

Abstract
Hepaciviruses and pegiviruses constitute two closely related sister genera of the family Flaviviridae. In the past five years, the known phylogenetic diversity of the hepacivirus genera has absolutely exploded. What was once an isolated infection in humans (and possibly other primates) has now expanded to include horses, rodents, bats, colobus monkeys, cows, and, most recently, catsharks, shedding new light on the genetic diversity and host range of hepaciviruses. Interestingly, despite the identification of these many animal and primate hepaciviruses, the equine hepaciviruses remain the closest genetic relatives of the human hepaciviruses, providing an intriguing clue to the zoonotic source of hepatitis C virus. This review summarizes the significance of these studies and discusses current thinking about the origin and evolution of the animal hepaciviruses as well as their potential usage as surrogate models for the study of hepatitis C virus.
Keywords: hepatitis C virus, pegivirus, hepegivirus, virome, virus evolution, animal models
INTRODUCTION
Hepacivirus is one of the four genera included in the positive-strand RNA virus family Flaviviridae, a genetically diverse group of human and animal pathogens known for causing clinically relevant diseases such as yellow fever, dengue, and Zika fever. Hepatitis C virus (HCV), identified in 1989 (1), is the type species of the genus Hepacivirus (2) and the etiological agent of hepatitis C in humans. Following acute infection, HCV establishes persistence in 60–80% of individuals (3). Approximately 160 million people worldwide are chronically infected with HCV and are therefore at significant risk for developing severe liver diseases such as progressive fibrosis, cirrhosis, and hepatocellular carcinoma (4, 5).
Despite tremendous advancements in HCV research over the past 25 years, the origin of HCV still remains a mystery. The vast majority of emerging infectious diseases are caused by viral zoonoses (6). Humans are constantly exposed to a plethora of genetically diverse animal viruses through direct contact with domestic and wild animal populations (coronaviruses, Ebola virus) and via vector intermediates such as arthropods (dengue virus, Zika virus) (7). Due to the presence of multiple biological and epidemiological transmission barriers, however, these exposure events infrequently lead to infection and disease (8). Nevertheless, zoonotic viruses do sporadically jump the species barrier, often causing immediate and significant diseases, as illustrated by the transmission of simian and human immunodeficiency viruses (SIV and HIV), coronaviruses, influenza viruses, and paramyxoviruses (8–11). Cross-species transmission of viruses often leads to accelerated viral evolution and altered virulence patterns, yet their basic biological characteristics remain unchanged (11). The identification and characterization of animal-derived viruses therefore warrant significant attention, as these viruses may represent lurking reservoirs of human disease as well as potential surrogates for the study of human viral homologs (12, 13).
HCV exhibits a restricted host range pattern, with natural infection occurring only in humans, although experimental infection of chimpanzees is possible. Although this might suggest that the HCV pandemic originated from a nonhuman primate reservoir, akin to SIV/HIV (9), no evidence for the existence of such a source has been identified (14, 15). Remarkably, in 2011, a novel HCV-like homolog was unexpectedly discovered in nasal samples collected from dogs with respiratory illness (16). This work provided the first evidence for a wider hepaciviral host range and helped pave the way for the subsequent identification of novel hepacivirus species in horses, rodents, bats, Old World primates, cows, and, most recently, the graceful catshark. This review summarizes the current understanding of the animal hepaciviruses, discusses their potential as surrogate HCV animal models, and offers hypotheses regarding their origin, evolution, and host range (see the Summary Figure). Figure 1 provides a timeline of discoveries related to the animal hepaciviruses.
Summary Figure.
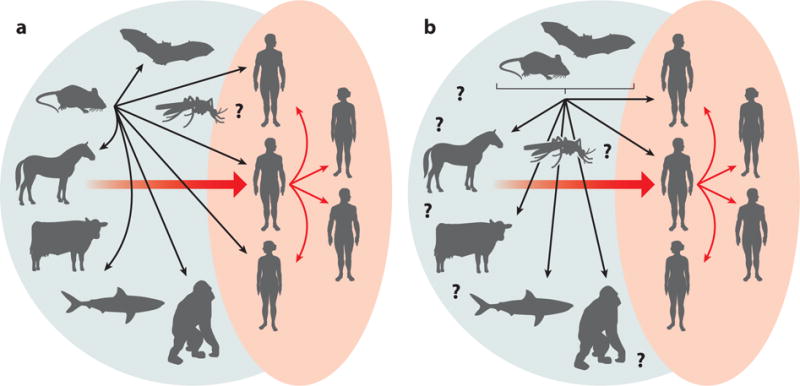
The expanded world of hepaciviruses (green background) and the different hypotheses of hepatitis C virus origins.
Figure 1.
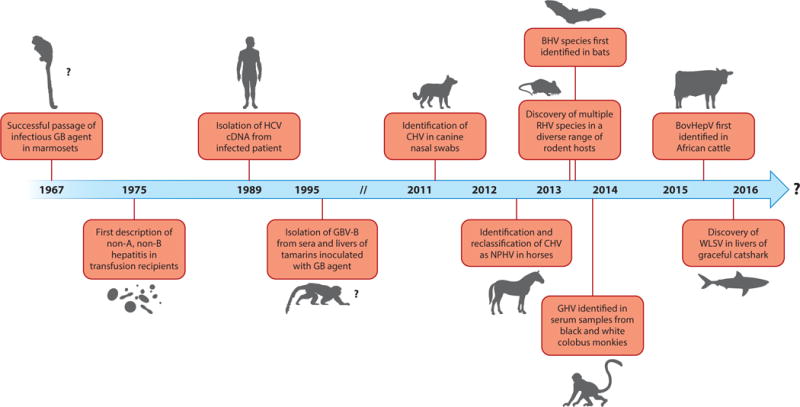
Timeline of discoveries related to the animal hepaciviruses. Abbreviations: BHV, bat hepacivirus; BovHepV, bovine hepacivirus; CHV, canine hepacivirus; GBV-B, GB virus B; GHV, guereza hepacivirus; HCV, hepatitis C virus; NPHV, nonprimate hepacivirus; RHV, rodent hepacivirus;WLSV, Wenling shark virus.
OVERVIEW OF ANIMAL HEPACIVIRUSES
Current Classification and Evidence for the Existence of a Closely Related Sister Genus
The genus Hepacivirus currently comprises one species of human, one species of unknown host, one species of horse, several species of rodent and bat, one species of bovine, one species of primate, and one species of shark virus (Figure 2). Prior to 2011, the only known member of the Hepacivirus genus besides HCV was the distantly related GB virus B (GBV-B). Deinhardt and colleagues (17) initially described GBV-B in 1967 following the inoculation of laboratory-housed tamarins with serum from a surgeon (initials G.B.) with acute hepatitis. Although GBV-B was the first hepacivirus associated with liver disease, the virus was not fully characterized until 1995 when Simons and colleagues (18) successfully isolated two distinct viral species, termed GBV-A and GBV-B, from the sera and livers of tamarins infected with passaged GB agent. Since then, GBV-B infection has not been observed in human populations, and extensive searches for the natural host of GBV-B in other primate species have been entirely unsuccessful (19). The origins of GBV-B and its obscure presence within a human host thus remain a tantalizing mystery.
Figure 2.
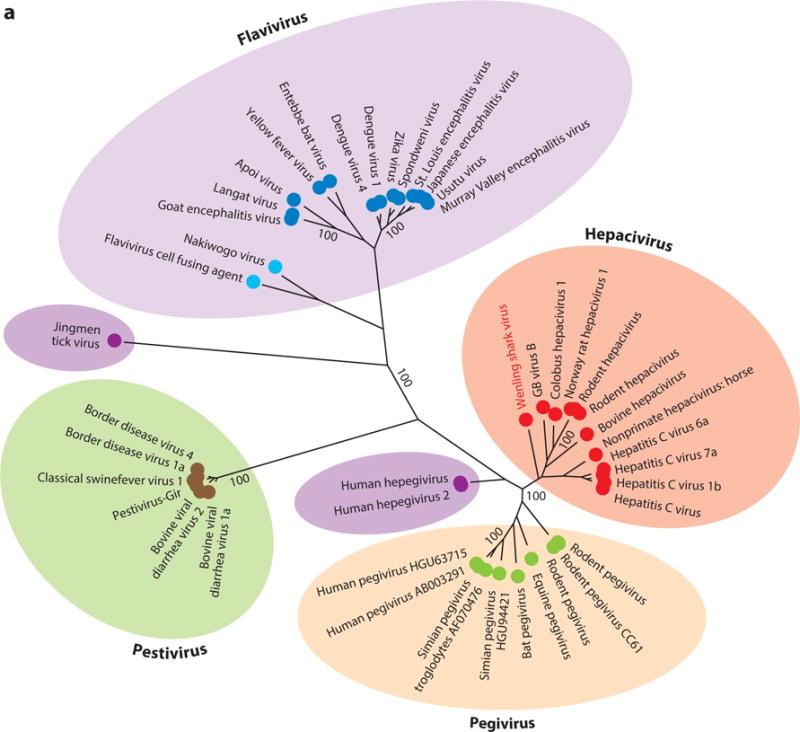
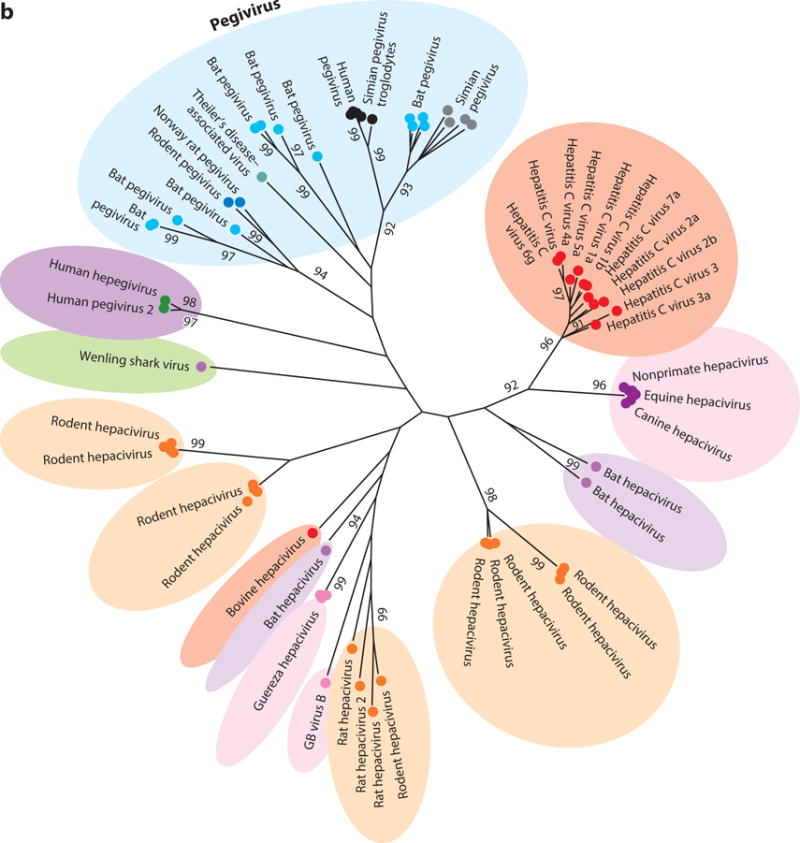
Phylogenetic analysis of helicase gene (motifs I to IV) of representative members of (a) all four genera of the family Flaviviridae plus newly identified viruses and (b) the two genera Hepacivirus and Pegivirus. The evolutionary history was inferred using the maximum likelihood method based on the Le and Gascuel 2008 model (112). The tree with the highest log likelihood (−47,019.0830) is shown. The percentage of trees in which the associated taxa clustered together is shown next to each branch. Initial trees for the heuristic search were obtained automatically by applying Neighbor-Join (NJ) and BIONJ algorithms to a matrix of pairwise distances estimated by using a JTT model and then selecting the topology with superior log likelihood value. A discrete gamma distribution was used to model evolutionary rate differences among sites (five categories, +G, parameter = 1.7148). The rate variation model allowed for some sites to be evolutionarily invariable (+I, 3.9855% sites). The tree is drawn to scale, with branch lengths measured in number of substitutions per site. The analysis involved 96 amino acid sequences. There were a total of 276 positions in the final data set. Evolutionary analyses were conducted in MEGA7 (113).
