

Abstract
Liposomes and nanoparticles that bind selectively to cell-surface receptors can target specific populations of cells. However, chemical conjugation of ligands to these particles is difficult to control, frequently limiting ligand uniformity and complexity. In contrast, the surfaces of living cells are decorated with highly uniform populations of sophisticated transmembrane proteins. Toward harnessing cellular capabilities, here we demonstrate that plasma membrane vesicles (PMVs) derived from donor cells can display engineered transmembrane protein ligands that precisely target cells on the basis of receptor expression. These multi-functional targeting proteins incorporate (i) a protein ligand, (ii) an intrinsically disordered protein spacer to make the ligand sterically accessible, and (iii) a fluorescent protein domain that enables quantification of the ligand density on the PMV surface. PMVs that displayed targeting proteins with affinity for the epidermal growth factor receptor (EGFR) bound at increasing concentrations to breast cancer cells that expressed increasing levels of EGFR. Further, as an example of the generality of this approach, PMVs expressing a single domain antibody against GFP bound to cells expressing GFP-tagged receptors with a selectivity of approximately 50:1. Our results demonstrate the versatility of PMVs as cell targeting systems, suggesting diverse applications from drug delivery to tissue engineering.
Keywords: extracellular vesicles, cell targeting, protein engineering, biomaterials
A multi-functional transmembrane protein that includes an extracellular fluorophore domain for tracking, an intrinsically disordered protein spacer for ligand accessiblity and an affinity domain for targeting was designed and expressed in donor cells. Plasma membrane vesicles extracted from these cells precisely and selectively target cells on the basis of target receptor expression profiles.
1. Introduction
Over the past decade extracellular vesicles such as exosomes, microvesicles, plasma membrane-derived vesicles, and other cell-derived particles1 have shown increasing promise in diverse therapeutic applications from delivery of drugs and siRNA2,3 to facilitating cellular interactions in regenerative medicine.4 As materials derived from living cells, these particles can incorporate a diverse range of complex biological macromolecules that are challenging to integrate into synthetic biomaterials such as conventional liposomes. For example, transmembrane proteins are key constituents of extracellular vesicles and are thought to underlie their ability to target specific cells, participate in cellular signaling,5 and even fuse with the membranes of cells.6
In order to direct extracellular vesicles to specific populations of target cells, it is desirable to display biochemical ligands on their surfaces that specifically recognize receptors overexpressed by the targeted cell population. This approach is inspired by the well-documented advantages of synthetic vesicle targeting using immunoglobulins or their fragments,7–9 vitamins,10,11 glycoproteins,12 and other peptides.13 Motivated by the success of these targeted synthetic vesicles, several groups have recently reported the display of targeting ligands on the surfaces of extracellular vesicles including delivery of siRNA to the brain using exosomes decorated with neuron-specific RVG peptide;14 expression of an engineered peptide on exosome surfaces to target microRNAs to EGFR positive tumor cells;15 and expression of an integrin-specific RGD peptide on the surfaces of exosomes to target αv-integrin positive breast cancer cells for delivery of doxorubicin.16 However, by employing simple peptide domains for targeting, these studies have not taken full advantage of the cell’s capacity to produce and display sophisticated molecules on membrane surfaces. Specifically, the ability of the cellular machinery to produce highly uniform populations of complex membrane proteins that contain multiple distinct functional domains creates the opportunity for targeting ligands that simultaneously achieve multiple goals including tunable biochemical affinity, greater steric accessibility, fluorescence visualization, and others.
To take advantage of this opportunity, here we develop and characterize a set of multi-domain transmembrane targeting proteins, which can be expressed by donor cells and biologically incorporated into cell-derived vesicles. Specifically, we have designed chimeric targeting proteins that consist of a transmembrane anchor and a multi-functional extracellular domain consisting of (i) an enhanced green fluorescence protein (eGFP) domain for visualizing the vesicles and quantifying the density of ligands on their surfaces; (ii) an intrinsically disordered linker domain to increase steric accessibility of the affinity domain, and (iii) an affinity domain consisting of a biochemical ligand or single domain camelid antibody.17 Our results demonstrate expression of these targeting proteins on the surfaces of donor cells as well as the extraction of plasma membrane vesicles (PMVs) from them. Specifically, PMVs display ligands for epidermal growth factor receptor (EGFR), either epidermal growth factor (EGF) or a single domain antibody,18 at surface densities comparable to synthetic liposomes.19 These PMVs bind to the surfaces of breast cancer cells in proportion with EGFR expression levels. Further, as an example of the generality of this approach we created PMVs displaying a single domain antibody against GFP20 and illustrated specific binding of the PMVs to GFP-tagged receptors on the surface of targeted cells.
Notably plasma membrane vesicles enable expression of complex proteins on the vesicle surface with preserved directionality and functionality,21,22 bypassing the limitations of conventional chemical conjugation approaches. In particular, synthetic attachment of targeting ligands to the surfaces of liposomes and other synthetic particles is a multi-step process in which protein ligands must withstand purification, derivatization,23 and conjugation reactions.24. In contrast, by employing the cell’s own machinery for protein production, our work uses transmembrane protein engineering to produce a uniform population of multi-domain proteins capable of targeting extracellular vesicles to specific populations of cells.
2. Results and Discussion
2.1. Design and Expression of Chimeric Transmembrane Proteins for Cellular Targeting
The first model receptor we chose to target was the Epidermal Growth Factor Receptor (EGFR). Multiple human cancers including breast, non-small cell lung cancer, ovarian and colorectal cancer,25 express EGFR at elevated levels, making EGFR a popular target for molecular delivery to tumors. To precisely target cells on the basis of EGFR expression level, we began by designing a chimeric targeting protein that consisted of the intracellular and transmembrane domains of the transferrin receptor. The ectodomain of the chimeric protein consisted of an eGFP domain followed by the first 289 amino acids of the intrinsically disordered C-terminal domain of the intracellular protein AP180,26 and finally a targeting moiety, either EGF or a single domain antibody against EGFR (Figure 1A). eGFP enables direct visualization and tracking of the targeting protein using a fluorescence microscope, while the intrinsically disordered domain, much like the polyethylene glycol (PEG) polymers on synthetic liposomes,27 provides a flexible linker and spacer between the targeting ligands and the surface of the lipid bilayer, enabling the ligands to interact with the targeted cell surface receptors. The expected hydrodynamic radius of the intrinsically disordered linker is approximately 3.8 nm,28 similar to a PEG 5000 – 10000 chain.29
Figure 1. Multi-functional targeting proteins expressed by the donor cells can be harvested through the extraction of plasma membrane vesicles.

A) Cartoon schematics showing the architecture of the targeting protein, consisting of the intracellular and transmembrane domain of transferrin receptor (Tf-R), an eGFP, a long stretch of intrinsically disordered amino acids (289 aa) and an EGF ligand domain. B) Confocal images of live CHO cells transiently expressing the EGF targeting protein (green) and incubated with red ATTO 594-labeled antibodies against EGF (red). The green fluorescent cell is an example of a cell expressing the targeting protein. The dotted line shows a cell with little or no expression, which clearly does not recruit the antibody. C) Cartoon schematics of extraction of giant plasma membrane vesicles (GPMV) (left) and a confocal image of a CHO cell stably expressing the targeting protein undergoing GPMV extraction (right). Membranes are in green to indicate the expression of the targeting protein throughout the donor cell. The arrowheads point to the growing GPMVs from this donor cell plasma membrane surface. The image is intentionally saturated to show GPMV formation. D) Donor cells after GPMV extraction (left and middle) have similar morphological appearance compared to healthy cells. Hoechst 33342 staining illustrated that the nuclei remained intact after the extraction process. All scale bars represent 10 µm.
To express and test the targeting proteins, 14–16,30,31we extracted giant plasma membrane vesicles (GPMVs), from donor cells in this study. GPMVs are micrometer-scale spherical protrusions from the surface of a cell, observed during normal physiological processes such as cytokinesis,32 apoptosis,33 and cell motility34 as a result of local contraction of the actin cortex.35,36 Protocols have been established to chemically induce the shedding of a substantial proportion of the plasma membrane in the form of GPMVs.37 Having similar lipid and protein profiles to the plasma membrane of donor cells, as well as the ability to preserve transmembrane protein orientation21,22 GPMVs are frequently used in biophysical studies as a model system to understand membrane phenomena, such as phase separation,37,38 membrane protein assembly,21 and viral membrane fusion.39 However, the potential of plasma membrane vesicles as targeted biomaterials has been largely unexplored to date. A key reason for choosing plasma membrane vesicles to demonstrate the potential of the transmembrane targeting proteins developed in this work is that the mechanisms of protein trafficking to the plasma membrane are well understood, giving us confidence that the transmembrane architecture we designed would appear in tact on the membrane surface. In contrast, the mechanisms by which protein are trafficked to the surface of exosomes are still emerging.40 However, despite the lack of complete mechanistic understanding, fusion to the transmembrane domain of the lamp2 protein14 as well as simple plasma membrane overexpression15 have both resulted in display of simple targeting peptides on exosome surfaces as cited above. These strategies could be used in the future to display the multi-domain targeting protein architectures developed in this work the surfaces of exosomes as well as nanoliposomes extracted from cells by other means, including serial extrusion of cells 30,31.
