand-assisted enhancement of CO2 capture in metal-organic frameworks. J. Am. Chem. Soc. 2012, 134, 6714–6719. 10.1021/ja2118943. [DOI] [PubMed] [Google Scholar]
- Lin L.-C.; Lee K.; Gagliardi L.; Neaton J. B.; Smit B. Force-field development from electronic structure calculations with periodic boundary conditions: applications to gaseous adsorption and transport in metal-organic frameworks. J. Chem. Theory Comput. 2014, 10, 1477–1488. 10.1021/ct500094w. [DOI] [PubMed] [Google Scholar]
- Mercado R.; Vlaisavljevich B.; Lin L.-C.; Lee K.; Lee Y.; Mason J. A.; Xiao D. J.; Gonzalez M. I.; Kapelewski M. T.; Neaton J. B.; et al. Force field development from periodic density functional theory calculations for gas separation applications using metal-organic frameworks. J. Phys. Chem. C 2016, 120, 12590–12604. 10.1021/acs.jpcc.6b03393. [DOI] [Google Scholar]
- Grajciar L.; Wiersum A. D.; Llewellyn P. L.; Chang J.-S.; Nachtigall P. Understanding CO2 adsorption in CuBTC MOF: comparing combined DFT-ab initio calculations with microcalorimetry experiments. J. Phys. Chem. C 2011, 115, 17925–17933. 10.1021/jp206002d. [DOI] [Google Scholar]
- Tiana D.; Hendon C. H.; Walsh A. Ligand design for long-range magnetic order in metal-organic frameworks. Chem. Commun. 2014, 50, 13990–13993. 10.1039/C4CC06433J. [DOI] [PubMed] [Google Scholar]
- Grajciar L.; Bludsky O.; Nachtigall P. Water adsorption on coordinatively unsaturated sites in CuBTC MOF. J. Phys. Chem. Lett. 2010, 1, 3354–3359. 10.1021/jz101378z. [DOI] [PubMed] [Google Scholar]
- Vogiatzis K. D.; Mavrandonakis A.; Klopper W.; Froudakis G. E. Ab initio Study of the Interactions between CO2 and N-Containing Organic Heterocycles. ChemPhysChem 2009, 10, 374–383. 10.1002/cphc.200800583. [DOI] [PubMed] [Google Scholar]
- Vogiatzis K. D.; Klopper W.; Mavrandonakis A.; Fink K. Magnetic properties of paddlewheels and trinuclear clusters with exposed metal sites. ChemPhysChem 2011, 12, 3307–3319. 10.1002/cphc.201100559. [DOI] [PubMed] [Google Scholar]
- Boyd P. G.; Moosavi S. M.; Witman M.; Smit B. On the force field prediction of materials properties in metal organic frameworks. J. Phys. Chem. Lett. 2017, 8, 357–363. 10.1021/acs.jpclett.6b02532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom C. R.; Bruno I. J.; Lightfoot M. P.; Ward S. C. The Cambridge structural database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72, 171–179. 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y. G.; Camp J.; Haranczyk M.; Sikora B. J.; Bury W.; Krungleviciute V.; Yildirim T.; Farha O. K.; Sholl D. S.; Snurr R. Q. Computation-ready, experimental metal-organic frameworks: A tool to enable high-throughput screening of nanoporous crystals. Chem. Mater. 2014, 26, 6185–6192. 10.1021/cm502594j. [DOI] [Google Scholar]
- Perdew J. P.; Ruzsinszky A.; Csonka G. I.; Vydrov O. A.; Scuseria G. E.; Constantin L. A.; Zhou X.; Burke K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. 10.1103/PhysRevLett.100.136406. [DOI] [PubMed] [Google Scholar]
- Giannozzi P.; Baroni S.; Bonini N.; Calandra M.; Car R.; Cavazzoni C.; Ceresoli D.; Chiarotti G. L.; Cococcioni M.; Dabo I.; et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 2009, 21, 395502. 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 1758. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Blöchl P. E. Projector augmented-wave method. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50, 17953. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Knowles P. J.; Andrews J. S.; Amos R. D.; Handy N. C.; Pople J. A. Restricted Møer-Plesset theory for open-shell molecules. Chem. Phys. Lett. 1991, 186, 130–136. 10.1016/S0009-2614(91)85118-G. [DOI] [Google Scholar]