Besides Hepacivirus, the Flaviviridae family includes three additional genera: Flavivirus, Pestivirus, and Pegivirus (Figure 2a). The previously unclassified GBV-A now belongs to the Pegivirus genus under its newly proposed name simian pegivirus (SPgV) (19). Also assigned to Pegivirus are human pegivirus (HPgV) and bat pegivirus (BPgV), formerly known as GBV-C/hepatitis G virus (HGV) and GBV-D, respectively, although neither shares a historical connection to the original GB agent. Despite harboring a close phylogenetic relationship to hepaciviruses, pegiviruses represent a distinct group of viruses that differ markedly in their genomic organization and pathogenicity (19). In contrast to HCV, pegiviruses are capable of establishing persistent infections without signs of clinical hepatitis or disease (20, 21). Additionally, molecular analysis of pegiviruses reveals that SPgV and certain HPgV strains do not encode a core protein (22–24). Because of these differences, Stapleton and colleagues (19) proposed to classify these viruses as members of a new genus termed Pegivirus (pe, persistent; g, GB or G) to better reflect their genetic and pathogenic features. Identification of BPgV in bats in 2010 was the first indication that pegiviruses were more widely distributed in mammals (25). More recently, this host range has expanded, with several reports demonstrating novel pegivirus species in equines (EPgV), rodents (RPgV), and bats (BPgV) (26–31). Theiler’s disease–associated virus, the putative etiological agent of Theiler’s disease in horses, is of particular importance as this is the first pegivirus species linked to acute hepatitis in mammals (26), suggesting pegiviruses do have pathogenic potential. Additional studies are needed to better characterize the host range of pegiviruses as well as their curious relationship to the hepaciviruses.
Human Hepacivirus: The Different Genotypes of Hepatitis C Virus
HCV is currently classified into seven major phylogenetic lineages, termed genotypes (1–7), that exhibit considerable heterogeneity in genomic sequence, transmission, and regional distribution (32). On average, HCV genotypes differ by up to 30% at the nucleotide level and 20% at the amino acid level (32). Within these genotypes exist numerous recognized subtypes (a, b, c, etc.) that differ by up to 20–25% at the nucleotide level. At present, there are 67 confirmed and 20 tentative subtypes (32). This remarkable level of genetic diversity is fueled by the inherent infidelity of the HCV RNA-dependent RNA polymerase coupled with a high in vivo replication rate (33). Evidence for low levels of viral RNA recombination has been reported as well (34), adding an additional mechanism for HCV diversification. Despite the considerable level of genetic diversity exhibited by HCV, all variants are currently classified as a single species under present International Committee on Taxonomy of Viruses classification (2). Moreover, all variants exhibit similar biological properties, including hepatotropism, propensity for persistence, and pathogenicity.
The global distribution and prevalence of the various HCV genotypes are unbalanced and extraordinarily complex. The vast majority of genotype 1 infections, which are the most common globally, concentrate within high-income areas such as North America and Europe, although genotypes 2 and 3 account for 10–25% of cases in these regions as well. In contrast, genotypes 3 and 6 are most common in patients in South and Southeast Asia, and genotype 4 is the major genotype within the Middle East and North Africa. Genotype 5 is far less prevalent, with the majority of cases clustering in Southern Africa (35). The single known isolate of genotype 7 was obtained from a Central African immigrant in Canada in 2007 (36). These differences in genotype distribution and prevalence are thought to reflect HCV’s epidemic spread during the past century (37). In support of this, genotypes 1a, 1b, 2a, and 3a, often designated the epidemic subtypes, are frequently observed in intravenous drug users or those exposed to contaminated blood products in high-income countries (38–40). In contrast, the so-called endemic strains of HCV are far more diverse and primarily predominate within sub-Saharan Africa and Southeast Asia, where they have likely circulated for hundreds of years (37, 38). Studies of the evolution and distribution of HCV variants have further revealed that current epidemic strains of HCV spread globally during the past 100 years (38, 40, 41). This global dissemination into new risk groups was no doubt aided by the emergence of new parenteral transmission routes created by immunizations, blood transfusions, and intravenous drug use after World War II. The ultimate source of the truly indigenous variants of HCV, nonetheless, remains to be resolved.
Canine and Equine Hepaciviruses
Since the discovery of HCV in 1989 (1), initial research endeavors undertaken to identify animal homologs of HCV were motivated by the prevailing hypothesis that HCV infections possessed a nonhuman primate origin (37). This intriguing idea was based on the observation that high-diversity areas of endemic HCV circulation in sub-Saharan Africa and Southeast Asia occurred where human, ape, and Old World monkey populations overlapped. This observation was further analogous to the recent SIV/HIV pandemic, for which a zoonotic origin from chimpanzees was ultimately identified (42). Numerous screenings of sera from Old World monkeys and apes, however, failed to identify the presence of HCV-like viruses (14, 15). Without direct evidence for the existence of a nonhuman primate counterpart, studies to identify the origins of HCV long remained at a frustrating standstill.
In 2011, however, the first evidence for the existence of a wider hepaciviral host range emerged, albeit in a peculiar fashion. While using high-throughput sequencing to identify viral causes of respiratory illness outbreaks in dogs kenneled in the United States, Kapoor and colleagues (43) made the serendipitous discovery of a novel hepacivirus species that genetically resembled HCV. Comparative phylogenetic analysis of this virus, initially termed canine hepacivirus (CHV), identified it to be the most genetically related viral homolog of HCV, with approximately 50% nucleotide divergence. Despite this remarkable discovery, the identification of CHV was met with some strange observations. Firstly, partial NS3 sequences obtained from animals of two independent outbreaks showed 99.2% sequence convergence, indicating a genetic homogeneity that is atypical for most RNA viruses. Secondly, high viral loads (>107 copies) of CHV RNA were detected in nasal swabs of most infected animals, a curious observation given that all known hepacivirus and pegivirus species exhibit strict hepatotropism and lymphotropism, respectively. Lastly, subsequent efforts to identify additional canines either infected with RNA or seropositive for CHV antibodies were largely unsuccessful (16, 44, 45).
Following the discovery of CHV, the same group used a serology-based discovery approach to further characterize the distribution and natural host range of CHV (16). Using a luciferase-based reporter assay to detect antibodies against the CHV NS3 helicase protein, the group determined the antibody prevalence for a wide range of mammals. In total, 60 dogs, 81 deer, 84 cows, 103 horses, and 14 rabbits were screened for the presence of anti-NS3 antibodies. To the group’s surprise, high-titered antibodies were consistently observed only in horses (35%), with the exception of a single cow that showed intermediate reactivity. Subsequent polymerase chain reaction (PCR) detection of viral RNA confirmed infection in 8 of the 36 seropositive horses. Genetic analysis of the isolated hepacivirus variants revealed moderate sequence diversity (~14% nucleotide divergence) over the complete length of the genome, with one variant (NZP1) showing significant homology to the original CHV isolate (99.7% at the nucleotide level). Because these variants were identified in a different natural host and exhibited a moderate degree of sequence divergence from each other compared to CHV, the investigators decided to broadly reclassify the viruses, including CHV, as nonprimate hepaciviruses (NPHV 1–8), although the term equine hepacivirus is often interchanged.