The first targeting protein we constructed used EGF as the ligand, chosen for its strong and well-characterized binding affinity with EGFR (Figure 1A).41 We began by testing the cell-surface expression of the EGF targeting protein using a live cell, fluorescence-based antibody-binding assay. CHO cells transiently expressing the EGF targeting protein showed a robust eGFP signal at the cellular plasma membrane (Figure 1B left and S1). Binding of ATTO 594 labeled-EGF antibodies to the cell surface demonstrated that EGF was expressed on the extracellular leaflet of the phospholipid bilayer (Figure 1B middle and S1). In addition, cells from the same culture dish with little or no expression of the eGFP-tagged targeting protein did not recruit antibodies against EGF, further confirming the specific binding between the antibodies and the chimeric targeting protein (Figure 1B right). Notably, the GFP-tagged EGF targeting proteins are translated and produced in the endoplasmic reticulum before being transported to the Golgi apparatus for post-translational modifications, and eventually trafficked onto the plasma membrane of the cells. Therefore, the fluorescence signal of the GFP-tagged targeting protein is expected to exist throughout the cell interior, as observed in the left panel of Figure 1B. In contrast, the antibody binds from the outside of the cell and is therefore expected to be present primarily on the outer cell surface, though uptake of the antibody during receptor recycling produces some internal antibody signal, as shown in the middle panel of Figure 1B.
Following the expression of functional targeting protein, we extracted GPMVs from these donor cells (Figure 1C). After the extraction process the donor cells remained attached to the culture dish and fluorescent images suggested that they had similar morphological appearances to normal donor cells, in agreement with prior studies.42 Hoechst 33342 staining showed that the nuclei of the donor cells remained intact (Figure 1D) and the GPMVs were free of nuclear contamination (Figure S2). Further, the proteins on the surface of the GPMVs are expected to remain active after vesiculation as demonstrated by several previous reports including studies on glycophorin A21 and hemagglutinin (HA) fusion proteins39 extracted by plasma membrane harvesting using the same protocol.
To test whether targeting proteins on GPMV surfaces are able to engage in molecular binding, ATTO 594 labeled antibodies against EGF were incubated with GPMVs. Anti-EGF bound to the surfaces of GPMVs that displayed the EGF targeting proteins (Figure 2A and Figure S3A). In contrast the labeled antibodies did not bind to GPMVs that lacked a significant eGFP signal, indicating lack of significant expression of the EGF targeting protein (Figure 2A, left and right). Fluorescence intensity analysis of the eGFP and ATTO 594 signals demonstrated a correlation between the display of the targeting protein and the extent of antibody binding (Figure S3B). Taken together, these data demonstrate that GPMVs extracted from donor cells expressing the EGF targeting protein displayed the targeting protein on their surfaces such that the ligand domain was accessible to the external solution.
Figure 2. Vesicles extracted from the plasma membranes of donor cells display functional targeting proteins on their surfaces at a high density.

A) Confocal images of GPMVs derived from CHO cells transiently expressing the EGF targeting protein (green) and incubated with red-labeled antibodies against EGF. Fluorescent GPMV displays the targeting protein while the brightfield image shows three other GPMVs that do not display the targeting protein, presumably because they came from cells with low expression levels. The GPMVs that do not display the targeting protein clearly did not recruit the antibody. Scale bar represents 10 µm. B) A calibration curve of GFP fluorescence. A linear fit to the curve was used to calculate the GFP content of a solution of GPMVs based on the intensity of eGFP fluorescence of the solution. The measured average of GPMV brightness represents 5 independent trials normalized to 2×107 GPMVs and the error bars represent the standard deviation. 7D12 protein is an alternative targeting protein we developed using a single chain variable domain only antibody, otherwise known as a nanobody, against EGFR as the ligand. A detailed discussion on the 7D12 protein can be found after the dicussion of Figure 4. C) Confocal z-stack images of vesicles were taken and the frame where the vesicles settled on the glass coverslip and appeared as a solid circle of relatively uniform intensity was chosen for analysis. The average fluorescence intensity of the vesicle was determined from the intensity profile (shown in white). Scale bar represents 10 µm. D) Copies of targeting proteins per square micrometer. The histogram shows the brightness distribution of 43 GPMVs. E) Transmission electron micrographs of plasma membrane vesicles showed that they have similar morphology to other liposomal particles.
2.2. Quantifying the density of targeting proteins on PMV surfaces
Several studies have shown that increasing the density of ligands on the surfaces of targeted particles can significantly increase nanoparticle binding to target cells, increasing the cell-particle binding affinity by as much as 10-fold.19,43,44 Therefore, having a sufficient density of ligands on the surfaces of targeted particles is critical to achieving high affinity binding. In particular, Nielsen et al have reported robust uptake of synthetic liposomes by target cells using a density of 10–30 ligands per 100-nm diameter liposome,19 a density of 300–1000 ligands per square micrometer of the particle surface. To estimate the density of targeting proteins displayed on the surfaces of GPMVs, we developed two distinct fluorescence-based approaches. The first is based on measuring the calibrated total fluorescence of the GPMV sample normalized by an estimate of its total membrane content, while the second is based on calibrated fluorescence intensity measurements of individual GPMVs. Conventional methods were used to produce a stable cell line expressing the EGF targeting protein. Notably, more than 80% of the stably transfected cells expressed significant levels of the targeting proteins, as demonstrated by elevated fluorescence intensity in the GFP channel during flow cytometry-based characterization (Figure S4). GPMVs were extracted from these cells as described in experimental section (Figure 1C). Expression of the EGF targeting protein was confirmed by immunoblotting GPMVs with an antibody against EGF (Figure S5).
First, based on the total fluorescence of GPMVs in solution and an average GPMV diameter of 11 µm (Figure S6, see methods), we determined that there were on average 400 copies of the EGF targeting proteins per square micrometer of the vesicle surface (Figure 2B red). We estimate that each targeting protein occupies an area of 50 nm2 on the membrane surface, based on a worm-like chain model of the intrinsically disordered domain.28,45 Combining this estimate of the area per protein with the measured density of targeting proteins on the membrane surface, the EGF targeting proteins cover approximately 2% of the total membrane surface. The auto-fluorescence of GPMVs derived from CHO cells without GFP expression was also measured and found to be small in comparison to the GFP signal (Figure S7).
As a second estimate of ligand density, we employed a quantitative fluorescence microscopy assay on individual GPMVs. In comparison to the bulk method described above, we expect a higher density of targeting proteins from this assay since GPMVs that lack significant eGFP fluorescence intensity cannot be clearly visualized on the basis of fluorescence and are thus under-represented in the analysis. To calculate the number of targeting proteins displayed per diffraction-limited unit of membrane area, we divided the mean fluorescence intensity of the GPMV surface (Figure 2C) by the integrated brightness of a single eGFP molecule. Forty total GPMVs from 3 independent sample preparations yielded an average of 1200 (400–2200) copies of the EGF targeting protein per square micrometer (Figure 2D). A detailed explanation of the targeting ligand density calculations can be found in experimental section of this manuscript. Notably, both measures of targeting protein density fall within or above the range cited above from the work of Nielsen et al and are therefore expected to provide robust targeting of plasma membrane vesicles. The substantial variation in the targeting protein density among GPMVs likely arises from variation in targeting protein expression among the donor cells, suggesting that sorting or gene editing of the donor cells would provide a more uniform targeting protein density.
2.3. EGFR Targeting is Sensitive to Cellular Receptor Expression
To evaluate cell targeting, GPMVs were extruded through one-micrometer polycarbonate filters to produce plasma membrane vesicles (PMVs). Vesicles of this size are convenient for targeting studies because they are small enough to avoid gravitational settling yet large enough to track easily using fluorescence microscopy. However, PMVs can be further extruded through 100 nm filters to produce a homogenous population of vesicles of the appropriate size for in vivo studies (Figure S8 and S9). Transmission electron micrograph images conveyed that PMVs have similar morphology to other liposomal particles (Figure 2E). To investigate the ability of PMVs to target specific cells (Figure 3A), PMVs expressing the EGF targeting protein were incubated with HeLa cells transiently expressing mRFP-tagged EGFR. At the end of the incubation following repeated washing of the cells, there was extensive colocalization of PMVs (eGFP signal) with cells overexpressing mRFP-tagged EGFR (Figure 3B). In contrast, PMVs bound much less strongly to cells in the same culture dish that lacked a significant mRFP-EGFP signal (Figure 3C). Notably, HeLa cells express EGFR endogenously,46 such that some binding of EGF-PMVs to all cells was expected.
