

Abstract
Background
Sex‐related differences in cardiac function and iron metabolism exist in humans and experimental animals. Male patients and preclinical animal models are more susceptible to cardiomyopathies and heart failure. However, whether similar differences are seen in iron‐overload cardiomyopathy is poorly understood.
Methods and Results
Male and female wild‐type and hemojuvelin‐null mice were injected and fed with a high‐iron diet, respectively, to develop secondary iron overload and genetic hemochromatosis. Female mice were completely protected from iron‐overload cardiomyopathy, whereas iron overload resulted in marked diastolic dysfunction in male iron‐overloaded mice based on echocardiographic and invasive pressure‐volume analyses. Female mice demonstrated a marked suppression of iron‐mediated oxidative stress and a lack of myocardial fibrosis despite an equivalent degree of myocardial iron deposition. Ovariectomized female mice with iron overload exhibited essential pathophysiological features of iron‐overload cardiomyopathy showing distinct diastolic and systolic dysfunction, severe myocardial fibrosis, increased myocardial oxidative stress, and increased expression of cardiac disease markers. Ovariectomy prevented iron‐induced upregulation of ferritin, decreased myocardial SERCA2a levels, and increased NCX1 levels. 17β‐Estradiol therapy rescued the iron‐overload cardiomyopathy in male wild‐type mice. The responses in wild‐type and hemojuvelin‐null female mice were remarkably similar, highlighting a conserved mechanism of sex‐dependent protection from iron‐overload‐mediated cardiac injury.
Conclusions
Male and female mice respond differently to iron‐overload‐mediated effects on heart structure and function, and females are markedly protected from iron‐overload cardiomyopathy. Ovariectomy in female mice exacerbated iron‐induced myocardial injury and precipitated severe cardiac dysfunction during iron‐overload conditions, whereas 17β‐estradiol therapy was protective in male iron‐overloaded mice.
Keywords: 17‐β‐estradiol, heart failure, hemojuvelin, iron overload, myocardial fibrosis, ovariectomy, oxidative stress, sex
Subject Categories: Cardiomyopathy, Heart Failure, Pathophysiology, Oxidant Stress
Introduction
Iron is an essential element in biological systems because of its ability to shuttle between 2 oxidative states and plays a key role in cell metabolism and homeostasis.1 Excess iron (iron overload) or lack of iron (iron deficiency) are the 2 major pathophysiological states of abnormal iron metabolism.2, 3 Under physiological conditions, iron transport is highly conserved and controlled by iron transporters including transferrin and its receptors via negative feedback regulatory mechanisms.4 However, in primary hemochromatosis and secondary iron overload, iron metabolism is perturbed, which leads to chronic iron overload and increased morbidity and mortality.3, 5 Iron‐overload cardiomyopathy is the most common cause of mortality in patients with secondary iron overload and is a major comorbidity in patients with primary hemochromatosis.5, 6, 7, 8 Uncontrolled iron absorption causes transferrin saturation and increased levels of non‐transferrin‐bound iron, which is highly reactive, toxic, and triggers oxidative stress.6 Iron‐induced oxidative stress is a key driver in the pathogenesis of myocardial tissue injury and progressive development of iron‐overload cardiomyopathy.7, 9, 10 Excess iron promotes oxidative stress via the Fenton reaction, which plays a key pathogenic role in myocardial injury and heart failure.9, 11, 12, 13
Sex‐related differences do exist in the pathophysiology of cardiac diseases, and sex‐specific pathways play a key role in the cardioprotection observed in cardiomyopathies and heart failure in preclinical models and patients.14, 15, 16, 17, 18, 19 Estrogen has several beneficial pleiotropic effects on the cardiovascular system.14, 20, 21 Functional estrogen receptors are expressed in cardiomyocytes and cardiac fibroblasts,22 and via the activation of these receptors, β‐estradiol attenuates cardiac hypertrophy, metabolic dysregulation, and cardiac apoptosis.14, 19, 23, 24 In contrast, lack of estrogen leads to adverse myocardial remodeling and precipitates heart failure.14, 19, 25 Sex‐related disparities in the regulation of iron metabolism may contribute to the differences in progression of iron‐overload heart disease.26, 27 Moreover, women have lower iron stores, which have been argued to confer protection in an iron‐overloaded state.28, 29 In this study we investigated the sex‐specific differences and mechanisms of iron‐overload cardiomyopathy.
Materials and Methods
Experimental Animal Protocols
Wild‐type (WT) C57BL6/J male and female mice (from Jackson Laboratory, Bar Harbor, ME) and hemojuvelin‐null (HJV−/−; HJVKO) male and female mice (kindly provided by Dr Nancy C. Andrews, Duke University, Durham, NC) were bred in house at the University of Alberta Health Sciences Laboratory Animal Services housing facility. All experiments were performed in accordance with University of Alberta institutional guidelines, which conformed to guidelines published by the Canadian Council on Animal Care and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (revised 2011). Intraperitoneal iron injections of 5 mg of iron dextran per 25 g body weight injected ip on a 5 day/week schedule for a total duration of 4 weeks followed by 1.25 mg/25 g body weight for 8 more weeks in WT mice generated the secondary iron‐overload model.9, 10, 30, 31 A separate group of male iron‐overloaded WT mice were randomized to receive vehicle or 17β‐estradiol treatment (0.04 mg/kg per day) during the terminal 21 days of iron overload using subcutaneously implanted continuous‐release pellets (Innovative Research of America, Sarasota, FL). Treatment with a high‐iron diet (Prolab®RHM 3000 with iron 380 ppm) to HJVKO mice for 6 months generated the murine model of genetic hemochromatosis.30, 32
Ovariectomy Surgery Protocol
Ten‐week‐old WT and HJVKO female mice were subjected to bilateral ovariectomy as described previously.24, 33 Briefly, mice were anesthetized by using isoflurane (1% to 1.5%) and maintained at 37°C on a heating pad. A midline abdominal incision was made in the skin and muscle layer, and the ovaries were identified and excised after ligation. Animals were carefully inspected after surgery. The removed ovaries were fixed in 10% buffered formalin and characterized by H&E staining to confirm complete removal of ovaries.