To further investigate the prevalence, clinical course of infection, and tissue tropism of NPHV, Pfaender and colleagues (13) analyzed NPHV infections in domestic horses in northern Germany. By screening for the presence of NPHV RNA and anti-NS3 antibodies, the researchers revealed evidence for acute and chronic stages of NPHV infection (13). Liver analyses of infected horses showed no evidence of severe disease, although clearance of the virus in acutely infected animals was correlated with transient elevations in blood liver-specific enzyme levels indicative of hepatic inflammation. Additionally, when various organ and tissue samples from a single NPHV-infected horse were analyzed, negative-strand viral RNA was detected only within hepatocytes. Consistent with these findings, Scheel and colleagues (46) showed that experimental inoculation of a single horse with NPHV RNA led to the development of acute liver disease, delayed seroconversion, and signs of hepatic inflammation. Recently, experimental inoculation of adult horses and foals by Ramsay and colleagues (47) resulted in acute and chronic liver disease as determined by elevations in blood liver-specific enzyme levels and signs of hepatocellular damage. Interestingly, infection in foals with severe combined immunodeficiency resulted in minimal liver disease compared with infection in fully immunocompetent foals, suggesting that adaptive immune responses contribute to the disease phenotype in horses. Taken together, these data indicate that the natural course of infection and tissue tropism of NPHV in horses closely parallel that of HCV in humans (48). Thus, NPHV may represent an exciting model for the study of hepacivirus-mediated disease, although additional studies are needed to further explore the differences in tropism, immunity, and pathogenesis exhibited by NPHV.
The high degree of sequence homology observed between a single NPHV strain (NZP1) and the original CHV isolate suggests two possibilities regarding the origin of CHV in dogs. First, it seems entirely plausible that CHV infection in dogs represents a recent and direct cross-species transmission event that results in low levels of hepatic replication without the clinical signs of inflammation or organ impairment. Second, CHV may simply represent a false transmission event facilitated by the feeding of animals with horsemeat or the usage of veterinary products that contained horse serum–derived components such as vaccines. Considering the lack of sequence diversity exhibited by CHV, the largely unsuccessful identification of additional hepaciviruses in canids, and the finding that commercial horse serum is contaminated with NPHV (46, 49), it is plausible that the discovery of CHV in dogs was the consequence of contamination rather than a true transmission event. Future studies should use serology to continue searching for evidence of hepacivirus infection in dogs to better resolve these two competing possibilities (50, 51).
Rodent Hepaciviruses
The discovery of naturally occurring NPHV infections in horses raised the possibility that additional homologs of HCV may be present in a broad range of mammalian host species. To better understand the host range of animal hepaciviruses and to identify potential surrogate small animal models for the study of HCV, Kapoor and colleagues (30) searched methodically for the presence of novel hepaciviruses in serum samples from 400 wild-caught rodents spanning four species of Rodentia. Using degenerate primer sets targeting conserved NS3 helicase domains of known hepaciviruses and pegiviruses, the group identified the presence of several genetically distinct hepaciviral species in deer mice (Peromyscus maniculatus), a widely distributed rodent species known for carrying a pathogenic strain of hantavirus. Additionally, two other rodent hepacivirus (RHV) species were identified in desert wood rats (Neotoma lepida) and hispid pocket mice (Chaetodipus hispidus). Genetic analysis of the various RHV species revealed a molecular divergence from HCV that was substantially greater than that observed between HCV and NPHV, indicating an absence of hepaciviral cospeciation (30, 51, 52).
Soon after RHV’s identification, Drexler and colleagues (27) independently reported the presence of RHV in European bank voles (Myodes glareolus) and South African four-striped mice (Rhabdomys pumilio) after screening sera and organ tissues from 4,770 rodents (Rodentia, 41 species) from Europe, Africa, Thailand, and Mexico. Genetic analysis of the isolates revealed three highly divergent hepacivirus clades. Histopathological and molecular examination of liver samples from RHV-infected bank voles showed evidence of viral RNA and low levels of lymphocytic invasion, indicating possible hepatotropism. Furthermore, serological investigations of two distinct hepacivirus lineages revealed the presence of anti-NS3 antibodies in several of the animals (8.3% and 12.4%). Interestingly, a low co-occurrence of viral RNA and anti-RHV antibodies was observed in PCR-positive animals (5.3%), suggesting that bank voles might be able to clear hepacivirus infections.
Overall, RHV exhibits a genomic organization similar to that of HCV, with a long open reading frame (ORF) encoding three structural and seven nonstructural proteins (Table 1). Distinct nucleotide and structural differences are found, however, in the 3′ and 5′ untranslated regions (UTRs) of RHV (27). In contrast to that of HCV, the 3′ UTR of RHV does not contain a poly(U) tract, although some studies indicate similarities in the terminal stem-loop of certain RHV isolates (27, 30). In contrast to other hepaciviruses, two of the RHV lineages obtained from European bank voles contain a pegivirus-like internal ribosome entry site (IRES) in the 5′ UTR (27). The functional significance of this structure is unknown, but it is possible that it originated during a novel recombination event between hepaciviruses and pegiviruses in a coinfected rodent host (34). RHV strains also exhibit a strain-dependent number of miR-122 binding sites ranging from one to two; whether these binding sites contribute to host specificity is entirely unknown, but it is an interesting observation nonetheless given the consistency in seed site number observed for HCV.
Table 1.
Molecular and epidemiological features of animal hepaciviruses
| HCV | GBV-B | NPHV | RHV | BHV | GHV | BovHepV | WLSV | HPgVa | HHpgV-la | |
|---|---|---|---|---|---|---|---|---|---|---|
| Natural host range | Humans | – | Horsesb | Rodentsc | Bats | (Nonhuman primates) | (Cattle) | (Cartilaginous fish) | Humans | – |
| Host for viral identification | Humans | Tamarins | Dogs, horses | Rodents | Bats | Black-and-white colobus monkeys | Domesticated cows | Graceful catshark | Humans | Humans |
| Experimental infection | Chimpanzees | Tamarins, marmosets | Single horse | – | – | – | – | – | – | – |
| Tissue tropism | Hepatocytes | Liver | Hepatocytes | Hepatocytes | – | – | Hepatocytes | (Liver) | Lymphocytes | – |
| Pathogenesis | Hepatitis, cirrhosis, carcinoma | Hepatitis | (Liver inflammation) | Liver Inflammation | – | – | – | – | – | – |
| Infection outcome | Acute/chronic | Acuted | Acute/chronic | Acute/chronicf | – | – | Acute/chronic | – | Acute/chronic | Acute/chronic |
| Seroprevalence | 2–4%, higher in risk groups | – | ~40% | ~20% | ≥10%e | – | – | – | ~25% | – |
| Nucleotide diversity | 7 genotypes, ~30% | One isolate | ~15% diversity | ≥8 clades, ≥80% | ≥3 clades, highly diverse | ~15% | ~8% | – | ~20% | <10% |
| ORF: number of predicted proteins | 10 | 10 (or 11)g | 10 | 10 | 10 | 10 | 10 | 10 | 9 | 10 |
| ORF: typical amino acid length | 3,008–3,033 | 2,864 | 2,942–2,949 | 2,748–3,007 | 2,901–3,024 | 3,334–3,336 | 2,779 | 3,086 | 2,873 | 3,058 |
| Predicted number of N-glycosylations (E1/E2) | 5–6/11 | 3/6 | 4/10 | 0–2/2–6 | 2/4–5 | 4/4 | – | – | 4 | 14 |
| 5′ UTR: predicted number of miR-122 seed sites | Two, required | Two, dispensableh | One | Variable | – | One | One | – | 0 | 0 |
| 3′ UTR: long internal poly(U) tract | Yes | No | – | No | – | Not foundi | – | – | No | No |
Entries in parentheses indicate preliminary findings with additional confirmation needed. Abbreviations: BHV, bat hepacivirus; BovHepV, bovine hepacivirus; GBV-B, GB virus B; GHV, guereza hepacivirus; HCV, hepatitis C virus; HHpgV-1, human hepegivirus; HPgV, human pegivirus; NPHV, nonprimate hepacivirus; ORF, open reading frame; RHV, rodent hepacivirus; UTR, untranslated region;WLSV, Wenling shark virus.
HPgV and HHpV are shown for comparison.
NPHV was originally identified in dogs (43), but subsequent confirmatory attempts have been unsuccessful with the exception of a single seropositive dog in the United Kingdom (115).
Identified in deer mice, hispid pocket mice, desert wood rats, European bank voles, South African four-striped mice, and rats (28, 30, 116).
Persistent infection has been reported with recombinant GBV-B (117).
Authors’ unpublished data.
Based on reactivity with HCV; may therefore be underestimated (116).
The p13 protein of GBV-B may be processed into a p7 protein (equivalent to HCV p7) and a p6 protein (117).
A recent study demonstrated miR-122 independent GBV-B replication in vitro (58).
Complete characterization remains to be done.
Phylogenetic analysis of all currently known RHV isolates, including those recently discovered in Norway rats (Rattus norvegicus) in New York City (28), has revealed a considerable level of genetic diversity among RHV species. In fact, the individual RHV species are as unrelated to each other as they are to other hepacivirus species identified in humans, horses, and bats (52). The order Rodentia comprises the most diverse group of mammals (40%), so this level of genetic diversity is not wholly unexpected. However, the lack of a phylogenetic clustering that mirrors the evolutionary history of mammals might suggest that the surprising diversity of RHV is the consequence of multiple cross-species transmission events. The question of whether hepaciviruses from other mammalian hosts are capable of crossing species and infecting rodents requires further exploration.
Bat Hepaciviruses
Genomic sequences of hepaciviruses infecting bats were first acquired by Quan and colleagues (31) in 2013. Using an unbiased high-throughput sequencing approach, the group screened serum specimens obtained from 415 bats representing 33 species across five countries (Guatemala, Cameroon, Nigeria, Democratic Republic of the Congo, and Kenya) for the presence of unique viral sequences. Their unbiased approach led to them to discover a diverse group of bat-derived viruses genetically related to hepaciviruses (bat hepaciviruses, or BHVs) and pegiviruses (bat pegiviruses, or BPgVs). To further characterize the host range and distribution of these viruses, the group screened an additional 34 bats representing 40 species from Nigeria, Bangladesh, and Mexico. In total, the group was able to detect 83 bat-derived viruses in six out of the eight families of bats tested. Genetic analysis of these isolates identified 22 potentially novel species of viruses (3 hepaciviruses and 19 pegiviruses). These findings, taken together with a report from another group identifying novel pegiviruses in bats (27), greatly expanded the genetic diversity of previously known BPgVs (25). Within the Hepacivirus genus, the viruses were derived solely from two species of African bats (Hipposideros vittatus and Otomops martiensseni) in Kenya. Although high levels of viremia were detected in the surveyed bats, no evidence of disease was observed, suggesting that BHV and BPgV may be nonpathogenic to their hosts.