Figure 3. PMVs displaying targeting proteins bound to EGFR-expressing cells.

A) Cartoon schematics of the targeting proteins. B) Confocal images of HeLa cells transiently overexpressing mRFP-tagged EGFR extensively recruited EGF-PMVs. C) Cells from the same culture dish, which had low levels of mRFP-tagged EGFR expression, recruited the PMVs to a much lesser extent. Both red and green fluorescent images from B and C are under identical brightness and contrast setting. The brightfield image was overlaid to show the cell.. D) Dosage response curve for EGF-PMVs binding to MDA-MB-468 cells, a cell line with high endogenous EGFR expression. Each point is the average of 3 independent trials and the error bars show the standard deviation. At low PMV to cell ratios the peak shifts were small and variable owing to variations in the cellular autofluorescence. However as the PMV to cell ratio increased, a clear correlation with increasing peak shift was observed. Example histograms from flow cytometry analysis showing the shift in GFP-channel fluorescence upon exposure of the cells to PMVs. From top to bottom, the histograms correspond to 100 PMVs per cell, 600 PMVs per cell and 1300 PMVs per cell respectively. The p values are derived from a one-tailed unpaired t-test on the mean fluorescence values with (+ vesicle) and without vesicles (− vesicle). All scale bars, 10 µm.
We also quantified the amount of PMV binding to cells with a high endogenous level of EGFR (MDA-MB-468 cells), as a function of increasing PMV concentration. Specifically, the shift in fluorescence intensity in a spectral region corresponding to eGFP was quantified using flow cytometry (Figure 3D right). Cells were incubated with EGF-PMVs at a range of concentrations for 4 hours at 37 °C. The cells were carefully washed 3 times with PBS to remove unbound PMVs and then trypsinized for flow cytometric analysis. As the concentration of PMVs increased, binding to MDA-MB-468 cells also increased (Figure 2D left), indicating a positive correlation between PMV dosage and binding.
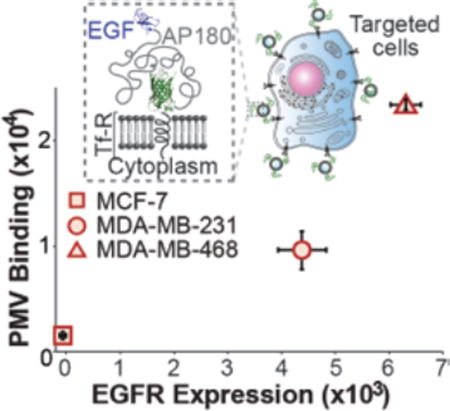
To further confirm the specificity of EGF-PMVs for EGFR expressing cells, we used three breast cancer lines (MCF-7, MDA-MB-231, and MDA-MB-468), which have increasing endogenous levels of EGFR expression.47 We confirmed this trend of increasing EGFR expression using flow cytometry studies on cells exposed to a fluorescent-labeled antibody against EGFR, obtaining a trend consistent with literature values (Figure S10). Following this confirmation, the three cell types were individually incubated with EGF-PMVs. PMV bound cells were first visualized using fluorescence confocal microscopy. As expected, the PMVs bound most abundantly to the MDA-MB-468 cells, which had the highest EGFR expression level, while they bound least to the MCF-7 cells, which had the lowest EGFR expression level (Figure 4A). To quantify the amount of binding, cells that had been incubated with PMVs were washed and analyzed using flow cytometry as described above. The results from these experiments confirmed an increasing level of EGF-PMV binding to the cells as the expression level of EGFR increased, demonstrating that EGF-PMVs are sensitive to EGFR expression level (Figure 4B and 4C; example scatterplots see Figure S11).
Figure 4. PMV binding to cells is correlated with cellular expression of EGFR.

A) Breast cancer cell lines with increasing EGFR expression level recruit increasing densities of EGF-PMVs. The green fluorescent images are maximum intensity Z projects. B) Example histograms from flow cytometry analysis showing an increase in the GFP-channel fluorescence upon exposure of cells to EGF and 7D12-PMVs. Breast cancer cells were incubated with PMVs at a concentration of 1300 PMVs per cell. The p values are derived from a one-tailed unpaired t-test on the mean fluorescence values with (+ vesicle) and without vesicles (− vesicle). C) Mean fluorescence analysis quantified using flow cytometry, showing PMV binding vs. EGFR expression level. The relative EGFR expression level of these cancer cells was also quantified using flow cytometry (Figure S10). The error bars represent the standard deviation of 3 independent measurements. All scale bars, 10 µm.
As an alternative to the EGF ligand, we also developed PMVs that used the 7D12 nanobody against EGFR as the targeting ligand.18 This choice of targeting ligand is more appropriate for therapeutic applications, since it lacks the potential mitogenicity of a growth factor. Specifically, nanobodies, single-domain antibodies derived from camelids,18,20,48 have nanomolar binding affinities and are much smaller in size, (~15 kDa18,20,49), in comparison to conventional antibodies, averaging around 150 kDa.50 They have emerged as a useful tool for cellular targeting, as studies have shown that gold nanoparticles chemically conjugated to nanobody against human epidermal growth factor receptor 2 (HER2) bound selectively to HER2 overexpressing cells.51 The 7D12 nanobody binds EGFR with high affinity and blocks downstream EGFR signaling.18 As such, the 7D12 nanobody provides an alternative to the EGF ligand for targeting EGFR positive cells (Figure 3A). 7D12-PMVs were prepared from a CHO cell line stably expressing the 7D12 targeting protein, using the same procedures used to prepare EGF-PMVs. The density of the 7D12 targeting protein on PMVs was somewhat lower in comparison to expression of the EGF targeting protein, with an average of over 300 copies per square micrometer (based on the ensemble assay), yielding a total surface coverage of approximately 1.6% (Figure 2B blue).
When incubated with each of the three breast cancer cell lines (MCF-7, MDA-MB-231, and MDA-MB-468), 7D12-PMVs behaved similarly to EGF-PMVs, showing a trend of increasing binding with increasing EGFR expression level (Figure 4B and 4C), though the absolute fluorescence values in the flow cytometry studies were somewhat lower. The reduced signal from 7D12-PMVs likely resulted from two factors. First, 7D12, has a reported dissociation constant for EGFR binding of 200 nM,18 which is substantially higher than the dissociation constant for EGF binding to EGFR, 5 nM,41 indicating weaker binding. Further, the density of the 7D12 targeting protein was approximately 30% less than the density of EGF targeting proteins, as noted above (Figure 2B). Nonetheless, 7D12-PMVs demonstrated clear sensitivity to EGFR expression level.
2.4 Targeting cells that express GFP-tagged receptors
Our work so far has demonstrated selective binding of PMVs on the basis of EGFR expression level using two different targeting ligands. To evaluate whether this strategy can be extended to an arbitrary receptor, we designed a targeting protein that selectively binds to any GFP-tagged receptor. The ligand domain of this targeting protein is a single domain antibody that specifically recognizes GFP (Figure 5A). For this targeting protein the fluorophore domain consisted of mRFP, rather than eGFP, so that the targeting protein and its ligand (GFP) would have distinct fluorescent signatures. Creating PMVs that target GFP-tagged receptors provides an opportunity to evaluate the absolute specificity of PMVs for target cells, since cells lack endogenous GFP expression. Further, the ability to target PMVs to cells that express GFP-tagged receptors could be useful for molecular delivery to engineered cell lines in complex contexts such as engineered tissues and cell implantation studies, where engineered cells are surrounded by other cell types. 52
Figure 5. Targeted PMVs provide a general strategy for specific binding to any cell-surface protein, including GFP-labeled receptors.

A) Cartoon schematics showing the GFPnb targeting protein binding to soluble eGFP. Here the ligand, GFPnb, is a nanobody against GFP. B) Confocal images of CHO wild type cells transiently expressing the GFPnb targeting protein. The single bright cell in the mRFP fluorescent channel is the only cell in this field of view that expresses the GFPnb targeting protein (compare brightfield and mRFP fluorescent images). As expected, only this cell recruits soluble eGFP from solution, demonstrating specific binding. C) Confocal images of a GPMV derived from CHO cells stably expressing the GFPnb targeting protein (red). Incubation of GPMVs with soluble eGFP shows binding. D) Display of the GFPnb targeting protein is correlated with recruitment of soluble eGFP based on fluorescence intensity analysis. A total of 39 GPMVs were analyzed, using two measurements per GPMV on opposite edges. All scale bars, 10 µm.