Plasma 17β‐Estradiol Measurement
We used a 17β‐estradiol kit (Cayman Chemical, Ann Arbor, MI) to measure plasma estrogen levels in female mice and in response to ovariectomy in accordance with the manufacturer's protocol as described previously.14
Echocardiography
Noninvasive transthoracic echocardiography was performed on anesthetized mice by using isoflurane (1% to 1.5%) as described previously.34, 35, 36 A Vevo 770 high‐resolution imaging system equipped with a 30‐MHz transducer (RMV‐707B; VisualSonics, Toronto, ON, Canada) was used, and systolic and diastolic cardiac functions were analyzed. Systolic function was assessed using B‐mode and motion (M)‐mode images of echocardiography. M‐mode images were obtained for measurements of left ventricular (LV) wall thickness, LV end‐diastolic diameter (LVEDD), and LV end‐systolic diameter (LVESD) (measures of LV dilation). LV fractional shortening (FS) and LV ejection fraction (EF) were calculated using the following equations: FS (%)=([LVEDD−LVESD]/LVEDD)×100 and EF (%)=([LVEDV−LVESV]/LVEDV)×100. Diastolic function was assessed using pulsed‐wave Doppler imaging of the transmitral filling pattern with the early transmitral filling wave (E‐wave) followed by the late filling wave due to atrial contraction (A‐wave). Isovolumetric relaxation time (IVRT) was calculated as the time from closure of the aortic valve to initiation of the E‐wave. The deceleration time of the E‐wave (DT) was determined by measuring the time needed for the downslope of the peak of the E‐wave to reach the baseline, and the E‐wave deceleration rate (EWDR) was calculated as the E‐wave divided by the DT. Tissue Doppler imaging (TDI) represents a novel and validated technique to assess systolic and diastolic function, with a reduction in E′ and an elevation in E/E′ being considered as valid markers of elevated LV filling pressure and diastolic dysfunction. TDI was carried out at the inferolateral region in the radial short axis at the base of the LV with the assessment of peak annular systolic (S′), early diastolic (E′), and late diastolic (A′) myocardial velocities as described previously.34, 35, 36
Invasive Hemodynamic Analysis
We performed invasive pressure‐volume (PV) loop analysis by using a 1.2F Scisense catheter connected to an amplifier (TCP‐500, Scisense Inc, London, ON, Canada).34 Mice were anesthetized by using isoflurane (1% to 1.5%) and maintained at 37°C by use of a heating pad. An incision was made in the right common carotid artery, and the catheter was carefully inserted into the incision, advanced through the aortic valve, and placed into the LV chamber. The position of the catheter was monitored by pressure along with the magnitude and phase using an ADvantage pressure volume system (Scisense Inc, London, ON, Canada) and iworx (iWorx Systems Inc, Dover, NH) data acquisition system connected to the catheter. After the magnitude was established in the desired range, the phase was adjusted to 4 to 8 by adjusting the position of the catheter in the LV where the phase represents the conductivity imparted by the LV tissue. Following baseline PV measurements, transient inferior vena cava occlusion was performed through the diaphragm to obtain the alteration in venous return to derive end‐diastolic pressure‐volume relationships; transient infrarenal aorta occlusion was used to derive the end‐systolic pressure‐volume relationship. Load‐dependent and load‐independent indices of LV functions were derived. By determining the instantaneous values of pressure and volume at different time points, we determined the end‐systolic pressure (ESP), end‐diastolic pressure (EDP), end‐systolic volume (ESV), and end‐diastolic volume (EDV). Heart rate (HR) was determined from the beat‐to‐beat cycle length. The stroke volume (SV)=EDV−ESV, cardiac output (CO)=SV×HR, ejection fraction (EF)=SV/EDV, and the stroke work (SW) is the area under a PV‐loop and was normalized to EDV to obtain preload‐recruitable stroke work (PRSW), which is also a load‐independent parameter. The preload‐independent +dP/dtmax, also called the Starling contractile index (SCI), was calculated by dividing +dP/dtmax/EDV, which is a better index of myocardial contractility. +dP/dtmax and −dP/dtmin are the first derivatives of the LV pressure, and their ratio (−dP/dtmin/+dP/dtmax) and tau (τ), the time constant of monoexponential pressure decay during isovolumic relaxation, reflect active relaxation. Systolic and diastolic cardiac performances were also assessed by the end‐systolic pressure‐volume relationship (ESPVR) and end‐diastolic pressure‐volume relationship (EDPVR), respectively, as described previously.34, 36, 37
Histology and Fluorescence Staining
Mouse hearts were removed and arrested in diastole by using 1 mol/L KCl, fixed with 10% buffered formalin, and embedded in paraffin. Thin sections (5 μm) were used for Prussian blue, picro‐sirius red (PSR), and Masson trichrome staining as described previously.30, 38 Briefly, tissue sections were deparaffinized in xylene and alcohol grades, rehydrated in water, and subjected to staining as described previously.30 The deposition of iron was visualized as blue depositions using bright‐field microscopy. Myocardial collagen content was evaluated by using PSR staining and visualization using an Olympus IX81 microscope and image analysis using MetaMorph software (Molecular Devices, Sunnyvale, CA) as described previously.30, 35, 38 4‐Hydroxynonenal (4‐HNE) immunofluorescence detected an important marker of iron‐induced lipid peroxidation as previously described.30 We also performed dihydroethidium (DHE) and dichlorodihydrofluorescein (DCF) fluorescence staining as described previously,19, 35 which were visualized using an Olympus IX81 fluorescent microscope and quantified using the MetaMorph software.
Terminal Deoxynucleotidyl Transferase–Mediated dUTP Nick End Labeling (TUNEL) Assay
The 5‐μm‐thick LV heart sections were subjected to TUNEL staining and visualized using an Olympus IX81 fluorescent microscope as described previously.30, 34 Briefly, OCT sections were fixed with 4% paraformaldehyde and then hydrated with PBS at room temperature. Permeabilization was followed by blocking with 1% BSA for 30 minutes followed by incubation with the DNA‐labeling solution for 1 hour at 37°C. Rinsing with buffer was followed by anti‐BrdU incubation at 37°C for 1 hour and counterstaining with propidium iodide for 30 minutes at 37°C; the sections were then mounted with antifade and coverslip and visualized using an Olympus IX81 fluorescent microscope.
Tissue Iron Levels
Frozen LV tissues (20 mg) were subjected to inductively coupled plasma resonance mass spectrometry to quantify tissue iron level in the Trace Metals Laboratory (London, ON, Canada) as described previously.9, 10, 30
Measurement of Lipid Peroxidation and Glutathione
Malondialdehyde (MDA), an indicator of lipid peroxidation, was measured in myocardial tissues (100‐150 mg) by using a commercially available kit (Bioxytech@ MDA‐586TM, OxiResearch, Percipio Biosciences Inc, Los Angeles, CA) as described previously.30, 31 Briefly, tissue samples were homogenized in potassium phosphate extraction (KPE) buffer pH 7.5 (0.1 mol/L potassium phosphate, 5 mmol/L EDTA, 0.1% Triton X‐100, and 0.6% sulfosalicylic acids) containing 5 mmol/L BHT. The samples were then allowed to react with N‐methyl‐2‐phenylindole (NMPI) in the acidic pH at 45°C for 1 hour. The clear supernatant was then collected and read at 586 nm using a plate reader (Spectramax M5, Molecular Devices, Sunnyvale, CA). MDA levels were estimated using a standard curve derived using 0.5 to 4.0 μmol/L of standard MDA. Myocardial reduced glutathione (GSH) as well as oxidized glutathione (GSSG) levels were measured by using a plate reader (Spectramax M5, Molecular Devices, Sunnyvale, CA) as described previously.30, 39
Taqman Real‐Time PCR
Taqman real‐time PCR for mRNA expression analysis was performed using the Taqman primers and probes as previously described.30, 31, 36 Briefly, 5 μL of suitable cDNA dilutions from unknown and standard (brain cDNA) samples and 8 μL Taqman master‐mix including primers and probes were loaded on white 384 Light cycler® 480 multiwell plates (Roche) with 18S rRNA as the internal control. Samples were loaded in triplicate, and the data were analyzed using the Light cycler® 480 system (Roche, Mannheim, Germany) as described previously.31, 38
Western Blotting
We homogenized flash‐frozen LV tissue by using Tissue Lyser II (Qiagen, Hilden, Germany) with special lysis buffer (CelLytic™ M) from Sigma supplemented with Protease/Phosphatase inhibitor cocktail from Roche as described previously.31, 34 Tissue lysates were processed and resolved on 8% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred to PVDF membranes using a Trans‐blot cell (Bio‐Rad laboratories, Hercules, CA) as previously described.30, 31, 35 Membranes were immunoblotted using primary antibodies against phospho‐Akt and total‐Akt (Cell Signaling, Danvers, MA), sarco/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) and sodium‐calcium exchanger 1 (NCX1) (ThermoScientific, Waltham, MA), ferritin (Abcam, Cambridge, UK), and estrogen receptor (ER)‐α and ER‐β (Santa Cruz Biotechnology, Dallas, TX) followed by blotting with their HRP‐conjugated secondary antibodies. We normalized our protein loading using MemCode™ protein stain on the PVDF membranes, and blots were scanned and quantified (ImageQuant LAS 4000, GE Health Care, Biosciences, Uppsala, Sweden).
Statistical Analysis
Statistical analysis was performed using SPSS software (version 23). Values are shown as mean±SEM. We performed the Shapiro‐Wilk test, which confirmed that our data are normally distributed. Normally distributed data were analyzed using the 2‐way ANOVA to evaluate the interactions between sex and iron diet (Figures 1, 2, 3, 4, 5 through 6) or ovariectomy and iron diet (Figures 7, 8, 9, 10, 11, 12, 13 through 14) as fixed factors. Main effects of sex/ovariectomy and iron were calculated accurately by excluding the interaction term. Normally distributed data from the 17β‐estradiol therapy experiments were analyzed using the 1‐way ANOVA followed by the Tukey post hoc test (Figures 15 and 16).
Figure 1.