Other Primate Hepaciviruses
Shortly following the discovery of BHV and RHV, Lauck and colleagues (53) reported the discovery of a novel hepacivirus in serum samples obtained from black-and-white colobus monkeys (Colobus guereza). Discovery of this virus, termed guereza hepacivirus (GHV), marked the first instance of documented hepacivirus infection in nonhuman primates. Despite the importance of this finding, subsequent analysis revealed that GHV shares a common ancestry with GBV-B, RHV, and a particular BHV strain, therefore separating it from the human hepacivirus grouping and arguing against hepacivirus cospeciation. Interestingly, despite possessing a genome organization typical of other hepaciviruses, GHV contains an exceptionally long NS5A gene, approximately twice the length of any other homologous NS5A gene, as well as an intrinsically disordered region of amino acids located within the C-terminal region. The functional consequence of this feature is currently unknown.
Bovine Hepaciviruses
Recently, within the span of a month, a group of investigators published two reports on the discovery of hepaciviruses in cattle (Bos taurus), first in Africa (54) and subsequently in Germany (55). Phylogenetic trees based on conserved NS3 and NS5B coding sequences support the basal grouping of these bovine hepaciviruses (BovHepV) next to GBV-B, BHV, and certain RHV strains. Estimation of the genetic diversity between BovHepV strains identified low intersequence variability but large genetic distances from other known hepaciviruses (54). Investigation into to the natural course of BovHepV infections in domesticated cattle from Germany by Baechlein and colleagues (55) revealed evidence for both acute and chronic stages of infection. Quantitative reverse transcriptase PCR data from multiple organ and tissue samples from these cattle showed high concentrations of viral RNA in liver tissue, which, together with the finding of predicted miR-122 binding sites in the viral 5′ UTR, indicate viral hepatotropism, although no clinical or postmortem signs of liver damage were observed. Additional studies should be undertaken to more thoroughly characterize the effect of BovHepV infections on cattle and the potential risks these viruses pose to farmers and breeders who work in close contact.
Shark Hepaciviruses
Until recently, the ability of hepaciviruses to infect nonmammalian hosts was entirely unknown. It was certainly reasonable to hypothesize the existence of hepacivirus species infecting additional classes of animals, especially given the enormous diversity exhibited by the Flaviviridae family, but absence of sampling data combined with technological limitations precluded the early scientific addressment of this proposition. While using RNA sequencing to survey a range of arthropod and vertebrate species for the presence of Flaviviridae-related viruses, however, Shi and colleagues (56) made the remarkable discovery of a novel hepacivirus, termed the Wenling shark virus (WLSV), in liver tissue samples taken from the graceful catshark (Proscyllium habereri). This discovery marked the first occasion of documented hepacivirus infection in a nonmammalian host. At the molecular level, the genomic organization of WLSV was typical of other viruses within the Hepacivirus genus (Table 1), although substantial differences were observed in the N terminus of the NS4 protein and the majority of the NS5A protein with the exception of a conserved zinc finger domain (NS5A-1a domain). The natural course of infection of WLSV and its pathogenicity, if any, to the shark species remain to be explored. Nevertheless, the discovery of WLSV in a cartilaginous fish greatly expands the potential host range of hepaciviruses and may lead to the discovery of additional novel hepaciviruses in other cold-blooded animals such as fish, amphibians, and reptiles.
GENETIC FEATURES OF ANIMAL HEPACIVIRUSES
Like other members of the Flaviviridae family, hepaciviruses possess a positive-strand RNA genome with a single large ORF encoding a multifunctional polyprotein that is flanked by UTRs (19). The 5′ UTR contains a type IV IRES structural element that directs cap-independent translation of the viral genomic RNA. Host- and viral-encoded proteases process the polyprotein precursor into ten distinct protein products: three structural proteins (core, E1, and E2) and seven nonstructural proteins (NS1, NS2, NS3, NS4a, NS4b, NS5a, and NS5b). These genetic features are in direct contrast to members of the Pegivirus genus, which do not encode a core protein and possess a type III rather than a type IV IRES structural element (19).
Although animal hepaciviruses share similar patterns of genomic organization and predicted protein structure, distinct differences do exist (Table 1). All hepaciviruses fully characterized at the genomic level possess putative seeding sites in the 5′ UTR for the binding of host-expressed miR-122. Most hepacivirus genomes (HCV, GBV-B, and certain RHV strains) contain two miR-122 binding sites within the 5′ UTR, but single sites have been identified in the BHV, NPHV, and various RHV genomes. In HCV, interaction between the viral 5′ UTR and human miR-122 promotes viral replication and accumulation of intracellular viral RNA (57). The requirement for a 5′ UTR–miR-122 interaction in other hepacivirus species is not fully known, although a single study demonstrated miR-122-independent replication of GBV-B in vitro (58). Given the close homology between HCV and NPHV, it is interesting that altered numbers of putative seeding sites are observed between the two viruses. This difference may indicate a role for the viral 5′ UTR in determining host and organ tropism, but additional studies are needed to address this hypothesis. In one study, however, Scheel and colleagues (46) demonstrated that construction of a chimeric NPHV/HCV clone in which the last stem-loop of the intermediate region of the NPHV 5′ UTR was replaced by the terminal stem-loop of HCV resulted in the disruption of NPHV viral RNA translation in vitro.
There are also distinct differences in the predicted number of glycosylation sites on the encoded envelope proteins (E1/E2) of animal hepaciviruses (Figure 3). Glycosylation of the HCV E1 and E2 proteins mediates protein folding and helps mask virions from the potential binding of host neutralizing antibodies (59), and it therefore has significant relevance to hepaciviral biology. The number of predicted glycosylation sites (totaled between E1 and E2) for hepaciviruses is highest for HCV, followed closely by NPHV. Given HCV’s propensity for establishing chronicity and the demonstration of chronic NPHV infections in horses (16), it would be interesting to assess whether any correlation exists between the number of predicted glycosylation sites within hepaciviral genomes and rates of establishing persistent infection in vivo.
Figure 3.
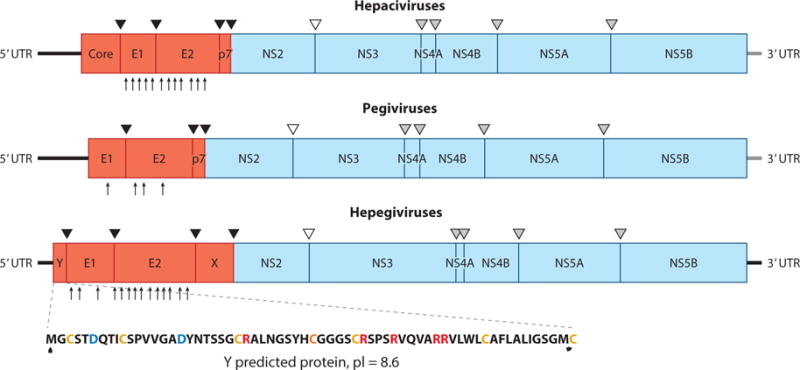
Genome organization and polyprotein cleavage map of hepaciviruses, pegiviruses, and hepegiviruses. The complete genome encodes a polyprotein that is co- and posttranslationally cleaved into individual viral proteins. The structural proteins include the core protein (C) and envelope glycoproteins (E1 and E2), and the nonstructural proteins are NS2, NS3, NS4A, NS4B, NS5A, and NS5B. Classical pegiviruses (human and simian pegiviruses) do not have a recognizable C protein. Structural proteins are cleaved by cellular signal peptidases (black triangles), and NS2-NS3 cleavage is accomplished by the NS2-NS3 autoprotease (empty triangle). The remaining nonstructural proteins are cleaved by the NS3-NS4A protease complex (gray triangles). Hepegiviruses encode a protein termed Y that is similar in location and properties to hepacivirus C. Glycosylation sites in E1 and E2 are shown by arrows at the bottom of the polyproteins for hepatitis C virus, GB virus C, and human hepegivirus (60, 114). Abbreviations: pI, protein isoelectric point; UTR, untranslated region.
HEPEGIVIRUSES CONNECT HEPACIVIRUSES AND PEGIVIRUSES
While using an unbiased approach to analyze the virome composition of patients receiving blood products, Kapoor and colleagues (60) identified a novel human virus, human hepegivirus (HHpgV-1), in the sera of four transfusion recipients that shares genetic features with both hepaciviruses and pegiviruses. Soon after, variants of HHpgV, named human pegivirus type 2 (HPgV-2), were described as coinfecting several HCV patients in the United States (61) and later in the United Kingdom (62). The HHpgV-1 and HPgV-2 variants show 1–7% nucleotide diversity over the entire length of their genomes, but like other hepaciviruses and pegiviruses, their UTRs are relatively more conserved. Presence of HHpgV-1 in several human serum samples from different geographies, together with the evidence of seroconversion in most infected individuals, confirms that HHpgV-1 causes an authentic human infection that plausibly transmits by the parenteral route (60–62).