Following the expression of the GFPnb targeting protein by donor cells, the external accessibility and functionality of the targeting ligand was tested. We transiently expressed the GFPnb targeting protein in CHO cells and then incubated these with soluble eGFP (Figure 5B). The soluble eGFP bound significantly only to cells expressing the GFPnb targeting protein and was not recruited by cells in the same dish that lacked significant expression of the targeting protein. Further, GPMVs derived from CHO cells stably expressing the GFPnb targeting protein were also capable of recruiting soluble eGFP from solution (Figure 5C), confirming that the GFP nanobody on the surfaces of GPMVs was accessible to the external environment and able to bind to eGFP. Fluorescence intensity analysis of the GPMVs and the soluble eGFP revealed a correlation between the expression of the GFPnb targeting proteins and the amount of soluble eGFP binding (Figure 5D).
Next, we evaluated the ability of GFPnb-PMVs to target eGFP-expressing cells. In particular, we conducted a competitive binding assay where CHO cells stably expressing eGFP on the cell surface (GFP positive cells) and CHO wild type cells (GFP negative cells) were co-cultured in a single culture dish (Figure 6A). At the end of incubation, extensive colocalization between PMVs and the cell membrane of GFP positive cells was observed. In contrast GFP negative cells bound significantly fewer PMVs (Figure 6B). This experiment was repeated using purified PMVs (Figure S12), which also selectively bind to GFP positive cells, in agreement with the results shown here. Detailed purification methods can be found in experimental section of this manuscript. To confirm this finding, flow cytometry analysis was conducted on the co-cultured cells. The significant difference in green channel fluorescence signal was used to distinguish GFP positive and control cells on a cell-by-cell basis (Figure 6C top). Only the GFP positive cells had a detectable increase in the mRFP fluorescence channel as a result of PMV binding (Figure 6C bottom). We calculated the mean fluorescence increase in the mRFP channel for the two cell populations (Figure 6D). The results indicated that the PMVs have a selectivity for GFP positive cells of approximately 50:1, which is comparable to the selectivity that chemically conjugated synthetic liposome particles can achieve for their target cells.7 Collectively, these results demonstrate that GFPnb-PMVs bind selectively to cells expressing GFP tagged transmembrane proteins.
Figure 6. GFPnb-PMVs bind to eGFP expressing cells with high specificity.

A) Cartoon schematics showing competitive binding assay. Only cells that express GFP-tagged receptors on their surfaces are expected to recruit GFPnb-PMVs. B) The specificity GFPnb-PMV binding to cells was evaluated by co-culturing GFP negative and GFP positive cells in a 1:1 ratio (brightfield, green) and then exposing them simultaneously (in the same culture dish) to GFPnb-PMVs (red). GFP positive cells recruited GFPnb-PMVs in substantially greater quantities. The red fluorescent image is a maximum intensity Z projection. C) Flow cytometry analysis of GFPnb-PMV binding to the co-cultured cells. Top: The green fluorescence signals of GFP positive and negative cells were used to set separate gates to distinguish these two co-cultured populations of cells. Bottom: The overlay of recipient cell fluorescence with (red) and without (blue) GFPnb-PMVs. Only the GFP positive cells (right) have a clearly detectable fluorescence shift upon PMV binding. D) Mean fluorescence increase owing to GFPnb-PMV binding. E) GFPnb PMVs were used to demonstrate the preservation of transmembrane protein topology. GPMVs extracted from CHO cells transiently express transmembrane receptors with an extracellular GFP recruited GFPnb PMVs (top) while those from CHO cells transiently express EGFR with an intracellular GFP did not (bottom). The line scans show the intensity of mRFP signal from GFPnb PMVs. These images were taken under same camera setting and displayed using identical brightness and contrast for direct comparison. All scale bars, 10 µm.
Lastly, while the preservation of membrane protein orientation in membrane blebs has been frequently demonstrated in the literature,21,22 we used GFPnb PMVs to explicitly demonstrate this principle for the transmembrane targeting proteins developed in this work. Specifically, we transiently transfected separate populations of CHO cells with plasmids encoding recombinant proteins, one of which displays an extracellular GFP domain (Figure 6E, top), while the other displays an intracellular GFP domain (Figure 6E, bottom). GPMVs were harvested from each population of transfected cells using the protocols described in the experimental section. Then the GPMVs were incubated with GFPnb PMVs. Line plots of the fluorescence intensity of the GPMV membranes showed that GFPnb PMVs bound to GPMVs displaying extracellular GFP. In contrast, no detectable binding was observed between GFPnb PMVs and GPMVs displaying intracellular GFP, indicating that a GFP domain expressed on the inner leaflet of the plasma membrane remains inaccessible on the surfaces of GPMVs. These results demonstrate that the process of harvesting GPMVs preserves the orientation of these model transmembrane proteins.
3. Conclusion
This work has demonstrated that plasma membrane vesicles (PMVs) can display a high density of multi-functional transmembrane targeting proteins, creating a versatile system for targeting cells on the basis of receptor expression. Specifically, we have extracted PMVs from donor cells expressing engineered transmembrane targeting proteins. These targeting proteins employ a modular architecture that combines fluorescence visualization with the display of a range of sterically accessible ligands including growth factors and nanobodies. PMVs displayed high densities of these targeting proteins, comparable to the density of antibodies conjugated to the surfaces of targeted synthetic liposomes.19 Quantitative flow cytometry-based studies demonstrated that cellular binding of PMVs was highly sensitive to the cellular level of receptor expression, with a selectivity of 50:1 over off-target cells, comparable to synthetic targeted particles.7 To our knowledge, this work represents the first example of extracellular vesicle targeting using multiple types of protein ligands for which the ligand density on the particle surface and the selectivity of cellular targeting have been quantified.
The unique ability of cells to manufacture sophisticated transmembrane proteins endows extracellular vesicles with some desirable features that are difficult to achieve using chemically-conjugated synthetic particles. In particular, by taking advantage of the cellular biosynthesis and trafficking machineries, the engineered proteins can have multiple domains and functions without loss of homogeneity. Beyond the basic demonstrations in this work, multi-domain transmembrane targeting proteins could be engineered to further improve the sophistication and precision of cellular targeting by several mechanisms. For example, the affinity of ligands for their targets can be increased by serially repeating copies of the targeting domain connected by short disordered linkers. Here the affinity could be tuned by varying the number of copies and the lengths of linkers between them. Additionally, combining several distinct ligand domains could be used to target multiple receptors and cell types, helping to address challenging problems such as tumor cell heterogeneity.53 Beyond cell targeting, PMVs can be engineered to display transmembrane proteins that drive other cellular processes including cell signaling and membrane fusion. Ultimately, the ability of cell-derived particles to display a diverse range of transmembrane protein architectures will enable the development of multi-functional biomaterials for a broad range of biomedical applications.
4. Experimental Section
Chemical Reagents
DTT (dithiothreitol), PFA (paraformaldehyde), NaCl, CaCl2, HEPES (4-(2-hydroxyehtyl)-1-piperazineethanesulfonic acid), imidazole, EDTA (ethylenediaminetetraacetic acid), sodium bicarbonate, TCEP-HCl solution (Tris(2-carboxyehtyl)phosphine hydrochloride) and ATTO 594-NHS ester, were purchased from Sigma-Aldrich (St. Louis, MO). β -ME (β-mercaptoethanol) was purchased from Fisher Scientific (Waltham, MA). Trypan blue was purchased from Life Technologies (Charlesbad, CA). Trypsin, penicillin, streptomycin, L-glutamine, PBS (phosphate buffered saline), Ham’s F-12, Ham’s F-12 without phenol red, DMEM (Dulbecco’s modified Eagle medium) and DMEM without phenol red were purchased from GE Healthcare (South Logan, UT). FBS (fetal bovine serum) was purchased from both GE Healthcare and Life Technologies. Geneticin (G418) was purchased from Corning (Corning, NY). All chemical reagents were used without further purification.