- Frisch M.; Trucks G.; Schlegel H.; Scuseria G.; Robb M.; Cheeseman J.; Scalmani G.; Barone V.; Mennucci B.; Petersson G.; et al. Gaussian 09, revision D.01; Gaussian, 2009. [Google Scholar]
- Dunning T. H. Jr Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Kendall R. A.; Dunning T. H. Jr; Harrison R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. 10.1063/1.462569. [DOI] [Google Scholar]
- Davidson E. R. Comment on Dunning’s correlation-consistent basis sets. Chem. Phys. Lett. 1996, 260, 514–518. 10.1016/0009-2614(96)00917-7. [DOI] [Google Scholar]
- Balabanov N. B.; Peterson K. A. Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3 d elements Sc-Zn. J. Chem. Phys. 2005, 123, 064107. 10.1063/1.1998907. [DOI] [PubMed] [Google Scholar]
- Boys S. F.; Bernardi F. D. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. 10.1080/00268977000101561. [DOI] [Google Scholar]
- Roos B. O.; Taylor P. R.; Sigbahn P. E. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. 10.1016/0301-0104(80)80045-0. [DOI] [Google Scholar]
- Andersson K.; Malmqvist P. A.; Roos B. O.; Sadlej A. J.; Wolinski K. Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 1990, 94, 5483–5488. 10.1021/j100377a012. [DOI] [Google Scholar]
- Aquilante F.; Autschbach J.; Carlson R. K.; Chibotaru L. F.; Delcey M. G.; De Vico L.; Ferré N.; Frutos L. M.; Gagliardi L.; Garavelli M.; et al. Molcas 8: new capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. 10.1002/jcc.24221. [DOI] [PubMed] [Google Scholar]
- Widmark P.-O.; Malmqvist P.-Å.; Roos B. O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. Theor. Chim. Acta 1990, 77, 291–306. 10.1007/BF01120130. [DOI] [Google Scholar]
- Reiher M.; Wolf A. Exact decoupling of the Dirac hamiltonian. II. The generalized Douglas-Kroll-Hess transformation up to arbitrary order. J. Chem. Phys. 2004, 121, 10945–10956. 10.1063/1.1818681. [DOI] [PubMed] [Google Scholar]
- Aquilante F.; Gagliardi L.; Pedersen T. B.; Lindh R. Atomic Cholesky decompositions: a route to unbiased auxiliary basis sets for density fitting approximation with tunable accuracy and efficiency. J. Chem. Phys. 2009, 130, 154107. 10.1063/1.3116784. [DOI] [PubMed] [Google Scholar]
- Dubbeldam D.; Calero S.; Ellis D. E.; Snurr R. Q. RASPA: Molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simul. 2016, 42, 81–101. 10.1080/08927022.2015.1010082. [DOI] [Google Scholar]
- Potoff J. J.; Siepmann J. I. Vapor-liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 2001, 47, 1676–1682. 10.1002/aic.690470719. [DOI] [Google Scholar]
- Rappé A. K.; Casewit C. J.; Colwell K.; Goddard W. III; Skiff W. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. 10.1021/ja00051a040. [DOI] [Google Scholar]
- Mayo S. L.; Olafson B. D.; Goddard W. A. DREIDING: a generic force field for Mol. Simul.s. J. Phys. Chem. 1990, 94, 8897–8909. 10.1021/j100389a010. [DOI] [Google Scholar]
- Campaná C.; Mussard B.; Woo T. K. Electrostatic potential derived atomic charges for periodic systems using a modified error functional. J. Chem. Theory Comput. 2009, 5, 2866–2878. 10.1021/ct9003405. [DOI] [PubMed] [Google Scholar]
- Otero-de-la Roza A.; Johnson E. R.; Luaña V. Critic2: A program for real-space analysis of quantum chemical interactions in solids. Comput. Phys. Commun. 2014, 185, 1007–1018. 10.1016/j.cpc.2013.10.026. [DOI] [Google Scholar]
- Fischer M.; Gomes J. R.; Jorge M. Computational approaches to study adsorption in MOFs with unsaturated metal sites. Mol. Simul. 2014, 40, 537–556. 10.1080/08927022.2013.829228. [DOI] [Google Scholar]
- Liechtenstein A.; Anisimov V.; Zaanen J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B: Condens. Matter Mater. Phys. 1995, 52, R5467. 10.1103/PhysRevB.52.R5467. [DOI] [PubMed] [Google Scholar]