Phylogenetic and molecular analysis indicates that HHpgV-1 is highly distant from known members of the Hepacivirus and Pegivirus genera (Figure 2). HHpgV-1 is predicted to encode a short, highly basic protein (termed Y) upstream of E1 that is comparable to the VR protein identified in rodent and bat pegivirus variants (60). The Y and VR proteins differ, however, in their predicted locations within the cell. Rodent and bat VR proteins possess a predicted signalase cleavage site at the N terminus that may function to relocate the protein to the endoplasmic reticulum. In contrast, the Y protein of HHpgV-1 possesses no such translocation sequence, suggestive of a cytoplasmic location akin to the core proteins of hepaciviruses. Although considerably shorter than the fully processed HCV core protein, the Y protein may share a similar function in RNA packaging and virion assembly. Additionally, the predicted E2 protein contains 11 potential N-linked glycosylation sites, a greater number than recorded for most pegiviruses (19) (Figure 3). This pattern of heavy glycosylation, comparable to that of HCV and other hepaciviruses, was distinct from the infrequent sites recorded for human and other group 1 pegiviruses (60). Furthermore, the HHpgV-1 5′ UTR is nonhomologous to that of most other pegiviruses, forming a type IV IRES, hitherto described only for members of the Hepacivirus and Pestivirus genera. Although highly divergent from all other known viral 5′ UTR sequences, certain motifs match those of members of the Hepacivirus genus, including the highly conserved sequence TACAGCCTGATAGGGT at position 274. Interestingly, however, the miR-122 seed site present in most hepacivirus species is absent from the 5′ UTR of HHpgV-1 and all other known pegiviruses (60). Presence of these unique features in the virus prompted the use of the name hepegivirus (a combination of hepacivirus and pegivirus), although it remains to be finalized by the International Committee on Taxonomy of Viruses.
ORIGIN AND EVOLUTION OF ANIMAL HEPACIVIRUSES
The discovery of novel hepacivirus species in previously unexplored hosts has greatly expanded our knowledge of the hepacivirus host range and provided new, intriguing insights into the origins and evolution of the Hepacivirus genus. Indeed, identification of a possible zoonotic source for HCV, which has eluded virologists since HCV’s discovery in 1989 (63), finally seems within our grasp (37, 51, 52, 64, 65). The hypotheses for HCV’s origin are based on the well-characterized genetic diversity of the HCV genotypes, their phylogenetic relatedness to other animal hepaciviruses (Figure 2), and observed differences in their geographical distribution.
A reasonable hypothesis is that HCV originated from a single cross-species transmission event between ancestral humans and a zoonotic host and then subsequently evolved as human populations dispersed globally into discrete geographical populations, resulting in the current global pattern of HCV diversity (12, 52). Despite the remarkable sequence variability observed among strains, the close genetic relatedness of the various HCV genotypes (Figure 2) supports this viewpoint. To date, NPHV remains the closest genetic relative of HCV (Figure 2), with similarity existing even between regions encoding the E2 envelope protein, a remarkable observation given the high rate of mutations that occur within these areas. Analysis of the number and position of cysteine residues within the encoded NPHV E2 protein even suggests a tertiary structure that is substantially more similar to HCV E2 than to E2 proteins from other known hepaciviruses (43, 66). Additional similarities have been reported for NPHV and HCV, including core protein processing and NS3/4A-mediated antagonism of the RIG-I antiviral pathway (67–69). Our analysis based on the evolutionary rates of HCV genotypes 1 and 6 (40) indicated that the HCV genotypes and NPHV shared a common ancestor 341 and 1,680 years ago, respectively (43). However, this should be regarded as a minimum estimate given the difficulties associated with extrapolating short-term substitution rates to longer evolutionary periods (70–73).
The close ecological relationship between horses and humans, culminating in the nineteenth century when domesticated horses were the primary means of human locomotion, certainly raises the possibility that horses are responsible for the initial transmission of HCV to humans, either directly or as intermediate hosts. How equine hepaciviruses could have been transmitted to humans is unclear, but advances in vaccine and antitoxin medicine during the twentieth century exposed humans to a vast array of horse serum–derived products, which certainly could have facilitated the transmission of viruses that normally do not breach the skin barrier. Although an equine origin for HCV is intriguing, the genetic distances between HCV and NPHV are nevertheless quite large (~50% at the nucleotide level), and compared with HCV, NPHV exhibits very low sequence diversity. Were horses the original source of HCV, then equine hepaciviruses would have had to transmit to humans, rapidly evolve and diversify, and then subsequently disperse into specific geographical regions, all the while remaining phylogenetically stagnant in horses, a scenario that is feasible but highly unlikely. Therefore, HCV may instead be the result of a cross-species transmission event between humans and an unidentified mammalian host, even possibly as an intermediate between horses and humans. Evidence for an extant mammalian species that harbors hepaciviruses more closely related to the HCV genotypes than NPHV would be consistent with this hypothesis.
Alternatively, the current genotypes of HCV could be the result of multiple, independent zoonotic transmission events. A limitation to this proposal, however, is that none of the currently identified animal hepaciviruses exhibit close phylogenetic clustering around any of the major HCV genotypes. This could be due to severe undersampling of the known mammalian diversity, or it could simply be an argument against multiple human-animal spillover events (52). Based on current phylogenetic analyses, RHVs are the most genetically diverse members of the Hepacivirus genus, comprising three separate lineages and eight different clades. In fact, depending on the strain, RHVs share common ancestors with BHVs, GHV, and, more distantly, the NPHV/HCV lineage. The basal positioning of RHVs in the phylogenetic trees of hepaciviruses (Figure 2) might further indicate a crucial evolutionary role for rodents as zoonotic transmitters of hepacivirus species. Whether rodents represent the original source of hepaciviruses or merely intermediate hosts is completely unknown, but considering rodents account for more than 40% of the observable diversity of mammal species, it seems very likely they would be responsible for a large proportion of hepacivirus cross-species transmission events between animals. The recent discovery of RHV in Norway rats in New York City is of particular significance, as these rodents share considerable ecological factors with humans and thus may be a potential source of foreign pathogens such as hepaciviruses (28).
The recent identification of WLSV in catsharks and hepegivirus species in humans poses new questions about the evolutionary relationship between hepaciviruses and pegiviruses. Despite significant divergence from the other known hepaciviruses, WLSV exhibits higher phylogenetic relatedness to mammalian hepaciviruses than to pegiviruses (Figure 2). This raises the interesting possibility that hepaciviruses may have infected different animals, including humans, throughout their evolution or even before the host speciation. Consistent with this scenario, the high species specificity of hepaciviruses, combined with their propensity for persistence, is suggestive of long-term coevolution and host adaptation. Furthermore, the identification of HHpgV-1 and HPgV-2 in human populations is significant not only because of their transmissibility between humans but also because their genomic organization argues against either pegivirus or hepacivirus classification (Figure 3). This might suggest that the Pegivirus and Hepacivirus genera recently diverged from a common hepegivirus-like ancestor, resulting in the observed low phylogenetic divergence between pegivirus and hepacivirus species compared with that of other Flaviviridae members. Identification of additional pegivirus and hepacivirus species will help resolve the obscure link between these two sister genera.
Despite the extraordinary expansion in knowledge of hepacivirus host range over the past few years, we cannot understate the importance of sampling diversity when speculating on the origin and evolution of animal hepaciviruses. Undersampling of hepaciviral diversity is the most likely cause of long phylogenetic branches separating various hepacivirus lineages, each of which contains very closely related species (51) (Figure 2). At present, only a few mammalian species have been screened for hepacivirus infection, and when extrapolating to all animal species, which is especially important considering the identification of WSLV in the graceful catshark, the result is even poorer. Further epidemiological and screening studies are thus necessary to more fully characterize the host range and genetic diversity among animal hepaciviruses and ultimately to solve the puzzle of hepacivirus evolution. We believe that the biological characterization of new hepaciviruses, specifically their mode of transmission and species specificity, will provide further clarity to the puzzle of hepaciviral origin and evolution.
ANIMAL HEPACIVIRUSES AS SURROGATE EXPERIMENTAL MODELS FOR HEPATITIS C VIRUS
HCV causes approximately one million deaths and two to three million new infections per year (74–77). The development of robust in vitro cell culture systems facilitated the study of the viral life cycle and ultimately aided in the generation of direct-acting antivirals that have transformed standard treatment regimens (78, 79). Development of a successful vaccine, however, remains a formidable challenge (80). Although cell culture systems permit functional and inhibition studies of HCV, detailed exploration into the host-viral interaction within the liver, particularly during early infection, when resolution or persistence is determined (81, 82), cannot be easily addressed with these platforms and thus remains a principal goal for in vivo models. Only chimpanzees can serve as immunocompetent hosts to study natural HCV infection (83–86), but new National Institutes of Health regulations completely restrict their usage in research. Development of a new tractable animal model therefore remains a top priority for HCV researchers (27, 30, 52, 65, 75, 87–96). Over the years, several models of immunodeficient mice engrafted with human liver cells have been developed (95, 97, 98). Recently, mice engineered to express human versions of the HCV entry factors were reported to sustain HCV replication when crossed to innate immune– deficient strains (STAT1, IRF, IRF7) (95, 99). Another mouse model ectopically expressing both human CD81 and occludin was shown to permit replication without the need for immune suppression (100). Collectively, these models have expanded our ability to study HCV infection in vivo; however, major drawbacks include low levels of infection and/or the increasing need to blunt innate and adaptive immune pathways to achieve HCV replication (89, 95). A relevant, tractable, and immunocompetent model for HCV is therefore needed if a more detailed analysis of HCV pathogenesis and immunity is to be achieved (89, 94–96).