Plasmid constructs
EGF (Tf-RΔEcto-eGFP AP180 EGF) and 7D12 (Tf-RΔEcto-eGFP AP180 7D12) DNA constructs. The cDNA of human EGF (BC093731), acquired from Mammalian Genome Collection, and the 7D12 plasmid (pET22b backbone), kindly provided by Dr. Kathryn M. Ferguson (University of Pennyslvania), were PCR amplified using primers containing MluI sites. PCR amplified products were restriction cloned into the Tf-RΔEcto-eGFP AP180 CTD plasmid, the construction of which we have previously described.28 GFP nanobody construct (Tf-RΔEcto-mRFP AP180 GFPnb). The Tf-RΔEcto-mRFP AP180 CTD plasmid was first constructed by excising eGFP from the Tf-RΔEcto-eGFP AP180 CTD plasmid above with BamH1 and SalI digestion and inserting the PCR amplified mRFP (Addgene plasmid #13032, pcDNA3 backbone), a generous gift from Dr. Douglas Golenbock (University of Massachusetts Medical School). The pOPINE GFP nanobody sequence, a gift from Brett Collins (Addgene plasmid #49172), was PCR amplified and restriction cloned into Tf-RΔEcto-mRFP AP180 CTD using primers containing MluI sites. EGFR-mRFP construct. The EGFR-mRFP plasmid was constructed by excising the GFP from the EGFR-GFP plasmid54 (Addgene #32751) kindly provided by Dr. Alexander Sorkin (University of Pittsburgh) with HindIII and NotI enzymes and inserting the PCR amplified mRFP above. Model transmembrane protein with extracellular eGFP construct (Tf-RΔEcto-eGFP EGF). The Tf-RΔEcto-GFP construct which we have described previously28 was modified without a stop codon. PCR ampliflied EGF was then inserted in frame with the modified Tf-RΔEcto-GFP backbone by digestion with NotI enzymes. Model transmembrane protein with intracellular eGFP construct (EGFR-GFP). As described above, this plasmid (Addgene #32751) was a generous gift from Dr. Alexander Sorkin (University of Pittsburgh). All constructs were confirmed by DNA sequencing.
Tissue culture and cell transfection
CHO-K1 cells were purchased from American Type Culture Collection (ATCC). These cells were cultured in Ham’s F-12 nutrients mixture supplemented with 10% Fetal Bovine Serum (FBS), 1% penicillin, 1% streptomycin, 1% L-glutamine (PSLG). Wild type HeLa cells were also purchased from ATCC. MDA-MB-468 and MDA-MB-231 cells were a kind gift from Dr. Tim Yeh (UT Austin). MCF-7 cells were a kind gift from Dr. Amy Brock (UT Austin). These cells were cultured in DMEM high glucose supplemented with 10% FBS and 1% PSLG. All cells were incubated at 37 °C with 5% CO2 and passaged every 48 to 72 hours. For transfection, wild type HeLa or CHO cells were grown for 24 hours on acid-cleaned 22 mm square glass coverslips (Fisherbrand) in 6 well plates (Corning) before being transfected with chimeric plasmids DNA using Fugene transfection reagent (Promega). For expression and binding studies, 1 µg of plasmid was used for transfection and the studies were conducted one day after transfection. For making stable expression cell lines, 2 µg of plasmid was used in the initial transfection.
Fluorescence Microscopy
A Zeiss Axio Observer Z1 Spinning Disc confocal microscope with Yokagawa CSU-X1M was used to image live cells and vesicles. Both fluorescent and brightfield images were collected using a Plan-Apochromat 100X, 1.4 NA oil immersion objective. Two laser wavelengths of 488 nm and 561 nm were used for excitation and three filters: 525 nm with a 50 nm width, 629 nm with a 62 nm width and a triple pass dichroic mirror 405/488/561 nm were used for emission. For spinning disk confocal and brightfield imaging, cells were cultured on acid-cleaned 22 mm square coverslips (Fisherbrand) or 35 mm collagen coated glass bottom dishes (MatTek). All experiments were imaged on a cooled (−70 °C) EMCCD iXon3 897 camera (Andor Technology; Belfast, UK). Fluorescence and brightfield images have been optimized for brightness and contrast using imageJ software (NIH). A Zeiss Axio Observer Z1 microscope with conventional epi-fluorescence capabilities was used to screen fluorescence of cell colonies in selecting stable expression cell lines.
Flow Cytometry
An Accuri C6 Flow Cytometer (BD Biosciences) with 488 nm and 551 nm lasers was used for all flow cytometry assays. Two band-pass filters centered at 530 nm with a 30 nm width, and 610 nm with a 20 nm width were used to establish the detecting channels. All data was collected at a flow rate of 35 µL/min and the gate was drawn on the majority population on the scatterplot. Once the appropriate gate was determined, the same gate was applied to all experiments of the same cell types. All flow cytometry data was analyzed using FlowJo software (Treestar).
Transmission Electron Microscopy
Three microliters of GFPnb PMVs were applied to glow-discharged 300 mesh carbon-coated copper grids (Electron Microscopy Science, Hatfield, PA). After PMVs had been allowed to be absorbed onto the grid for 1 minute, the grids were negatively stained with 2% uranyl acetate (Electron Microscopy Science, Hatfield, PA). Electron micrographs were recorded with a FEI Tecnai TEM (Technai, Hillsboro, OR).
Production of Stable Cell Lines
Wild type CHO-K1 cells were transfected with chimeric protein plasmids as described above. One day following transfection, cells were washed once with F-12 media before being cultured in the same media with an additional 0.4 mg/mL geneticin (Corning). Cell media was changed and fresh geneticin added every 48 hours until confluency. Cells were then transferred to a 100 mm tissue culture treated dish (Corning) and cultured in F-12 media with geneticin. Once cells were confluent, they were plated onto a 96 well plate (Corning) at a density of 50 cells/well with geneticin free F-12 media. All 96 wells were screened for expression of fluorescent proteins 4 to 6 days after plating. Wells with the brightest cell colonies were selected and cells transferred into 6 well plates and subsequently 100 mm dishes until confluency. Cell media was changed at least every 48 hours and fresh geneticin added at a concentration of 0.4 mg/mL. Expanded cells were once again plated onto fresh 96 well plates at a density of 1 cell/well. Fluorescence of cell colonies was screened 7–10 days following plating. Wells with a single cell colony and strong fluorescence were selected and expanded as a stable expression line of chimeric constructs. Following selection, cells were propagated in geneticin-free F-12 media. The fluorescence of the stable expression line was characterized using flow cytometry and compared to the fluorescence of the CHO wildtype cells. Both cells were resuspended in 50 µL of phosphate buffered saline for analysis.
Preparation of Cell-derived Vesicles and Purification
According to published protocols,37 giant plasma membrane vesicles (GPMVs) were derived from donor cell lines that stably expressed the targeting proteins. These donor cells were rinsed twice with GPMV buffer (10 mM HEPES, 2 mM CaCl2, 150 mM NaCl, pH 7.4) and once with active buffer (10 mM HEPES, 2 mM CaCl2, 150 mM NaCl, 25 mM PFA, 1 mM DTT). The cells were then incubated in active buffer at 37 °C for 6 hours. Supernatants containing GPMVs were collected and concentrated by centrifugation at 17,000 × g for 30 minutes at 4 °C. The GPMV pellet was then resuspended in 100 µL fresh DMEM phenol red free media. Notably, the concentration of fixative (PFA) used in the GPMV extraction procedure is 100 times lower than the concentration used for fixation reactions, such that very limited protein fixation is likely to occur, allowing most proteins to remain active. To test whether the GPMVs were free of contamination from the nuclei, both the donor cells after vesicles extraction and the resulting GPMVs were stained with Hoechst 33342 (Life Technologies) at a concentration of 6 µg/mL for 30 minutes prior to imaging. To determine the average size of the GPMVs, the diameters of 100 GPMVs from 3 independent sample preparations were analyzed using images taken in the green fluorescent channel. To prepare vesicles for targeting assays, resuspended GPMVs were extruded to PMVs through 1.0 µm pore polycarbonate filters (VWR) with 11 passes. The efficiency of extrusion was measured using a Cytation 3 fluorimeter (BioTek). Fluorescence intensities with excitation of 485 nm and emission of 515 nm were collected before and after the extrusion process. The percentage of GPMVs surviving the extrusion process was then estimated by dividing the background subtracted fluorescence intensity after extrusion by the background subtracted fluorescence intensity before extrusion. The extruded vesicles were pelleted using a Sorvall MX150+ ultracentrifuge (Thermo Fisher Scientific) at 100,000g for 1 hour at 4°C and washed once with phenol red free DMEM media.
eGFP purification
The pRSET vector containing the non-dimerizable hexa-his-tagged eGFP (hisGFP A206K) was generously shared by Dr. Adam Arkin (University of California, Berkeley). Following published purification protocols,55,56 his-eGFP was expressed in BL21(DE3) pLysS cells overnight at 18 °C and purified from bacterial extracts by incubating with Ni-NTA agarose beads in 25 mM HEPES, pH 7.4, 150 mM NaCl, 1mM imidazole, and 1 mM β-ME. After extensive washing with 25 mM HEPES pH 7.4, 150–300 mM NaCl, 15mM imidazole and 1 mM β-ME, proteins were eluted via gradient imidazole wash to a final concentration of 250 mM. Eluted proteins were concentrated and dialyzed in 2 L 25 mM HEPES, pH 7.4, 150 mM NaCl, 1mM EDTA and 1 mM β-ME at 4 °C overnight and again for 2 hours in fresh buffer at 4 °C.