- Mann G. W.; Lee K.; Cococcioni M.; Smit B.; Neaton J. B. First-principles Hubbard U approach for small molecule binding in metal-organic frameworks. J. Chem. Phys. 2016, 144, 174104. 10.1063/1.4947240. [DOI] [PubMed] [Google Scholar]
- Wang L.; Maxisch T.; Ceder G. Oxidation energies of transition metal oxides within the GGA+ U framework. Phys. Rev. B: Condens. Matter Mater. Phys. 2006, 73, 195107. 10.1103/PhysRevB.73.195107. [DOI] [Google Scholar]
- Thonhauser T.; Zuluaga S.; Arter C.; Berland K.; Schröder E.; Hyldgaard P. Spin signature of nonlocal correlation binding in metal-organic frameworks. Phys. Rev. Lett. 2015, 115, 136402. 10.1103/PhysRevLett.115.136402. [DOI] [PubMed] [Google Scholar]
- Hamada I. van der Waals density functional made accurate. Phys. Rev. B: Condens. Matter Mater. Phys. 2014, 89, 121103. 10.1103/PhysRevB.89.121103. [DOI] [Google Scholar]
- Klimeš J.; Michaelides A. Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory. J. Chem. Phys. 2012, 137, 120901. 10.1063/1.4754130. [DOI] [PubMed] [Google Scholar]
- Řezáč J.; Hobza P. Benchmark calculations of interaction energies in noncovalent complexes and their applications. Chem. Rev. 2016, 116, 5038–5071. 10.1021/acs.chemrev.5b00526. [DOI] [PubMed] [Google Scholar]
- Chen B.; Eddaoudi M.; Reineke T.; Kampf J.; O’keeffe M.; Yaghi O. Cu2 (ATC) 6H2O: Design of Open Metal Sites in Porous Metal- Organic Crystals (ATC: 1, 3, 5, 7-Adamantane Tetracarboxylate). J. Am. Chem. Soc. 2000, 122, 11559–11560. 10.1021/ja003159k. [DOI] [Google Scholar]
- Maurice R.; Sivalingam K.; Ganyushin D.; Guihéry N.; De Graaf C.; Neese F. Theoretical determination of the zero-field splitting in copper acetate monohydrate. Inorg. Chem. 2011, 50, 6229–6236. 10.1021/ic200506q. [DOI] [PubMed] [Google Scholar]
- Hu J.; Liu J.; Liu Y.; Yang X. Improving carbon dioxide storage capacity of metal organic frameworks by lithium alkoxide functionalization: a Mol. Simul. study. J. Phys. Chem. C 2016, 120, 10311–10319. 10.1021/acs.jpcc.6b01119. [DOI] [Google Scholar]
- Aprea P.; Caputo D.; Gargiulo N.; Iucolano F.; Pepe F. Modeling carbon dioxide adsorption on microporous substrates: Comparison between Cu-BTC metal- organic framework and 13X zeolitic molecular sieve. J. Chem. Eng. Data 2010, 55, 3655–3661. 10.1021/je1002225. [DOI] [Google Scholar]
- Chowdhury P.; Mekala S.; Dreisbach F.; Gumma S. Adsorption of CO, CO2 and CH4 on Cu-BTC and MIL-101 metal organic frameworks: Effect of open metal sites and adsorbate polarity. Microporous Mesoporous Mater. 2012, 152, 246–252. 10.1016/j.micromeso.2011.11.022. [DOI] [Google Scholar]
- Krishna R. Adsorptive separation of CO 2/CH 4/CO gas mixtures at high pressures. Microporous Mesoporous Mater. 2012, 156, 217–223. 10.1016/j.micromeso.2012.02.034. [DOI] [Google Scholar]
- Li B.; Zhang Z.; Li Y.; Yao K.; Zhu Y.; Deng Z.; Yang F.; Zhou X.; Li G.; Wu H.; et al. Enhanced binding affinity, remarkable selectivity, and high capacity of CO2 by dual functionalization of a rht-Type metal-organic framework. Angew. Chem., Int. Ed. 2012, 51, 1412–1415. 10.1002/anie.201105966. [DOI] [PubMed] [Google Scholar]
- Spanopoulos I.; Bratsos I.; Tampaxis C.; Vourloumis D.; Klontzas E.; Froudakis G.; Charalambopoulou G.; Steriotis T.; Trikalitis P. Exceptional gravimetric and volumetric CO2 uptake in a palladated NbO-type MOF utilizing cooperative acidic and basic, metal-CO2 interactions. Chem. Commun. 2016, 52, 10559–10562. 10.1039/C6CC04790D. [DOI] [PubMed] [Google Scholar]
- Wilmer C. E.; Kim K. C.; Snurr R. Q. An extended charge equilibration method. J. Phys. Chem. Lett. 2012, 3, 2506–2511. 10.1021/jz3008485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.