Considering the strict species tropism of HCV and the absence of immunocompetent animal models permissive to robust infection, the animal hepaciviruses should be evaluated as potential surrogate models for the study of HCV. Indeed, remarkable insights into viral pathogenesis and immunity were gleaned from the use of animal homologs as surrogates for the study of human noroviruses, HIV, herpesviruses, and several others (101). GBV-B is a well-recognized HCV surrogate, but unlike HCV, it usually causes only acute resolving infections in tamarins (88, 92). The initial identification of animal hepaciviruses therefore bodes well for the development of a fully immunocompetent surrogate for HCV study (27, 30, 43, 46, 51, 52). However, so far only genetic properties of these viruses are known, and an ideal surrogate should resemble HCV, including its hepatotropism, propensity for establishing persistent infection, and associated immunity and pathogenesis (52, 97). However, the two crucial questions that need to be answered are which host and which virus should be targeted to develop an informative HCV surrogate. Recently, a reverse genetics system was developed for NPHV, the use of which revealed that NPHV parallels HCV in its hepatotropism and ability to cause persistent infection (46). However, horses are not genetically amenable or tractable models. Moreover, the limited genetic diversity observed among NPHV variants restricts studies of heterologous immunity and pathogenesis. Similar limitations exist for bovine or Colobus sp. models and to an extent for bats due to laboratory intractability and a lack of host-specific reagents.
Ideally, the identification of novel hepaciviruses in house mice would simplify the choice of an HCV surrogate, but so far no such virus has been discovered. Although it may be argued that hepacivirus studies in primates would provide the most relevant insights into human HCV infection, currently only rodents can be easily manipulated by targeted mutagenesis, gene knockout, and transgenic technologies such as closely interspersed short palindromic repeats (CRISPRs). Furthermore, phylogenetic analysis of all known hepaciviruses reveals that HCV is more closely related to hepaciviruses found in rodents than to the other primate hepaciviruses (GBV-B and GHV) (51, 52, 65) (Figure 2). For these reasons, RHVs found in deer mice and rats are most appropriate, as both these host species are tractable and well characterized. Deer mice were extensively used as an experimental model for hantaviruses and are available as inbred and outbred strains (102–108). Additionally, the genetic diversity observed among deer mouse and rat hepacivirus variants would be extremely useful for the study of heterologous immunity following virus challenge. Although much is to be learned from the study of animal hepaciviruses, one major focus should be to identify the molecular determinants of viral clearance and persistence after primary and secondary infections, with particular emphasis on host immunity. A full understanding of the role of innate, humoral, and cellular immunity in controlling RHV infection and persistence in immunocompetent rat models would provide a molecular basis for the development of novel HCV vaccination strategies in humans. Finally, the development of reverse genetics systems for RHV would enable a detailed exploration into the precise role of various genetic elements (such as 3′ and 5′ UTRs) in determining viral persistence, the findings of which would facilitate the rational design of attenuated hepacivirus vaccines and antiviral therapeutics.
CONCLUSIONS
The identification of animal hepaciviruses has undeniably increased our understanding of hepaciviral origin and host range, but only their full biological characterization will ultimately aid in our understanding of how these viruses cross species barriers and disseminate into new animal populations, including humans. It is plausible that, like other flaviviruses, a few of these hepacivirus species are readily transmitted via insect or airborne routes. However, with the exception of CHV in dogs, hepacivirus species have not been detected in respiratory swabs, and adequate sampling of arthropod vectors has not been fully pursued. Understanding how hepacivirus species flow into new animal populations, and thereby how HCV was initially introduced into humans, requires additional clues. Nevertheless, it seems clear that the zoonotic source of HCV should be a virus remarkably similar to at least one of the HCV genotypes and should infect a host phylogenetically close to humans. Despite identification of several new hepaciviruses in various mammalian species and a shark, the horse hepaciviruses remain the closest genetic relatives of HCV and therefore the most plausible zoonotic source of HCV. However, the high genetic diversity and geographical clustering of HCV genotypes are discordant with the relatively homogeneous horse virus populations worldwide. The only, but very unlikely, possibility is a single introduction of horse virus in humans followed by faster adaptation, rapid diversification, and robust dissemination into human populations globally. We believe that biological characterization of animal hepaciviruses and their mode of infection, transmission, and epidemiology is therefore necessary to gain a more definitive understanding of HCV’s origin.
In addition, biological characterization of animal hepaciviruses is crucial to estimate their significance as surrogate models for HCV. The viral and host determinants of HCV infection outcomes after primary and secondary infections remain largely unknown (48, 81, 82, 92, 96, 109, 110). Similarly, the breadth and nature of protective immunity are not clearly defined, posing major challenges to current HCV vaccine design (111). HCV is unique in its ability to establish lifelong persistence with robust viremia in the infected host. The only two animal hepaciviruses that are biologically characterized in some depth, GBV-B and NPHV, share biological properties with HCV, such as mode of transmission and hepatotropism, but their propensity to establish persistence varies. A diverse array of RHVs is now known. RHVs share several genetic features with HCV, including the folding pattern of their UTRs, their NS5B structure, their polyprotein cleavage sites, and the presence of a miR-122 seed site. Moreover, there is some evidence of their hepatotropism and ability to persist after seroconversion. Together, these findings indicate that the biological characterization of these rodent viruses will pave the way for development of fully immunocompetent and informative surrogate models for delineating the mechanisms of HCV persistence and immunity.
Acknowledgments
We thank Dr. Christopher M. Walker for useful comments. A.H. is supported by the Susan L. Huntington Dean’s Distinguished University Fellowship from The Ohio State University Graduate School. Research in the Kapoor laboratory is supported by National Institutes of Health grants HL119485 and AI107631, the US Department of Agriculture, and The Research Institute at Nationwide Children’s Hospital.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–62. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Simmonds PB, Becher P, Collett MS, Gould EA, Heinz FX, et al. Flaviviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses. London: Elsevier; 2012. pp. 1003–20. [Google Scholar]
- 3.Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002;36:S21–29. doi: 10.1053/jhep.2002.36227. [DOI] [PubMed] [Google Scholar]
- 4.Yamane D, McGivern DR, Masaki T, Lemon SM. Liver injury and disease pathogenesis in chronic hepatitis C. Curr Top Microbiol Immunol. 2013;369:263–88. doi: 10.1007/978-3-642-27340-7_11. [DOI] [PubMed] [Google Scholar]
- 5.Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect. 2011;17:107–15. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- 6.Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, et al. Global trends in emerging infectious diseases. Nature. 2008;451:990–93. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mackenzie JS, Jeggo M. Reservoirs and vectors of emerging viruses. Curr Opin Virol. 2013;3:170–79. doi: 10.1016/j.coviro.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parrish CR, Holmes EC, Morens DM, Park EC, Burke DS, et al. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol Mol Biol Rev. 2008;72:457–70. doi: 10.1128/MMBR.00004-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharp PM, Hahn BH. Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med. 2011;1:a006841. doi: 10.1101/cshperspect.a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flanagan ML, Parrish CR, Cobey S, Glass GE, Bush RM, Leighton TJ. Anticipating the species jump: surveillance for emerging viral threats. Zoonoses Public Health. 2012;59:155–63. doi: 10.1111/j.1863-2378.2011.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morse SS, Mazet JA, Woolhouse M, Parrish CR, Carroll D, et al. Prediction and prevention of the next pandemic zoonosis. Lancet. 2012;380:1956–65. doi: 10.1016/S0140-6736(12)61684-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfaender S, Brown RJ, Pietschmann T, Steinmann E. Natural reservoirs for homologs of hepatitis C virus. Emerg Microbes Infect. 2014;3:e21. doi: 10.1038/emi.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfaender S, Cavalleri JM, Walter S, Doerrbecker J, Campana B, et al. Clinical course of infection and viral tissue tropism of hepatitis C virus-like non-primate hepaciviruses. Hepatology. 2014;61:447–59. doi: 10.1002/hep.27440. [DOI] [PubMed] [Google Scholar]
- 14.Makuwa M, Souquiere S, Telfer P, Leroy E, Bourry O, et al. Occurrence of hepatitis viruses in wild-born non-human primates: a 3 year (1998–2001) epidemiological survey in Gabon. J Med Primatol. 2003;32:307–14. doi: 10.1046/j.1600-0684.2003.00042.x. [DOI] [PubMed] [Google Scholar]
- 15.Makuwa M, Souquiere S, Telfer P, Bourry O, Rouquet P, et al. Hepatitis viruses in non-human primates. J Med Primatol. 2006;35:384–87. doi: 10.1111/j.1600-0684.2006.00163.x. [DOI] [PubMed] [Google Scholar]