Anti-EGFR antibody labeling
Anti-EGFR (Clone E225, Sigma Aldrich) was first buffer exchanged to 150 mM sodium bicarbonate, 140 mM NaCl, 1 mM TCEP, pH 8.2 using Centri-spin 20 size exclusion columns (Princeton Separations). The labeling reactions were performed using amine-reactive, NHS-ester functionalized dyes ATTO 594 (ATTO-TEC). The dye was added to protein solution and the reactions proceeded at room temperature for 30 minutes. The amount of dye added was optimized to achieve a labeling ratio of approximately 1:1. The unconjugated dye was then removed using another Centri-spin 20 size exclusion column (Princeton Separations). The final antibody concentration was measured on a Nanodrop 2000 spectrophotometer (Thermo Scientific) and the labeling ratio estimated according to the published formula by ATTO-TEC.
Quantification of targeting ligands on vesicles
Ensemble fluorescence measurements. All fluorescence intensity readings in this study were measured using a Cytation 3 fluorimeter (BioTek). Purified eGFP was serially diluted to generate a calibration curve of fluorescence intensity vs. protein concentration. eGFP concentrations were determined by measuring the absorbance at 280 nm using a Nanodrop 2000 spectrophotometer (Thermo Scientific). The total number of GPMVs in a sample preparation as described above was estimated by counting the GPMVs on a hemacytometer under a 20x objective. Based on the estimated average diameter and the number of GPMVs, the total lipid surface area was calculated. The fluorescence of the GPMV samples were measured and plotted against the calibration curve to calculate the molar concentration of eGFP in the GPMV sample. This concentration was converted to estimate the number of eGFP molecules and then divided by the total lipid surface area to yield an average number of eGFP molecules per 1 square micron on the vesicle surface. Since each targeting construct molecule contains only 1 eGFP molecule, the calculated number of eGPF molecules is equivalent to the number of targeting protein molecules. To determine the percent surface area coverage, each targeting protein was 2 assumed to have a 50 nm projected area on the membrane based on polymer theory. The average surface area of one GPMV was calculated based on the average diameter. A division between the area covered by the transmembrane targeting proteins on one GPMV and the total surface area of this GPMV yielded the percent surface area coverage. Quantitative imaging measurements. We divided the brightness of the GPMV membrane by the integrated28 total brightness of individual eGFP molecules. As described previously, the brightness of individual fluorescent molecules was determined by adding a dilute solution (~50 pM) of eGFP to an ultraclean coverslips hydrated with PBS followed by imaging the surface of the coverslips. The acquired images contained sparse fluorescent puncta having uniform intensities, which correspond to individual eGFP molecules. These puncta were selected and a 2D Gaussian function was fit to the background subtracted pixel intensity. This function was integrated to determine the brightness of individual eGFP molecules. Confocal images of GPMVs were then acquired using the same laser power and camera gain settings. The confocal slice where the GPMVs rested on the coverslip and formed a region of approximately uniform fluorescent intensity was selected. The average intensity of this region was calculated. The number of targeting proteins was estimated simply by dividing the background subtracted average intensity of the GPMV membrane by the integrated brightness of individual eGFP molecules.
Targeting Protein Expression and Function
Western blot analysis. GPMVs derived from one confluent 100 mm dish of wild type CHO cells as well as from one dish of CHO cells stably expressing the EGF targeting protein were collected separately and boiled at 100 °C for 10 minutes for western blotting. Protein extracts, separated by SDS-PAGE and transferred onto nitrocellulose membranes, were probed with antibodies against EGF (sc-166779, 1:200, Santa Cruz Biotechnology, Santa Cruz, CA). Antibody binding was detected with HRP conjugated goat anti-mouse IgG (sc-2005, 1:2000, Santa Cruz Biotechnology, Santa Cruz, CA) and visualized with SuperSignal west femto maximum sensitivity substrates (ThermoFisher Scientific). Live cell and vesicle antibody binding. To demonstrate protein expression on the cell surface and functional binding, CHO cells transiently expressing the EGF targeting protein were incubated with ATTO 594 labeled EGF antibody at a concentration of 220 nM. Fluorescence images were collected 20 minutes after antibody incubation. The same study was conducted on GPMVs derived from CHO cells transiently transfected with the same plasmid DNA. Soluble eGFP binding. CHO cells transiently expressing the GFPnb targeting protein were cultured for 24 hours before co-incubated with ~250 nM soluble eGFP for 20 minutes at 37 °C. Cells were rinsed twice with PBS and then imaged. To test the expression and functionality of the GFPnb targeting protein, GPMVs collected from GFPnb stable expression cell lines were incubated with 1 µM purified eGFP. Images were taken after 20 minutes of incubation at 37 °C. Expression and binding correlation analysis. A line was drawn across the GPMVs bound to either ATTO 594 labeled antibody (EGF-PMVs) or soluble eGFP (GFPnb-PMVs) (Figure 1D and Figure 4C) and the fluorescence intensity profile of the line was plotted. The two points where the line intersected the membranes of GPMVs (green fluorescence for EGF-PMVs and red fluorescence for GFPnb-PMVs) had significantly higher intensities than the background. The intensity of GPMV was then determined by subtracting the background fluorescence from these two points respectively. Similarly, the intensities of the GPMV-bound antibody or eGFP were determined with the same line scan before the background fluorescence was subtracted. A total of 23 GPMVs were analyzed with two distinct points per GPMV. All images were taken under the same camera gain setting and the intensity was linearly converted onto the same scale based on exposure times before plotting.
Targeting Study
HeLa EGFR overexpression study. Wild type HeLa cells were transfected with mRFP tagged EGFR plasmid as described above. GPMVs were collected from 2 confluent 100 mm dishes of cells stably expressing the EGF targeting protein and were extruded to 1.0 µm. The extruded vesicles (PMVs) were diluted to a total volume of 700 µL with DMEM phenol red free media. The media was aspirated away from confluent HeLa cells and the PMVs were added to the well and coincubated with the cells for 4 hours at 37 °C. Cell samples were imaged on the spinning disc confocal microscope at the end of the incubation after being washed 3 times with PBS to remove unbound PMVs. Cells with a range of EGFR expression levels could be found in the same dish, as is the nature of transient overexpression. Cellular binding studies. A similar approach was used to demonstrate binding of i) EGF-PMVs to 3 breast cancer cell lines, MDA-MB-468, MDA-MB-231, and MCF-7 and ii) the GFPnb-PMVs to GFP expressing cells and wild type CHO cells. For imaging experiments, recipient cells were plated on a six well plate one day before the experiment. On the day of the study, total cell number in the well was determined by live cell counting on a hemocytometer with trypan blue stain. GPMVs were also counted before extrusion and the PMVs were coincubated with each of those three cell lines at a PMV to cell ratio of 1300. The PMV number was estimated using the measured diameter of GPMVs and efficiency of extrusion under the assumption that all the PMVs have a diameter of 1.0 µm. The coincubation condition and duration were the same as the HeLa EGFR overexpression study. To quantify the amount of binding, we prepared samples for flow cytometery analysis following the same protocol above. MDA-MB-468, MDA-MB-231, and MCF-7 cells were plated in a 96 well plate (Corning) one day before the study at the following densities, 30,000, 15,000, and 32,000 cells/well respectively. CHO cells stably expressing the model transmembrane protein with an extracellular eGFP domain (GFP positive cells) and CHO cells (GFP negative cells) were plated in the same well at 16,000 cells/well respectively and co-cultured for the competitive binding experiment. The plating densities were determined based on their growth rates so that the recipient cells would be confluent by the time of the study. The average cell yield per well of a 96 well plate was used to estimate the number of PMVs needed. GPMVs were extruded and co-incubated with breast cancer cells at a PMV to cell ratio of 1058 for 4 hours. An internal negative control sample was set up for every experiment. Instead of coincubating with targeted vesicles, negative controls cells were incubated with 100 µL of DMEM phenol red free media without PMVs under the same condition. At the end of the incubation, cells were trypsinized and resuspended in 50 µL of PBS for flow cytometery analysis. In all flow cytometry analysis, each sample consisted of two wells of a 96 well plate with the same incubation conditions. EGFR expression measurements. To determine the relative EGFR expression levels of MDA-MB-468, MDA-MB-231, and MCF-7, these cells were plated on 96 wells plates one day before the study with densities reported above. ATTO 594 labeled monoclonal EGFR antibody (Clone E225, Sigma Alridge) with a dye: antibody labeling ratio of 1.8:1 was incubated with all three cell lines at a final antibody concentration of 264 nM for 2 hours at 37 °C. At the end of the incubation, cell samples were prepared for flow cytometry analysis.