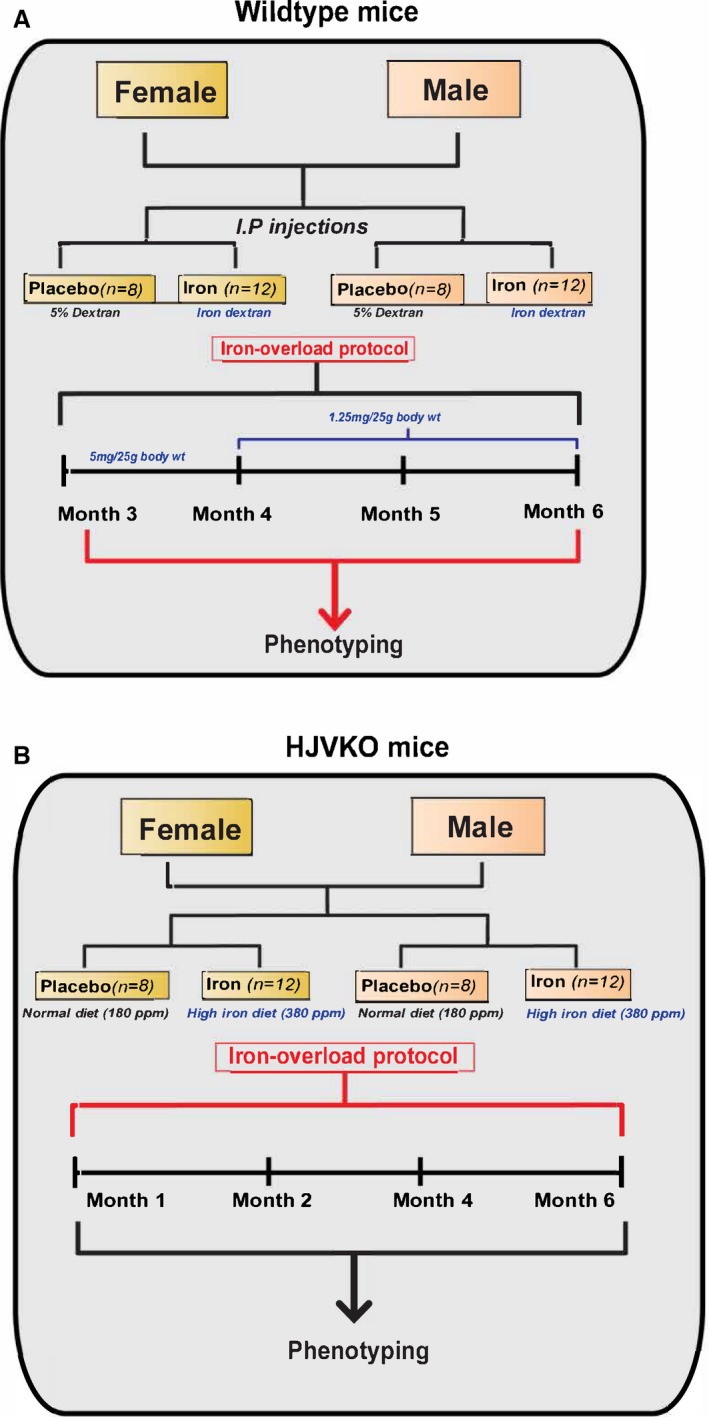
Experimental protocol to study the effects of sex on iron‐overload cardiomyopathy in WT (A) and HJV knockout (B) mice. HJVKO indicates hemojuvelin; WT, wild‐type.
Figure 2.
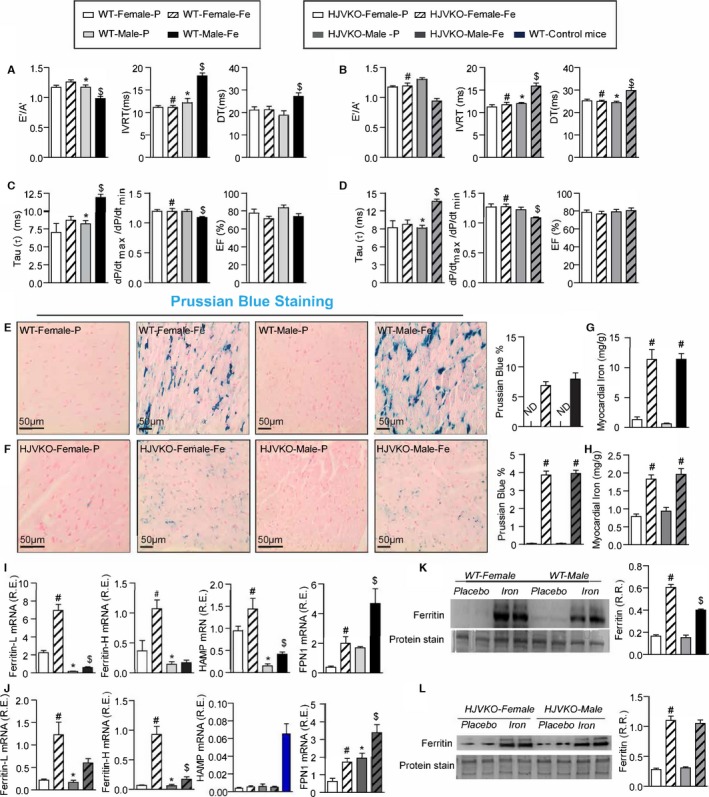
Marked sex differences in iron‐overload cardiomyopathy. Noninvasive echocardiographic assessment of heart function by tissue Doppler imaging and transmitral filling pattern and invasive hemodynamic assessment using pressure‐volume loops in WT (A and B) and HJVKO mice (C and D) showing preserved cardiac function in female mice and heart failure with preserved ejection fraction in male mice in response to iron overload. Representative Prussian blue staining images (E and F) and quantification of myocardial tissue iron levels (G and H) in WT and HJVKO mice showing equivalent cardiac iron deposition in male and female mice in response to iron overload. Taqman real‐time PCR analysis of ferritin light (L) and heavy (H) chain, hepcidin (HAMP) and ferroportin (FPN1) myocardial mRNA expression in WT (I) and HJVKO (J) mice. Western blot analysis for total ferritin in WT (K) and HJVKO (L) hearts showing a smaller increase in ferritin levels in WT male mice in response to iron overload. The blue bar indicates the data from WT controls. A′ indicates tissue Doppler due to atrial contraction; DT, deceleration time; E′, early tissue Doppler velocity; EF, ejection fraction; IVRT, isovolumetric relaxation time; ND, not detected; R.R., relative ratio; n=8 to 12 for functional studies; n=8 for gene expression analysis; n=4 for Western blot analysis. HJVKO indicates hemojuvelin‐null; WT, wild‐type. *P<0.05 for effect of sex; # P<0.05 for effect of iron; $ P<0.05 for interaction.
Figure 3.