- 16.Burbelo PD, Dubovi EJ, Simmonds P, Medina JL, Henriquez JA, et al. Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J Virol. 2012;86:6171–78. doi: 10.1128/JVI.00250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deinhardt F, Holmes AW, Capps RB, Popper H. Studies on the transmission of human viral hepatitis to marmoset monkeys. I. Transmission of disease, serial passages, and description of liver lesions. J Exp Med. 1967;125:673–88. doi: 10.1084/jem.125.4.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simons JN, Pilot-Matias TJ, Leary TP, Dawson GJ, Desai SM, et al. Identification of two flavivirus-like genomes in the GB hepatitis agent. PNAS. 1995;92:3401–5. doi: 10.1073/pnas.92.8.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stapleton JT, Foung S, Muerhoff AS, Bukh J, Simmonds P. The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J Gen Virol. 2011;92:233–46. doi: 10.1099/vir.0.027490-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alter MJ, Gallagher M, Morris TT, Moyer LA, Meeks EL, et al. Sentin. Cties. Viral Hepat. Study Team Acute non-A–E hepatitis in the United States and the role of hepatitis G virus infection. N Engl J Med. 1997;336:741–46. doi: 10.1056/NEJM199703133361101. [DOI] [PubMed] [Google Scholar]
- 21.Schlauder GG, Pilot-Matias TJ, Gabriel GS, Simons JN, Muerhoff AS, et al. Origin of GB-hepatitis viruses. Lancet. 1995;346:447–48. doi: 10.1016/s0140-6736(95)92821-9. [DOI] [PubMed] [Google Scholar]
- 22.Kim JP, Fry KE. Molecular characterization of the hepatitis G virus. J Viral Hepat. 1997;4:77–79. doi: 10.1111/j.1365-2893.1997.tb00208.x. [DOI] [PubMed] [Google Scholar]
- 23.Leary TP, Muerhoff AS, Simons JN, Pilot-Matias TJ, Erker JC, et al. Sequence and genomic organization of GBV-C: a novel member of the Flaviviridae associated with human non-A-E hepatitis. J Med Virol. 1996;48:60–67. doi: 10.1002/(SICI)1096-9071(199601)48:1<60::AID-JMV10>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 24.Xiang J, Klinzman D, McLinden J, Schmidt WN, LaBrecque DR, et al. Characterization of hepatitis G virus (GB-C virus) particles: evidence for a nucleocapsid and expression of sequences upstream of the E1 protein. J Virol. 1998;72:2738–44. doi: 10.1128/jvi.72.4.2738-2744.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Epstein JH, Quan PL, Briese T, Street C, Jabado O, et al. Identification of GBV-D, a novel GB-like flavivirus from old world frugivorous bats (Pteropus giganteus) in Bangladesh. PLOS Pathog. 2010;6:e1000972. doi: 10.1371/journal.ppat.1000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandriani S, Skewes-Cox P, Zhong W, Ganem DE, Divers TJ, et al. Identification of a previously undescribed divergent virus from the Flaviviridae family in an outbreak of equine serum hepatitis. PNAS. 2013;110:E1407–15. doi: 10.1073/pnas.1219217110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drexler JF, Corman VM, Muller MA, Lukashev AN, Gmyl A, et al. Evidence for novel hepaciviruses in rodents. PLOS Pathog. 2013;9:e1003438. doi: 10.1371/journal.ppat.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York City. mBio. 2014;5:e01933–14. doi: 10.1128/mBio.01933-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapoor A, Simmonds P, Cullen JM, Scheel TK, Medina JL, et al. Identification of a pegivirus (GB virus-like virus) that infects horses. J Virol. 2013;87:7185–90. doi: 10.1128/JVI.00324-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapoor A, Simmonds P, Scheel TK, Hjelle B, Cullen JM, et al. Identification of rodent homologs of hepatitis C virus and pegiviruses. mBio. 2013;4:e00216–13. doi: 10.1128/mBio.00216-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quan PL, Firth C, Conte JM, Williams SH, Zambrana-Torrelio CM, et al. Bats are a major natural reservoir for hepaciviruses and pegiviruses. PNAS. 2013;110:8194–99. doi: 10.1073/pnas.1303037110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318–27. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-α therapy. Science. 1998;282:103–7. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 34.Galli A, Bukh J. Comparative analysis of the molecular mechanisms of recombination in hepatitis C virus. Trends Microbiol. 2014;22:354–64. doi: 10.1016/j.tim.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 35.Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015;61:77–87. doi: 10.1002/hep.27259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy DG, Willems B, Deschenes M, Hilzenrat N, Mousseau R, Sabbah S. Use of sequence analysis of the NS5B region for routine genotyping of hepatitis C virus with reference to C/E1 and 5 untranslated region sequences. J Clin Microbiol. 2007;45:1102–12. doi: 10.1128/JCM.02366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simmonds P. The origin of hepatitis C virus. Curr Top Microbiol Immunol. 2013;369:1–15. doi: 10.1007/978-3-642-27340-7_1. [DOI] [PubMed] [Google Scholar]
- 38.Smith DB, Pathirana S, Davidson F, Lawlor E, Power J, et al. The origin of hepatitis C virus genotypes. J Gen Virol. 1997;78(Pt. 2):321–28. doi: 10.1099/0022-1317-78-2-321. [DOI] [PubMed] [Google Scholar]
- 39.Pybus OG, Cochrane A, Holmes EC, Simmonds P. The hepatitis C virus epidemic among injecting drug users. Infect Genet Evol. 2005;5:131–39. doi: 10.1016/j.meegid.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 40.Magiorkinis G, Magiorkinis E, Paraskevis D, Ho SY, Shapiro B, et al. The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis. PLOS Med. 2009;6:e1000198. doi: 10.1371/journal.pmed.1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gray RR, Tanaka Y, Takebe Y, Magiorkinis G, Buskell Z, et al. Evolutionary analysis of hepatitis C virus gene sequences from 1953. Philos Trans R Soc B. 2013;368:20130168. doi: 10.1098/rstb.2013.0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999;397:436–41. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- 43.Kapoor A, Simmonds P, Gerold G, Qaisar N, Jain K, et al. Characterization of a canine homolog of hepatitis C virus. PNAS. 2011;108:11608–13. doi: 10.1073/pnas.1101794108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bexfield NH, Watson PJ, Heaney J, Heeney JL, Tiley L. Canine hepacivirus is not associated with chronic liver disease in dogs. J Viral Hepat. 2014;21:223–28. doi: 10.1111/jvh.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyons S, Kapoor A, Sharp C, Schneider BS, Wolfe ND, et al. Nonprimate hepaciviruses in domestic horses, United Kingdom. Emerg Infect Dis. 2012;18:1976–82. doi: 10.3201/eid1812.120498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheel TK, Kapoor A, Nishiuchi E, Brock KV, Yu Y, et al. Characterization of nonprimate hepacivirus and construction of a functional molecular clone. PNAS. 2015;112:2192–97. doi: 10.1073/pnas.1500265112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramsay JD, Evanoff R, Wilkinson TE, Jr, Divers TJ, Knowles DP, Mealey RH. Experimental transmission of equine hepacivirus in horses as a model for hepatitis C virus. Hepatology. 2015;61:1533–46. doi: 10.1002/hep.27689. [DOI] [PubMed] [Google Scholar]
- 48.Park SH, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity. 2014;40:13–24. doi: 10.1016/j.immuni.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Postel A, Cavalleri JM, Pfaender S, Walter S, Steinmann E, et al. Frequent presence of hepaci and pegiviruses in commercial equine serum pools. Vet Microbiol. 2016;182:8–14. doi: 10.1016/j.vetmic.2015.10.032. [DOI] [PubMed] [Google Scholar]
- 50.Lyons S, Kapoor A, Schneider BS, Wolfe ND, Culshaw G, et al. Viraemic frequencies and seroprevalence of non-primate hepacivirus and equine pegiviruses in horses and other mammalian species. J Gen Virol. 2014;95:1701–11. doi: 10.1099/vir.0.065094-0. [DOI] [PubMed] [Google Scholar]
- 51.Theze J, Lowes S, Parker J, Pybus OG. Evolutionary and phylogenetic analysis of the hepaciviruses and pegiviruses. Genome Biol Evol. 2015;7:2996–3008. doi: 10.1093/gbe/evv202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheel TK, Simmonds P, Kapoor A. Surveying the global virome: identification and characterization of HCV-related animal hepaciviruses. Antivir Res. 2015;115:83–93. doi: 10.1016/j.antiviral.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lauck M, Sibley SD, Lara J, Purdy MA, Khudyakov Y, et al. A novel hepacivirus with an unusually long and intrinsically disordered NS5A protein in a wild Old World primate. J Virol. 2013;87:8971–81. doi: 10.1128/JVI.00888-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corman VM, Grundhoff A, Baechlein C, Fischer N, Gmyl A, et al. Highly divergent hepaciviruses from African cattle. J Virol. 2015;89:5876–82. doi: 10.1128/JVI.00393-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baechlein C, Fischer N, Grundhoff A, Alawi M, Indenbirken D, et al. Identification of a novel hepacivirus in domestic cattle from Germany. J Virol. 2015;89:7007–15. doi: 10.1128/JVI.00534-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi M, Lin XD, Vasilakis N, Tian JH, Li CX, et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J Virol. 2015;90:659–69. doi: 10.1128/JVI.02036-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science. 2005;309:1577–81. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 58.Sagan SM, Sarnow P, Wilson JA. Modulation of GB virus B RNA abundance by microRNA-122: dependence on and escape from microRNA-122 restriction. J Virol. 2013;87:7338–47. doi: 10.1128/JVI.00378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hundt J, Li Z, Liu Q. Post-translational modifications of hepatitis C viral proteins and their biological significance. World J Gastroenterol. 2013;19:8929–39. doi: 10.3748/wjg.v19.i47.8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kapoor A, Kumar A, Simmonds P, Bhuva N, Singh Chauhan L, et al. Virome analysis of transfusion recipients reveals a novel human virus that shares genomic features with hepaciviruses and pegiviruses. mBio. 2015;6:e01466–15. doi: 10.1128/mBio.01466-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berg MG, Lee D, Coller K, Frankel M, Aronsohn A, et al. Discovery of a novel human pegivirus in blood associated with hepatitis C virus co-infection. PLOS Pathog. 2015;11:e1005325. doi: 10.1371/journal.ppat.1005325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonsall D, Gregory WF, Ip CL, Donfield S, Iles J, et al. Evaluation of viremia frequencies of a novel human pegivirus by using bioinformatic screening and PCR. Emerg Infect Dis. 2016;22:671–78. doi: 10.3201/eid2204.151812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kubo Y, Takeuchi K, Boonmar S, Katayama T, Choo QL, et al. A cDNA fragment of hepatitis C virus isolated from an implicated donor of post-transfusion non-A, non-B hepatitis in Japan. Nucleic Acids Res. 1989;17:10367–72. doi: 10.1093/nar/17.24.10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pybus OG, Gray RR. The virus whose family expanded. Nature. 2013;498:310–11. doi: 10.1038/498310b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pybus OG, Theze J. Hepacivirus cross-species transmission and the origins of the hepatitis C virus. Curr Opin Virol. 2016;16:1–7. doi: 10.1016/j.coviro.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 66.Krey T, d’Alayer J, Kikuti CM, Saulnier A, Damier-Piolle L, et al. The disulfide bonds in glyco-protein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLOS Pathog. 2010;6:e1000762. doi: 10.1371/journal.ppat.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parera M, Martrus G, Franco S, Clotet B, Martinez MA. Canine hepacivirus NS3 serine protease can cleave the human adaptor proteins MAVS and TRIF. PLOS ONE. 2012;7:e42481. doi: 10.1371/journal.pone.0042481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patel MR, Loo YM, Horner SM, Gale M, Jr, Malik HS. Convergent evolution of escape from hepaciviral antagonism in primates. PLOS Biol. 2012;10:e1001282. doi: 10.1371/journal.pbio.1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tanaka T, Kasai H, Yamashita A, Okuyama-Dobashi K, Yasumoto J, et al. Hallmarks of hepatitis C virus in equine hepacivirus. J Virol. 2014;88:13352–66. doi: 10.1128/JVI.02280-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Worobey M, Telfer P, Souquiere S, Hunter M, Coleman CA, et al. Island biogeography reveals the deep history of SIV. Science. 2010;329:1487. doi: 10.1126/science.1193550. [DOI] [PubMed] [Google Scholar]