Transmembrane Protein Orientation Study
CHO wildtype cells were transfected with Tf-RΔEcto-eGFP EGF plasmid (extracellular eGFP, see plasmid constructs for details) and EGFR-GFP plasmid (intracellular eGFP, see plasmid constructs) respectively. The transfected cells were selected using geneticin (Corning) at a concentration of 0.4 mg/mL for two days after transfection. GPMVs were then extracted from these cells following the protocol described above and mixed with 50 µL of GFPnb PMVs produced from one confluent 100mm dish of cells with stable expression of GFPnb targeting proteins. Free GFPnb PMVs were removed by spinning at 17,000xg for 20 minutes and resuspending the GPMV pellet in fresh buffer prior to imaging.
Supplementary Material
Acknowledgments
We thank Dr. Amy Brock and Dr. Janet Zoldan (University of Texas at Austin) as well as the members of our laboratory for discussion of the manuscript. We acknowledge the NSF (DMR1352487 to Stachowiak) and NIH (GM112065 to Stachowiak) for research funding. We thank Dr. Kathryn Ferguson of the University of Pennsylvania for the EGFR nanobody plasmid, Dr. Brett Collins of the University of Queensland, Australia for the GFP nanobody plasmid, Dr. Adam Arkin of University of California, Berkeley for the eGFP plasmid, Dr. Douglas Golenbock of University of Massachusetts Medical School for the mRFP plasmid. We also thank Dr. Tim Yeh and Dr. Amy Brock (University of Texas at Austin) for MDA-MB-468, MDA-MB-231 and MCF-7 cell lines.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Raposo G, Stoorvogel W. Extracellular Vesicles: Exosomes, Microvesicles, and Friends. The Journal of cell biology. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batrakova EV, Kim MS. Using Exosomes, Naturally-Equipped Nanocarriers, for Drug Delivery. Journal of Controlled Release. 2015;219:396–405. doi: 10.1016/j.jconrel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Dommelen SM, Vader P, Lakhal S, Kooijmans SA, van Solinge WW, Wood MJ, Schiffelers RM. Microvesicles and Exosomes: Opportunities for Cell-Derived Membrane Vesicles in Drug Delivery. Journal of Controlled Release. 2012;161:635–644. doi: 10.1016/j.jconrel.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 4.De Jong OG, Van Balkom BW, Schiffelers RM, Bouten CV, Verhaar MC. Extracellular Vesicles: Potential Roles in Regenerative Medicine. Frontiers in immunology. 2014;5:608. doi: 10.3389/fimmu.2014.00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simpson RJ, Jensen SS, Lim JW. Proteomic Profiling of Exosomes: Current Perspectives. Proteomics. 2008;8:4083–4099. doi: 10.1002/pmic.200800109. [DOI] [PubMed] [Google Scholar]
- 6.Hemler ME. Tetraspanin Proteins Mediate Cellular Penetration, Invasion, and Fusion Events and Define a Novel Type of Membrane Microdomain. Annual review of cell and developmental biology. 2003;19:397–422. doi: 10.1146/annurev.cellbio.19.111301.153609. [DOI] [PubMed] [Google Scholar]
- 7.Mamot C, Drummond DC, Greiser U, Hong K, Kirpotin DB, Marks JD, Park JW. Epidermal Growth Factor Receptor (Egfr)-Targeted Immunoliposomes Mediate Specific and Efficient Drug Delivery to Egfr- and Egfrviii-Overexpressing Tumor Cells. Cancer Research. 2003;63:3154–3161. [PubMed] [Google Scholar]
- 8.Mamot C, Drummond DC, Noble CO, Kallab V, Guo ZX, Hong KL, Kirpotin DB, Park JW. Epidermal Growth Factor Receptor-Targeted Immunoliposomes Significantly Enhance the Efficacy of Multiple Anticancer Drugs in Vivo. Cancer Research. 2005;65:11631–11638. doi: 10.1158/0008-5472.CAN-05-1093. [DOI] [PubMed] [Google Scholar]
- 9.Park JW, Hong KL, Kirpotin DB, Colbern G, Shalaby R, Baselga J, Shao Y, Nielsen UB, Marks JD, Moore D, Papahadjopoulos D, Benz CC. Anti-Her2 Immunoliposomes: Enhanced Efficacy Attributable to Targeted Delivery. Clinical Cancer Research. 2002;8:1172–1181. [PubMed] [Google Scholar]
- 10.Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor Cell Targeting of Liposome-Entrapped Drugs with Phospholipid-Anchored Folic Acid-Peg Conjugates. Advanced Drug Delivery Reviews. 2004;56:1177–1192. doi: 10.1016/j.addr.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Chen S, Zhao X, Chen J, Chen J, Kuznetsova L, Wong SS, Ojima I. Mechanism-Based Tumor-Targeting Drug Delivery System. Validation of Efficient Vitamin Receptor-Mediated Endocytosis and Drug Release. Bioconjugate Chemistry. 2010;21:979–987. doi: 10.1021/bc9005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatakeyama H, Akita H, Maruyama K, Suhara T, Harashima H. Factors Governing the in Vivo Tissue Uptake of Transferrin-Coupled Polyethylene Glycol Liposomes in Vivo. International Journal of Pharmaceutics. 2004;281:25–33. doi: 10.1016/j.ijpharm.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 13.Kullberg EB, Bergstrand N, Carlsson J, Edwards K, Johnsson M, Sjoberg S, Gedda L. Development of Egf-Conjugated Liposomes for Targeted Delivery of Boronated DNA-Binding Agents. Bioconjugate Chemistry. 2002;13:737–743. doi: 10.1021/bc0100713. [DOI] [PubMed] [Google Scholar]
- 14.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of Sirna to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nature Biotechnology. 2011;29:341–U179. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 15.Ohno S-i, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, Mizutani T, Ohgi T, Ochiya T, Gotoh N, Kuroda M. Systemically Injected Exosomes Targeted to Egfr Deliver Antitumor Microrna to Breast Cancer Cells. Molecular Therapy. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian YH, Li SP, Song J, Ji TJ, Zhu MT, Anderson GJ, Wei JY, Nie GJ. A Doxorubicin Delivery Platform Using Engineered Natural Membrane Vesicle Exosomes for Targeted Tumor Therapy. Biomaterials. 2014;35:2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- 17.Muyldermans S. Nanobodies: Natural Single-Domain Antibodies. Annual Review of Biochemistry, Vol 82. 2013;82:775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 18.Schmitz KR, Bagchi A, Roovers RC, Henegouwen PMPvBE, Ferguson KM. Structural Evaluation of Egfr Inhibition Mechanisms for Nanobodies/Vhh Domains. Structure. 2013;21:1214–1224. doi: 10.1016/j.str.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen UB, Kirpotin DB, Pickering EM, Hong KL, Park JW, Shalaby MR, Shao Y, Benz CC, Marks JD. Therapeutic Efficacy of Anti-Erbb2 Immunoliposomes Targeted by a Phage Antibody Selected for Cellular Endocytosis. Biochimica Et Biophysica Acta-Molecular Cell Research. 2002;1591:109–118. doi: 10.1016/s0167-4889(02)00256-2. [DOI] [PubMed] [Google Scholar]
- 20.Kubala MH, Kovtun O, Alexandrov K, Collins BM. Structural and Thermodynamic Analysis of the Gfp:Gfp-Nanobody Complex. Protein Science. 2010;19:2389–2401. doi: 10.1002/pro.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, Novicky L, Merzlyakov M, Hristov T, Hristova K. Measuring the Energetics of Membrane Protein Dimerization in Mammalian Membranes. Journal of the American Chemical Society. 2010;132:3628–3635. doi: 10.1021/ja910692u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards MJ, Hsia C-Y, Singh RR, Haider H, Kumpf J, Kawate T, Daniel S. Membrane Protein Mobility and Orientation Preserved in Supported Bilayers Created Directly from Cell Plasma Membrane Blebs. Langmuir : the ACS journal of surfaces and colloids. 2016;32:2963–2974. doi: 10.1021/acs.langmuir.5b03415. [DOI] [PubMed] [Google Scholar]