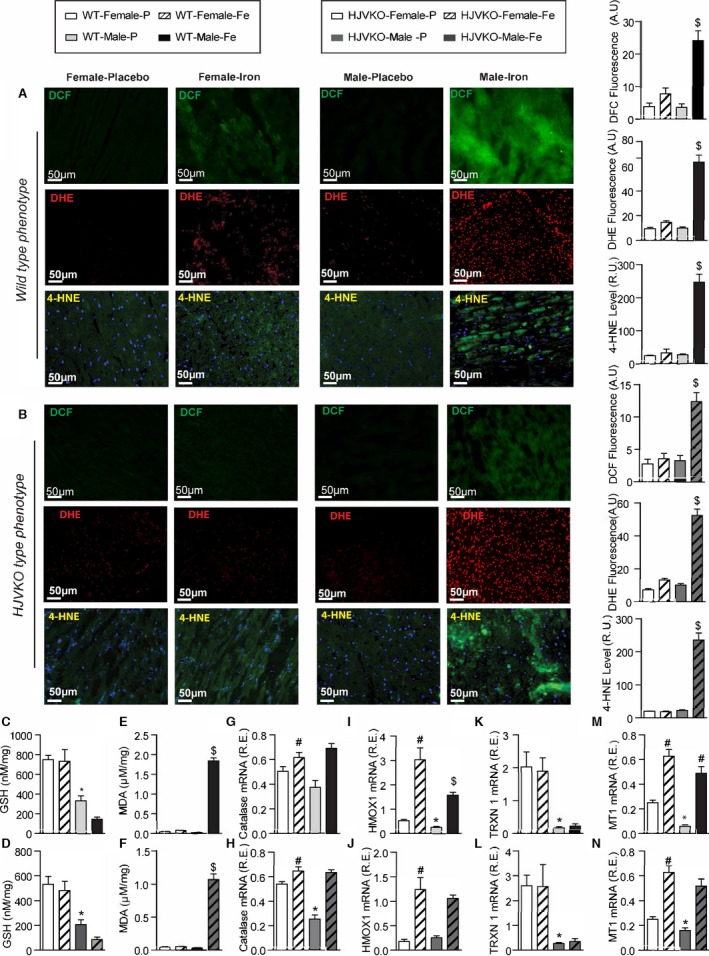
Female mice are protected against iron‐overload‐induced oxidative stress. Representative dichlorodihydrofluorescein (DCF) (green), dihydroethidium (DHE) fluorescence (red), 4‐hydroxynonenal (4‐HNE) immunofluorescence (green), and quantification showing a relative lack of iron‐induced myocardial oxidative stress in female WT (A) and HJVKO (B) mice, whereas iron overload resulted in increased oxidative stress in male mice. Biochemical analysis of myocardial reduced glutathione (GSH) (C and D) and lipid peroxidation product, malondialdehyde (MDA) (E and F) levels in WT (C and E) and HJVKO hearts (D and F) showing increased oxidative stress in male mice in contrast to unchanged oxidative stress in female mice in response to iron overload. Myocardial gene expression analysis using Taqman real‐time PCR showing sex‐specific and iron‐overloaded related alteration in mRNA expression for catalase (G and H), heme oxygenase 1 (HMOX1) (I and J), thioredoxin 1 (TRXN1) (K and L), and metallothionein 1 (MT1) (M and N) in WT and HJVKO mice, respectively; n=4 for histology; n=8 for gene expression and biochemical analyses. HJVKO indicates hemojuvelin‐null; WT, wild‐type. *P<0.05 for effect of sex; # P<0.05 for effect of iron; $ P<0.05 for interaction.
Figure 4.
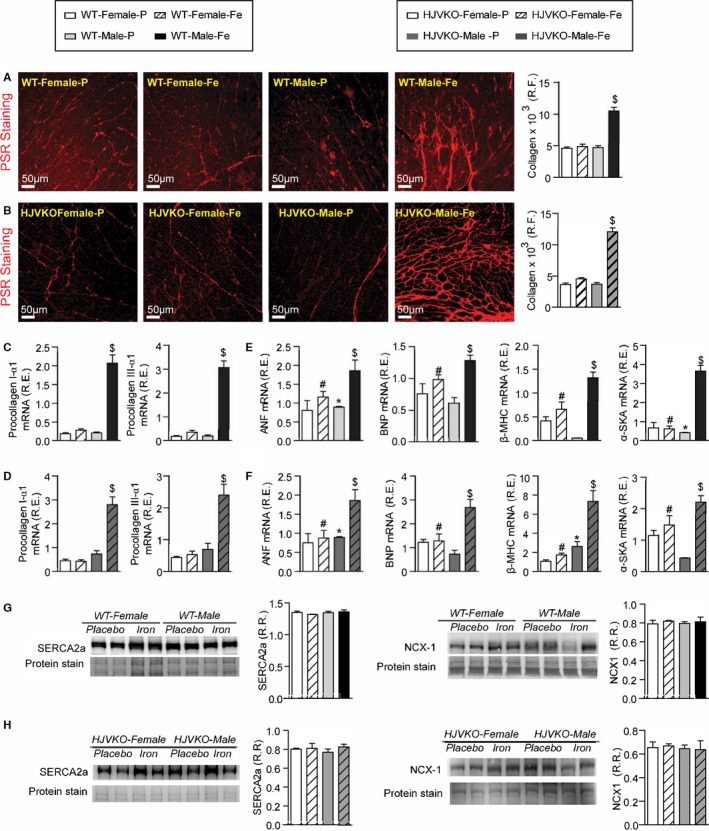
Female mice are protected against iron‐overload‐induced myocardial fibrosis and heart disease. Representative picro‐sirius red (PSR) staining and quantification of myocardial fibrosis (A and B), and Taqman real‐time PCR expression analysis of procollagen type Iα1 and procollagen type IIIα1 mRNA (C and D) in male and female WT and HJVKO mice showing a clear protection against iron‐overload‐induced myocardial fibrosis in female mice. Expression analysis of cardiac disease markers, atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), and β‐myosin heavy chain (β‐MHC) and α‐skeletal actin (α‐SkA) in WT (E) and HJVKO mice (F), showing a potential intrinsic cardioprotective effect against iron‐overload cardiomyopathy in female mice. Western blot analyses in male and female WT and HJVKO mice showing no change in myocardial sarco/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) and sodium‐calcium exchanger 1 (NCX1) levels in WT (G) and HJVKO (H) mice in response to iron overload. R.E. indicates relative expression; R.F., relative fraction; R.R., relative ratio; n=4 for histology and Western blot analyses; n=8 for gene expression analysis. HJVKO indicates hemojuvelin‐null; WT, wild‐type. *P<0.05 for effect of sex; # P<0.05 for effect of iron; $ P<0.05 for interaction.
Figure 5.
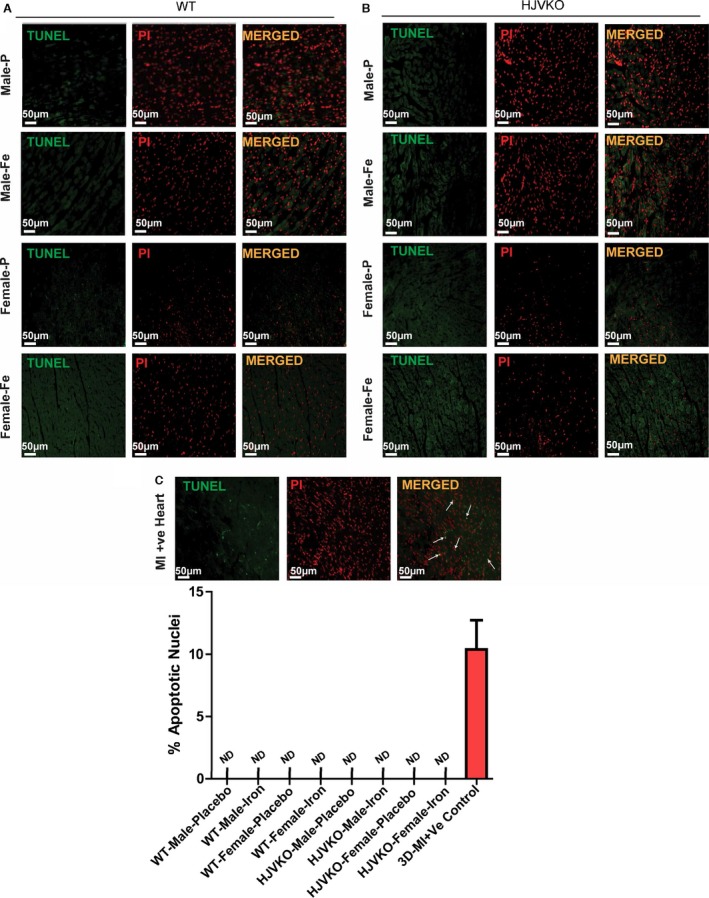
Lack of myocardial apoptosis in chronic iron‐overloaded mice based on terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in WT (A) and HJVKO (B) female hearts (n=3 each). Two sections from each heart were stained and imaged and 5 fields examined from each section. Positive TUNEL staining (white arrows) is shown from 3‐day postmyocardial infarction murine hearts using the LAD ligation technique (C). LAD indicates left anterior descending; HJVKO, hemojuvelin‐null; WT, wild‐type.
Figure 6.