- 71.Katzourakis A, Gifford RJ. Endogenous viral elements in animal genomes. PLOS Genet. 2010;6:e1001191. doi: 10.1371/journal.pgen.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kapoor A, Simmonds P, Lipkin WI. Discovery and characterization of mammalian endogenous parvoviruses. J Virol. 2010;84:12628–35. doi: 10.1128/JVI.01732-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kapoor A, Simmonds P, Dubovi EJ, Qaisar N, Henriquez JA, et al. Characterization of a canine homolog of human Aichivirus. J Virol. 2011;85:11520–25. doi: 10.1128/JVI.05317-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gravitz L. Introduction: a smouldering public-health crisis. Nature. 2011;474:S2–4. doi: 10.1038/474S2a. [DOI] [PubMed] [Google Scholar]
- 75.Verstrepen BE, Boonstra A, Koopman G. Immune mechanisms of vaccine induced protection against chronic hepatitis C virus infection in chimpanzees. World J Hepatol. 2015;7:53–69. doi: 10.4254/wjh.v7.i1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29(Suppl. 1):74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 77.Hagan LM, Wolpe PR, Schinazi RF. Treatment as prevention and cure towards global eradication of hepatitis C virus. Trends Microbiol. 2013;21:625–33. doi: 10.1016/j.tim.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lindenbach BD, Rice CM. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol. 2013;11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scheel TK, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19:837–49. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walker CM, Grakoui A. Hepatitis C virus: Why do we need a vaccine to prevent a curable persistent infection? Curr Opin Immunol. 2015;35:137–43. doi: 10.1016/j.coi.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Raghuraman S, Park H, Osburn WO, Winkelstein E, Edlin BR, Rehermann B. Spontaneous clearance of chronic hepatitis C virus infection is associated with appearance of neutralizing antibodies and reversal of T-cell exhaustion. J Infect Dis. 2012;205:763–71. doi: 10.1093/infdis/jir835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Investig. 2009;119:1745–54. doi: 10.1172/JCI39133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–26. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 84.Pietschmann T, Lohmann V, Kaul A, Krieger N, Rinck G, et al. Persistent and transient replication of full-length hepatitis C virus genomes in cell culture. J Virol. 2002;76:4008–21. doi: 10.1128/JVI.76.8.4008-4021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–96. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dolgin E. Research technique: the murine candidate. Nature. 2011;474:S14–15. doi: 10.1038/474S14a. [DOI] [PubMed] [Google Scholar]
- 87.Barth H, Robinet E, Liang TJ, Baumert TF. Mouse models for the study of HCV infection and virus-host interactions. J Hepatol. 2008;49:134–42. doi: 10.1016/j.jhep.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Beames B, Chavez D, Lanford RE. GB virus B as a model for hepatitis C virus. ILAR J. 2001;42:152–60. doi: 10.1093/ilar.42.2.152. [DOI] [PubMed] [Google Scholar]
- 89.Billerbeck E, de Jong Y, Dorner M, de la Fuente C, Ploss A. Animal models for hepatitis C. Curr Top Microbiol Immunol. 2013;369:49–86. doi: 10.1007/978-3-642-27340-7_3. [DOI] [PubMed] [Google Scholar]
- 90.Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol. 2004;5:971–74. doi: 10.1038/ni1004-971. [DOI] [PubMed] [Google Scholar]
- 91.Lanford RE, Bigger C, Bassett S, Klimpel G. The chimpanzee model of hepatitis C virus infections. ILAR J. 2001;42:117–26. doi: 10.1093/ilar.42.2.117. [DOI] [PubMed] [Google Scholar]
- 92.Lanford RE, Evans MJ, Lohmann V, Lindenbach B, Gale M, Jr, et al. The accelerating pace of HCV research: a summary of the 15th International Symposium on Hepatitis C Virus and Related Viruses. Gastroenterology. 2009;136:9–16. doi: 10.1053/j.gastro.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu L, Fisher BE, Thomas DL, Cox AL, Ray SC. Spontaneous clearance of primary acute hepatitis C virus infection correlated with high initial viral RNA level and rapid HVR1 evolution. Hepatology. 2012;55:1684–91. doi: 10.1002/hep.25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scull MA, Shi C, de Jong YP, Gerold G, Ries M, et al. Hepatitis C virus infects rhesus macaque hepatocytes and simianized mice. Hepatology. 2015;62:57–67. doi: 10.1002/hep.27773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.von Schaewen M, Ploss A. Murine models of hepatitis C: What can we look forward to? Antivir Res. 2014;104:15–22. doi: 10.1016/j.antiviral.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li K, Lemon SM. Innate immune responses in hepatitis C virus infection. Semin Immunopathol. 2013;35:53–72. doi: 10.1007/s00281-012-0332-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bukh J. Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology. 2012;142:1279–87. doi: 10.1053/j.gastro.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 98.Vercauteren K, de Jong YP, Meuleman P. HCV animal models and liver disease. J Hepatol. 2014;61:S26–33. doi: 10.1016/j.jhep.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 99.Dorner M, Rice CM, Ploss A. Study of hepatitis C virus entry in genetically humanized mice. Methods. 2013;59:249–57. doi: 10.1016/j.ymeth.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen J, Zhao Y, Zhang C, Chen H, Feng J, et al. Persistent hepatitis C virus infections and hepatopathological manifestations in immune-competent humanized mice. Cell Res. 2014;24:1050–66. doi: 10.1038/cr.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Karst SM, Wobus CE. Viruses in rodent colonies: lessons learned from murine noroviruses. Annu Rev Virol. 2015;2:525–48. doi: 10.1146/annurev-virology-100114-055204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Herbst MM, Prescott J, Palmer AD, Schountz T. Sequence and expression analysis of deer mouse interferon-γ, interleukin-10, tumor necrosis factor, and lymphotoxin-α. Cytokine. 2002;17:203–13. doi: 10.1006/cyto.2001.0998. [DOI] [PubMed] [Google Scholar]
- 103.Oko L, Aduddell-Swope B, Willis D, Hamor R, Coons TA, et al. Profiling helper T cell subset gene expression in deer mice. BMC Immunol. 2006;7:18. doi: 10.1186/1471-2172-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Root JJ, Black WC, III, Calisher CH, Wilson KR, Mackie RS, et al. Analyses of gene flow among populations of deer mice (Peromyscus maniculatus) at sites near hantavirus pulmonary syndrome case-patient residences. J Wildl Dis. 2003;39:287–98. doi: 10.7589/0090-3558-39.2.287. [DOI] [PubMed] [Google Scholar]
- 105.Schountz T, Acuna-Retamar M, Feinstein S, Prescott J, Torres-Perez F, et al. Kinetics of immune responses in deer mice experimentally infected with Sin Nombre virus. J Virol. 2012;86:10015–27. doi: 10.1128/JVI.06875-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schountz T, Green R, Davenport B, Buniger A, Richens T, et al. Cloning and characterization of deer mouse (Peromyscus maniculatus) cytokine and chemokine cDNAs. BMC Immunol. 2004;5:1. doi: 10.1186/1471-2172-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schountz T, Quackenbush S, Rovnak J, Haddock E, Black WC, IV, et al. Differential lymphocyte and antibody responses in deer mice infected with Sin Nombre or Andes hantaviruses. J Virol. 2014;88:8319–31. doi: 10.1128/JVI.00004-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schountz T, Shaw TI, Glenn TC, Feldmann H, Prescott J. Expression profiling of lymph node cells from deer mice infected with Andes virus. BMC Immunol. 2013;14:18. doi: 10.1186/1471-2172-14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spada E, Mele A, Berton A, Ruggeri L, Ferrigno L, et al. Multispecific T cell response and negative HCV RNA tests during acute HCV infection are early prognostic factors of spontaneous clearance. Gut. 2004;53:1673–81. doi: 10.1136/gut.2003.037788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, et al. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology. 2007;132:667–78. doi: 10.1053/j.gastro.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 111.Dahari H, Feinstone SM, Major ME. Meta-analysis of hepatitis C virus vaccine efficacy in chimpanzees indicates an importance for structural proteins. Gastroenterology. 2010;139:965–74. doi: 10.1053/j.gastro.2010.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–20. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 113.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–74. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mohr EL, Stapleton JT. GB virus type C interactions with HIV: the role of envelope glycoproteins. J Viral Hepat. 2009;16:757–68. doi: 10.1111/j.1365-2893.2009.01194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lyons S, Kapoor A, Schneider BS, Wolfe ND, Culshaw G, et al. Viraemic frequencies and sero-prevalence of non-primate hepacivirus and equine pegiviruses in horses and other mammalian species. J Gen Virol. 2014;95:1701–11. doi: 10.1099/vir.0.065094-0. [DOI] [PubMed] [Google Scholar]
- 116.Drexler JF, Corman VM, Muller MA, Lukashev AN, Gmyl A, et al. Evidence for novel hepaciviruses in rodents. PLOS Pathog. 2013;9:e1003438. doi: 10.1371/journal.ppat.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Takikawa S, Engle RE, Emerson SU, Purcell RH, St Claire M, Bukh J. Functional analyses of GB virus B p13 protein: development of a recombinant GB virus B hepatitis virus with a p7 protein. PNAS. 2006;103:3345–50. doi: 10.1073/pnas.0511297103. [DOI] [PMC free article] [PubMed] [Google Scholar]