- 23.Manjappa AS, Chaudhari KR, Venkataraju MP, Dantuluri P, Nanda B, Sidda C, Sawant KK, Murthy RSR. Antibody Derivatization and Conjugation Strategies: Application in Preparation of Stealth Immunoliposome to Target Chemotherapeutics to Tumor. Journal of Controlled Release. 2011;150:2–22. doi: 10.1016/j.jconrel.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Bronshtein T, Toledano N, Danino D, Pollack S, Machluf M. Cell Derived Liposomes Expressing Ccr5 as a New Targeted Drug-Delivery System for Hiv Infected Cells. Journal of Controlled Release. 2011;151:139–148. doi: 10.1016/j.jconrel.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 25.Herbst RS. Review of Epidermal Growth Factor Receptor Biology. International Journal of Radiation Oncology Biology Physics. 2004;59:21–26. doi: 10.1016/j.ijrobp.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 26.Morgan JR, Prasad K, Hao W, Augustine GJ, Lafer EM. A Conserved Clathrin Assembly Motif Essential for Synaptic Vesicle Endocytosis. The Journal of neuroscience. 2000;20:8667–8676. doi: 10.1523/JNEUROSCI.20-23-08667.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Immordino ML, Dosio F, Cattel L. Stealth Liposomes: Review of the Basic Science, Rationale, and Clinical Applications, Existing and Potential. International journal of nanomedicine. 2006;1:297–315. [PMC free article] [PubMed] [Google Scholar]
- 28.Busch DJ, Houser JR, Hayden CC, Sherman MB, Lafer EM, Stachowiak JC. Instrinsically Disordered Proteins Drive Membrane Curvature. Nature Communications NCOMMS8875. 2015 doi: 10.1038/ncomms8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holyst R, Bielejewska A, Szymanski J, Wilk A, Patkowski A, Gapinski J, Zywocinski A, Kalwarczyk T, Kalwarczyk E, Tabaka M, Ziebacz N, Wieczorek SA. Scaling Form of Viscosity at All Length-Scales in Poly(Ethylene Glycol) Solutions Studied by Fluorescence Correlation Spectroscopy and Capillary Electrophoresis. Physical Chemistry Chemical Physics. 2009;11:9025–9032. doi: 10.1039/b908386c. [DOI] [PubMed] [Google Scholar]
- 30.Jang SC, Kim OY, Yoon CM, Choi D-S, Roh T-Y, Park J, Nilsson J, Lotvall J, Kim Y-K, Gho YS. Bioinspired Exosome-Mimetic Nanovesicles for Targeted Delivery of Chemotherapeutics to Malignant Tumors. Acs Nano. 2013;7:7698–7710. doi: 10.1021/nn402232g. [DOI] [PubMed] [Google Scholar]
- 31.Oh K, Kim SR, Kim D-K, Seo MW, Lee C, Lee HM, Oh J-E, Choi EY, Lee D-S, Gho YS, Park KS. In Vivo Differentiation of Therapeutic Insulin-Producing Cells from Bone Marrow Cells Via Extracellular Vesicle-Mimetic Nanovesicles. Acs Nano. 2015;9:11718–11727. doi: 10.1021/acsnano.5b02997. [DOI] [PubMed] [Google Scholar]
- 32.Paluch E, Sykes C, Prost J, Bornens M. Dynamic Modes of the Cortical Actomyosin Gel During Cell Locomotion and Division. Trends in Cell Biology. 2006;16:5–10. doi: 10.1016/j.tcb.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Mills JC, Stone NL, Pittman RN. Extranuclear Apoptosis: The Role of the Cytoplasm in the Execution Phase. Journal of Cell Biology. 1999;146:703–707. doi: 10.1083/jcb.146.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedl P, Wolf K. Tumour-Cell Invasion and Migration: Diversity and Escape Mechanisms. Nature Reviews Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 35.Charras GT, Coughlin M, Mitchison TJ, Mahadevan L. Life and Times of a Cellular Bleb. Biophysical Journal. 2008;94:1836–1853. doi: 10.1529/biophysj.107.113605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charras GT, Hu C-K, Coughlin M, Mitchison TJ. Reassembly of Contractile Actin Cortex in Cell Blebs. Journal of Cell Biology. 2006;175:477–490. doi: 10.1083/jcb.200602085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sezgin E, Kaiser H-J, Baumgart T, Schwille P, Simons K, Levental I. Elucidating Membrane Structure and Protein Behavior Using Giant Plasma Membrane Vesicles. Nature Protocols. 2012;7:1042–1051. doi: 10.1038/nprot.2012.059. [DOI] [PubMed] [Google Scholar]
- 38.Baumgart T, Hammond AT, Sengupta P, Hess ST, Holowka DA, Baird BA, Webb WW. Large-Scale Fluid/Fluid Phase Separation of Proteins and Lipids in Giant Plasma Membrane Vesicles. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3165–3170. doi: 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Costello DA, Hsia C-Y, Millet JK, Porri T, Daniel S. Membrane Fusion-Competent Virus-Like Proteoliposomes and Proteinaceous Supported Bilayers Made Directly from Cell Plasma Membranes. Langmuir. 2013;29:6409–6419. doi: 10.1021/la400861u. [DOI] [PubMed] [Google Scholar]
- 40.Roucourt B, Meeussen S, Bao J, Zimmermann P, David G. Heparanase Activates the Syndecan-Syntenin-Alix Exosome Pathway. Cell research. 2015;25:412–428. doi: 10.1038/cr.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozcan F, Klein P, Lemmon MA, Lax I, Schlessinger J. On the Nature of Low- and High-Affinity Egf Receptors on Living Cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5735–5740. doi: 10.1073/pnas.0601469103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott RE, Perkins RG, Zschunke MA, Hoerl BJ, Maercklein PB. Plasma-Membrane Vesiculation in 3t3-Cells, Sv3t3-Cells.1. Morphological and Biochemical Characterization. Journal of Cell Science. 1979;35:229–243. doi: 10.1242/jcs.35.1.229. [DOI] [PubMed] [Google Scholar]
- 43.Choi CHJ, Alabi CA, Webster P, Davis ME. Mechanism of Active Targeting in Solid Tumors with Transferrin-Containing Gold Nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1235–1240. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Tian S, Petros RA, Napier ME, DeSimone JM. The Complex Role of Multivalency in Nanoparticles Targeting the Transferrin Receptor for Cancer Therapies. Journal of the American Chemical Society. 2010;132:11306–11313. doi: 10.1021/ja1043177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bright JN, Woolf TB, Hoh JH. Predicting Properties of Intrinsically Unstructured Proteins. Progress in Biophysics & Molecular Biology. 2001;76:131–173. doi: 10.1016/s0079-6107(01)00012-8. [DOI] [PubMed] [Google Scholar]
- 46.Yu C, Hale J, Ritchie K, Prasad NK, Irudayaraj J. Receptor Overexpression or Inhibition Alters Cell Surface Dynamics of Egf-Egfr Interaction: New Insights from Real-Time Single Molecule Analysis. Biochemical and Biophysical Research Communications. 2009;378:376–382. doi: 10.1016/j.bbrc.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 47.Reilly RM, Kiarash R, Sandhu J, Lee YW, Cameron RG, Hendler A, Vallis K, Gariepy J. A Comparison of Egf and Mab 528 Labeled with in-111 for Imaging Human Breast Cancer. Journal of Nuclear Medicine. 2000;41:903–911. [PubMed] [Google Scholar]
- 48.Hamerscasterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. Naturally-Occurring Antibodies Devoid of Light-Chains. Nature. 1993;363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 49.Fridy PC, Li Y, Keegan S, Thompson MK, Nudelman I, Scheid JF, Oeffinger M, Nussenzweig MC, Fenyo D, Chait BT, Rout MP. A Robust Pipeline for Rapid Production of Versatile Nanobody Repertoires. Nature Methods. 2014;11:1253. doi: 10.1038/nmeth.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic Antibodies: Successes, Limitations and Hopes for the Future. British journal of pharmacology. 2009;157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van de Broek B, Devoogdt N, D’Hollander A, Gijs H-L, Jans K, Lagae L, Muyldermans S, Maes G, Borghs G. Specific Cell Targeting with Nanobody Conjugated Branched Gold Nanoparticles for Photothermal Therapy. ACS Nano. 2011;5:4319–4328. doi: 10.1021/nn1023363. [DOI] [PubMed] [Google Scholar]
- 52.Tu C, Das S, Baker AB, Zoldan J, Suggs LJ. Nanoscale Strategies: Treatment for Peripheral Vascular Disease and Critical Limb Ischemia. ACS Nano. 2015;9:3436–3452. doi: 10.1021/nn507269g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of Epidermal Growth Factor Receptor Signalling Networks in Glioblastoma. Nature Reviews Cancer. 2015;15:302–310. doi: 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carter RE, Sorkin A. Endocytosis of Functional Epidermal Growth Factor Receptor-Green Fluorescent Protein Chimera. The Journal of biological chemistry. 1998;273:35000–35007. doi: 10.1074/jbc.273.52.35000. [DOI] [PubMed] [Google Scholar]
- 55.Scheve CS, Gonzales PA, Momin N, Stachowiak JC. Steric Pressure between Membrane-Bound Proteins Opposes Lipid Phase Separation. Journal of the American Chemical Society. 2013;135:1185–1188. doi: 10.1021/ja3099867. [DOI] [PubMed] [Google Scholar]
- 56.Stachowiak JC, Schmid EM, Ryan CJ, Ann HS, Sasaki DY, Sherman MB, Geissler PL, Fletcher DA, Hayden CC. Membrane Bending by Protein-Protein Crowding. Nature Cell Biology. 2012;14:944. doi: 10.1038/ncb2561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.