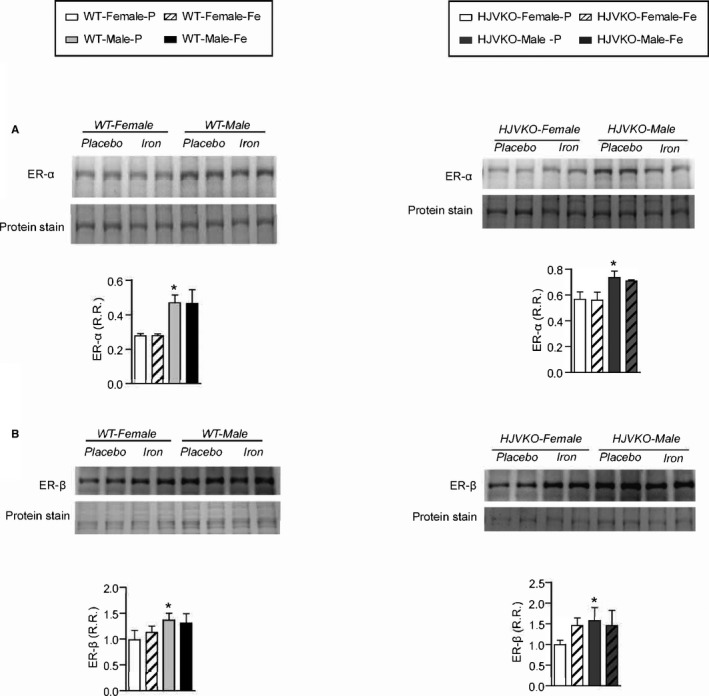
Western blot analysis of myocardial estrogen receptor‐α (ER‐α) (A) and estrogen receptor‐β (ER‐β) (B) levels showing increased ER‐α in male WT hearts with no changes observed in response to iron‐overload. n=6 per group. HJVKO indicates hemojuvelin‐null; WT, wild‐type. *P<0.05 for effect of sex.
Figure 7.
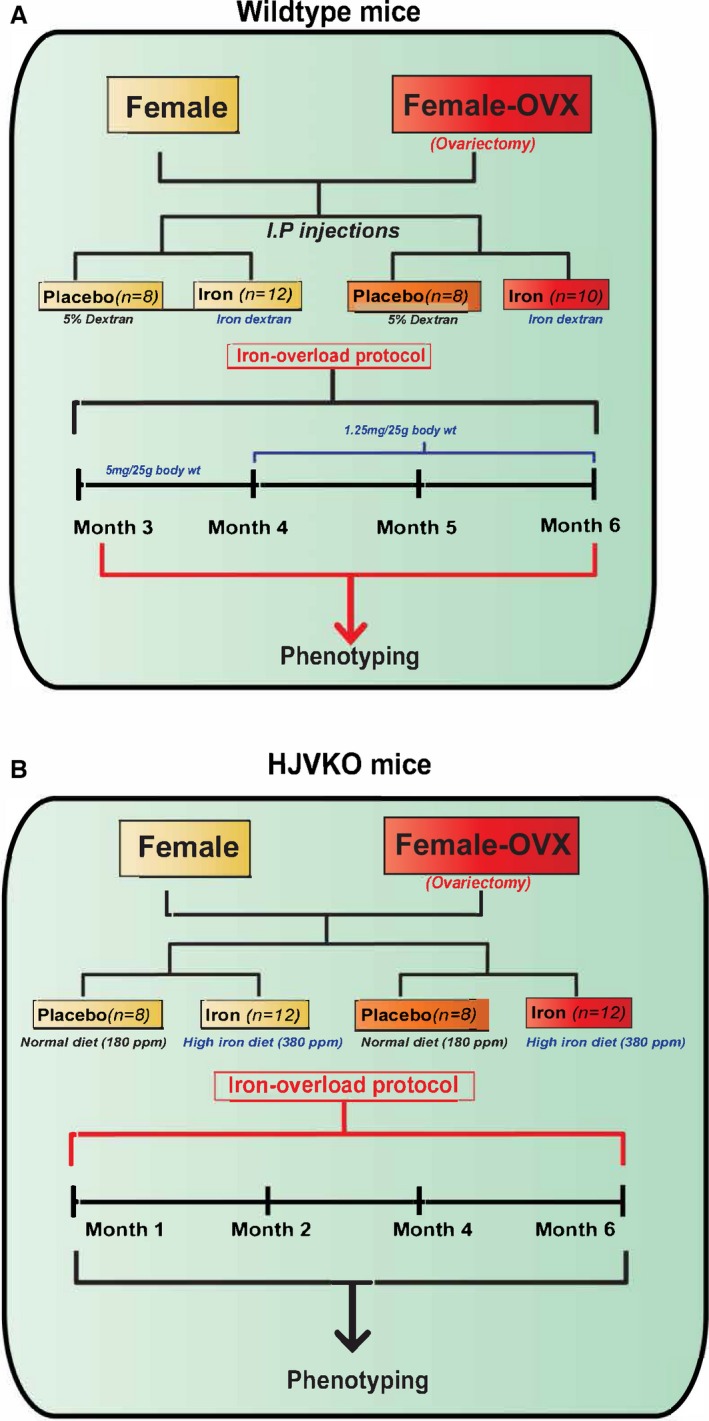
Experimental protocol to study the effects of ovariectomy (OVX) on iron‐overload cardiomyopathy in WT (A) and HJV knockout (B) mice. HJV indicates hemojuvelin; WT, wild‐type.
Figure 8.
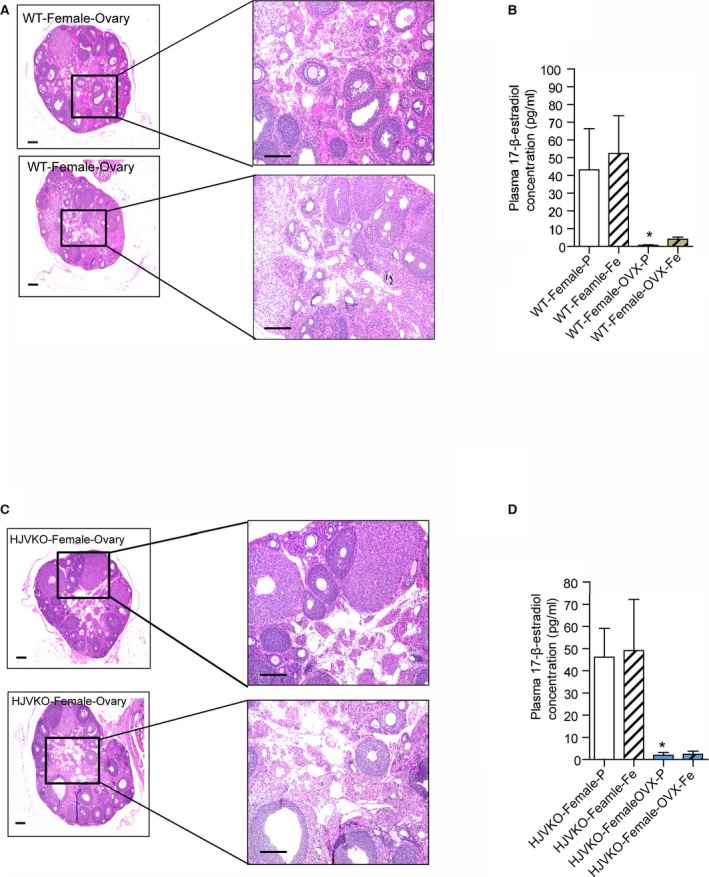
Histological analysis of ovaries and plasma 17β‐estradiol levels in WT mice and HJVKO mice. Hematoxylin and eosin staining of surgically removed ovaries from a WT (A) and HJVKO (C) mouse and plasma 17β‐estradiol levels in WT (B) and HJVKO (D) mice illustrating the effectiveness of ovariectomy (OVX). Scale bars represent 250 μm (left) and 50 μm (right); n=8 per group. HJVKO indicates hemojuvelin‐null; WT, wild‐type. *P<0.05 for effect of OVX.
Figure 9.
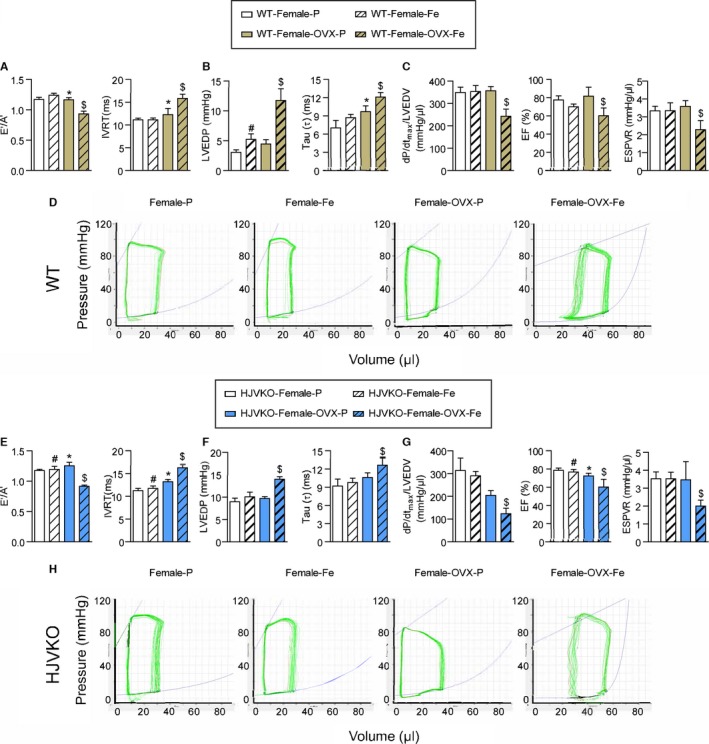
Ovariectomy (OVX) precipitates diastolic and systolic dysfunction in response to myocardial iron overload. Noninvasive echocardiographic assessment of heart function illustrated by tissue Doppler (E′/A′) and transmitral filling (IVRT) (A), invasive hemodynamics illustrated by LVEDP and LV pressure exponential decay constant (τ) (B) showing diastolic dysfunction in OVX WT females with iron overload. Invasive hemodynamic assessment based on dP/dtmax/LVEDV, EF, and ESPVR (C) showing systolic dysfunction in OVX WT female iron‐overloaded mice. Representative pressure‐volume loops illustrating diastolic and systolic dysfunction in OVX WT female iron‐overloaded mice (D). Noninvasive echocardiographic assessment of heart function illustrated by tissue Doppler (E′/A′) and transmitral filling (IVRT) (E), invasive hemodynamics illustrated by LVEDP and LV pressure exponential decay constant (τ) (F) showing diastolic dysfunction in OVX HJVKO females with iron overload. Invasive hemodynamic assessment based on dP/dtmax/LVEDV, EF, and ESPVR (G) showing systolic dysfunction in OVX HJVKO female iron‐overloaded mice. Representative pressure‐volume loops illustrating diastolic and systolic dysfunction in OVX WT female iron‐overloaded mice (H). A′ indicates tissue Doppler due to atrial contraction; E′, early tissue Doppler velocity; EF, ejection fraction; ESPVR, end‐systolic pressure‐volume relationship; HJVKO, hemojuvelin‐null; IVRT, isovolumetric relaxation time; LVEDP, LV end‐diastolic pressure; LVEDV, LV end‐diastolic volume; WT, wild‐type; n=8 for placebo and n=10 for iron‐overload groups. *P<0.05 for effect of OVX; # P<0.05 for effect of iron; $ P<0.05 for interaction.
Figure 10.
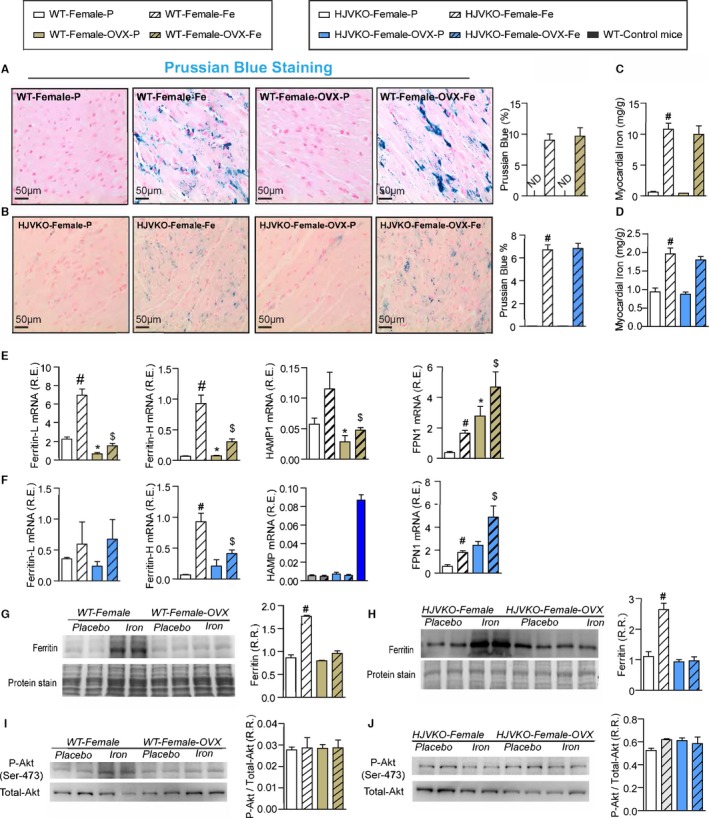
Ovariectomy (OVX) does not modulate myocardial iron deposition but suppresses the upregulation of ferritin expression. Prussian blue staining (A and B) and quantification of myocardial tissue iron levels (C and D) in WT (A and C) and HJVKO (B and D) female hearts and in response to OVX, confirming the presence of myocardial iron deposition without a differential response to OVX. Taqman real‐time PCR analysis of ferritin light (L) and heavy (H) chain, hepcidin (HAMP) and ferroportin (FPN1) myocardial mRNA expression in female WT (E) and HJVKO (F) mice in response to OVX and iron overload. Western blot analysis of myocardial ferritin levels in WT (G) and HJVKO (H) females in response to iron overload showing a complete lack of an increase in myocardial ferritin level in OVX mice. Western blot analysis of phospho‐Akt/total‐Akt in WT (I) and HJVKO (J) female hearts showing no change in myocardial phospho‐Akt/PKB(Ser‐473) level in response to OVX and iron overload. ND indicates not detected; R.E., relative expression; R.R., relative ratio; n=8 for iron quantification and gene expression analysis; n=4 for histology and Western blot analysis. HJVKO indicates hemojuvelin‐null; WT, wild‐type. *P<0.05 for effect of ovariectomy; # P<0.05 for effect of iron; $ P<0.05 for interaction.
Figure 11.
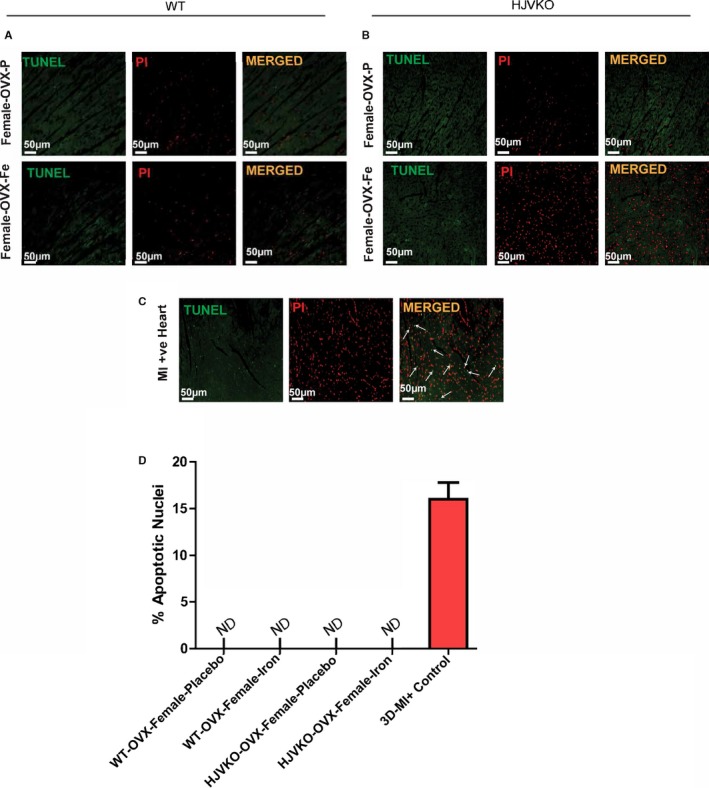
Lack of myocardial apoptosis in chronic iron‐overloaded mice based on terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in WT (A) and HJVKO (B) female hearts (n=3 each). Two sections from each heart were stained and imaged, and 5 fields were examined from each section. Positive TUNEL staining is shown (white arrows) from 3‐day post–myocardial infarction murine hearts using the LAD ligation technique (C). Quantification of TUNEL images did not show TUNEL‐positive nuclei in WT and HJVKO female hearts. Positive control showed markedly increased TUNEL‐positive nuclei (D). LAD indicates left anterior descending; HJVKO, hemojuvelin‐null; MI, myocardial infarction; ND, not detectable; OVX, ovariectomy; PI: propidium iodide; WT, wild‐type.
Figure 12.
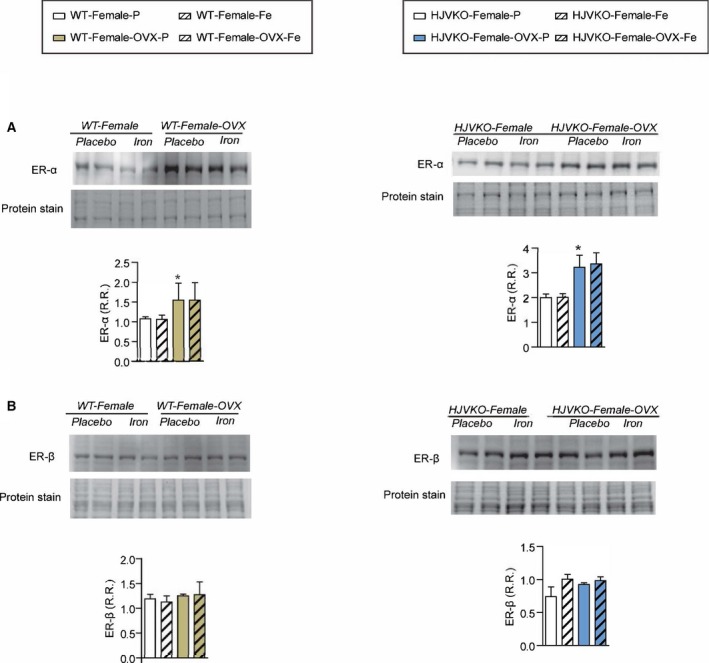
Western blot analysis of myocardial estrogen receptor‐α (ER‐α) (A) and estrogen receptor‐β (ER‐β) (B) levels showing increased ER‐α in male WT hearts with no changes observed in response to iron overload; n=6 per group; *P<0.05 for effect of ovariectomy. HJVKO indicates hemojuvelin‐null; OVX, ovariectomy; R.R., relative ratio; WT, wild‐type.
Figure 13.
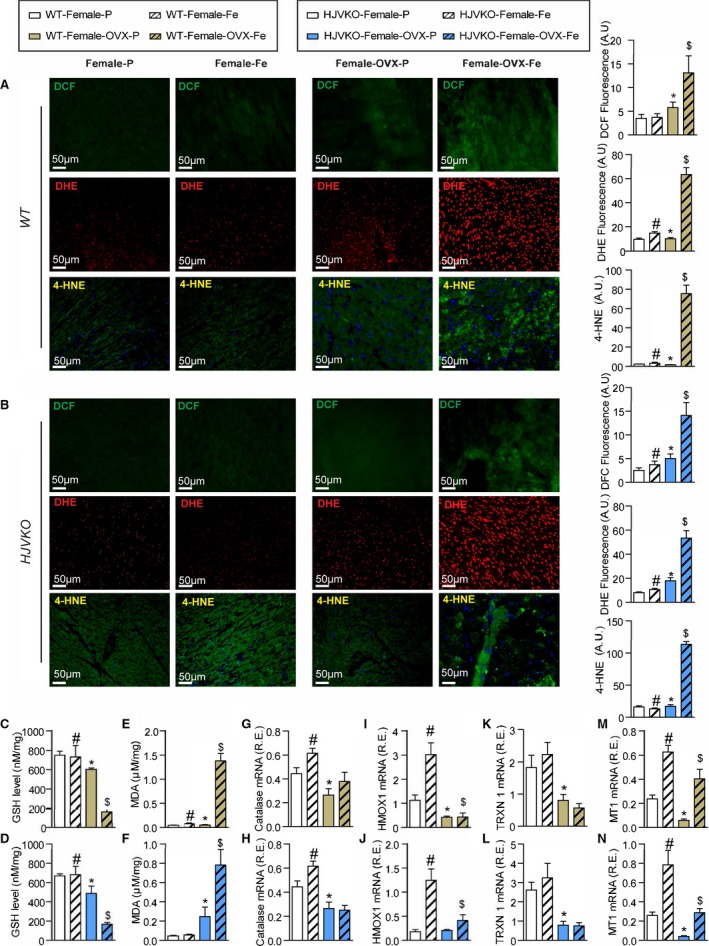
Iron‐overload‐induced myocardial oxidative stress is potentiated in ovariectomized (OVX) female mice. Increased iron‐induced myocardial oxidative stress in OVX females with iron overload detecting reactive oxygen species (ROS) by dichlorodihydrofluorescein (DCF) fluorescence (green), dihydroethidium (DHE) fluorescence (red), 4‐hydroxyneonal (4‐HNE) immunofluorescence (green), and quantification in wild‐type (WT) (A) and hemojuvelin‐null (HJVKO) (B) females in response to OVX and iron overload. Biochemical analysis of myocardial reduced glutathione (GSH) (C and D) and lipid peroxidation product malondialdehyde (MDA) (E and F) levels in female WT (C and E) and HJVKO hearts (D and F), clearly illustrating increased myocardial oxidative injury in OVX female iron‐overloaded hearts. Gene expression analysis in hearts showing a marked downregulation in key antioxidant enzymes catalase (G and H) and heme oxygenase 1 (HMOX1) (I and J) and antioxidant molecules thioredoxin 1 (TRXN1) (K and L), and metallothionein 1 (MT1) (M and N) in WT and HJVKO mice, respectively, following OVX and a lack of upregulation of their expression following iron overload; n=4 for histology analysis; n=8 for gene expression and biochemical analysis. R.E. indicates relative expression; *P<0.05 for effect of ovariectomy; # P<0.05 for effect of iron; $ P<0.05 for interaction.
Figure 14.
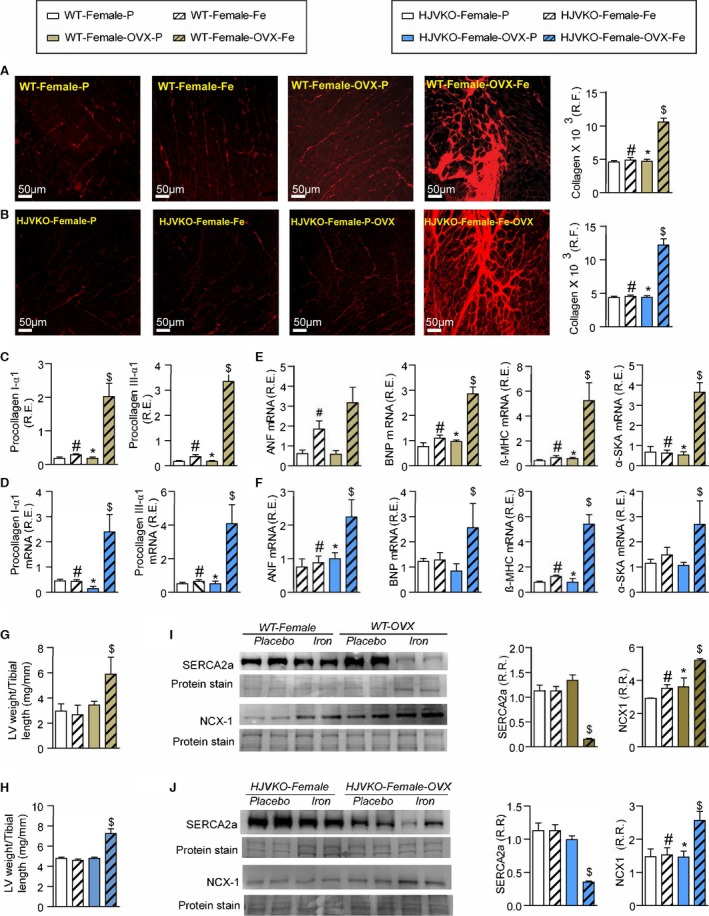
Exacerbation of pathological myocardial remodeling in iron‐overloaded ovariectomized (OVX) female mice. Picro‐sirius red (PSR) staining and quantification of myocardial fibrosis (A and B) and gene expression of procollagen type Iα1 and procollagen type IIIα1 (C and D) in wild‐type (WT) and hemojuvelin‐null (HJVKO) females clearly demonstrating that OVX potentiates iron‐overload‐mediated myocardial fibrosis. Expression of disease markers, atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), β‐myosin heavy chain (β‐MHC), and α‐skeletal actin (α‐SkA) in WT (E) and HJVKO (F) mice illustrating pathological myocardial remodeling in iron‐overloaded OVX female hearts. Morphometric assessment of hypertrophy showing increased LV weights in iron‐overloaded OVX female WT (G) and HJVKO (H) hearts. Western blot analysis and quantification clearly showed significant downregulation in myocardial sarco/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) and sodium‐calcium exchanger 1 (NCX1) levels in female WT (I) and HJVKO (J) iron‐overloaded hearts following OVX. R.E. indicates relative expression; R.F., relative fraction; R.R., relative ratio; n=8 for gene expression analysis; n=4 for histology and Western blot analyses. *P<0.05 for effect of OVX; # P<0.05 for effect of iron; $ P<0.05 for interaction.
Figure 15.
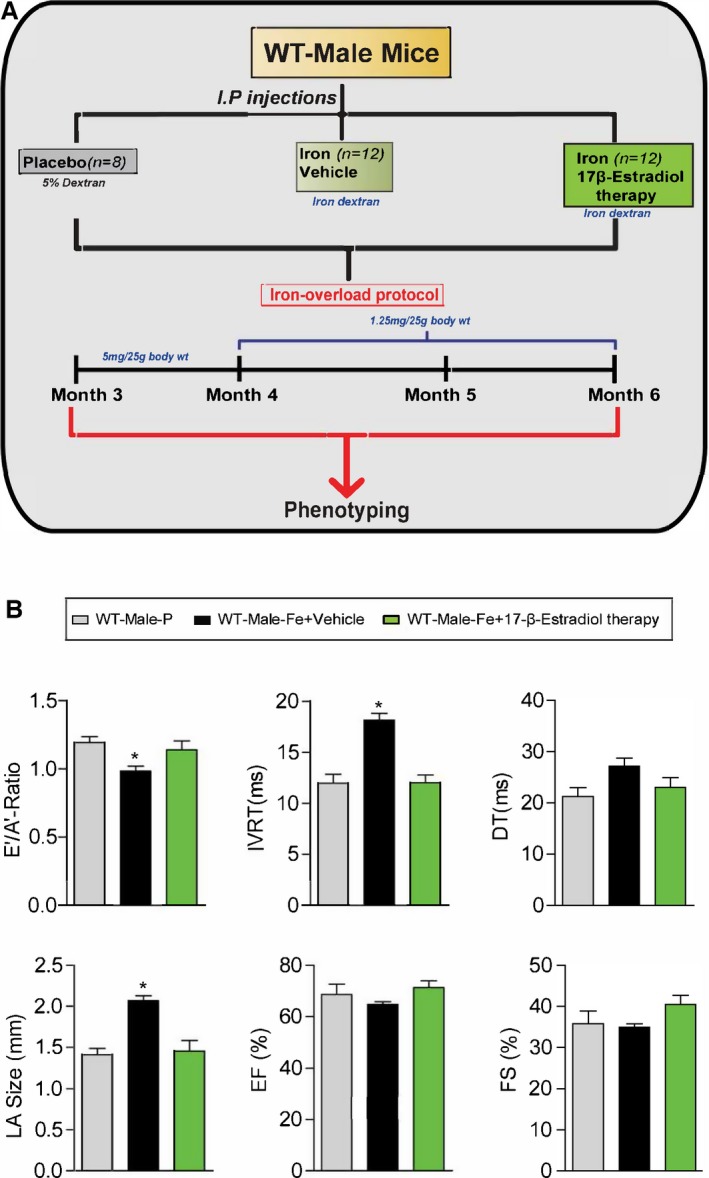
Experimental protocol for exogenous 17β‐estradiol therapy (0.04 mg/kg per day) in male iron‐overloaded WT mice (A) and functional assessment showing a complete rescue of iron‐overload‐induced diastolic dysfunction in response to 17β‐estradiol therapy (B). A′ indicates tissue Doppler due to atrial contraction; DT, deceleration time; E′, early tissue Doppler velocity; EF, ejection fraction; FS, fractional shortening; IVRT, isovolumetric relaxation time; LA, left atrial; WT, wild‐type; n=8 to 12 for functional studies. *P<0.05 compared to all the groups.
Figure 16.
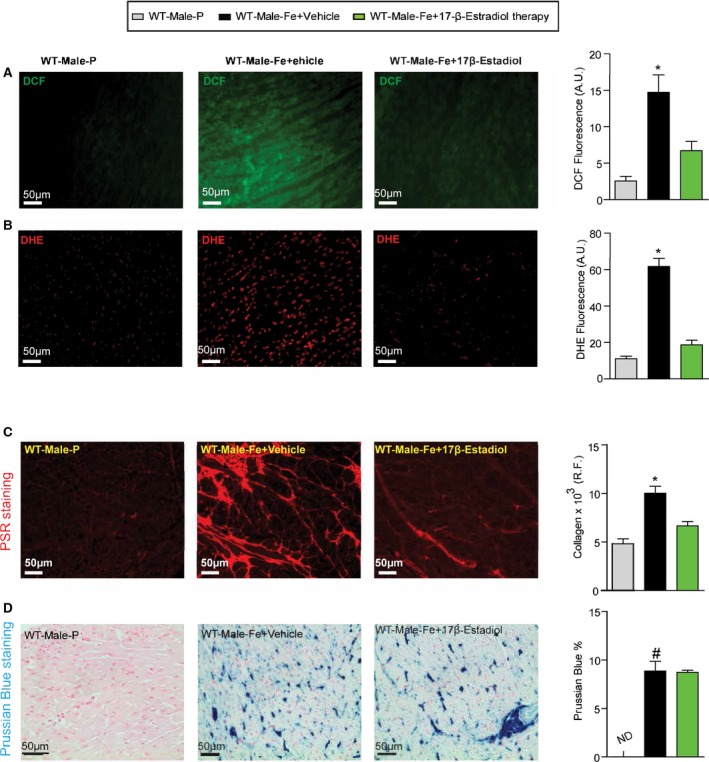
Reduction in myocardial oxidative stress and fibrosis in male iron‐overloaded mice in response to 17β‐estradiol therapy. Representative dichlorodihydrofluorescein (DCF) fluorescence (green) (A) and dihydroethidium (DHE) fluorescence (red) (B) and quantification showing a marked suppression of myocardial oxidative stress in male WT iron‐overloaded mice in response to 17β‐estradiol therapy, with a similar change seen in myocardial fibrosis as determined by picro‐sirius red (PSR) staining (C) without suppression of myocardial iron deposition as illustrated by Prussian blue staining (D); n=4 for histology analysis. R.F. indicates relative fraction; WT, wild‐type. *P<0.05 compared with all the groups; # P<0.05 compared with the placebo group.
Results
Sex Difference in Iron‐Overload‐Induced Heart Disease
Chronic iron injections in WT and high‐iron diet in HJVKO murine models are well‐established models of iron overload recapitulating secondary iron overload and genetic hemochromatosis, respectively (Figure 1).9, 10, 31, 32 Effects of iron overload on cardiac function were assessed noninvasively by transthoracic echocardiography and invasively by pressure‐volume loop analysis. Iron‐overloaded male WT and HJVKO mice resulted in diastolic dysfunction with preserved EF characterized by decreased E′/A′ ratio, prolonged IVRT, and deceleration time (DT) (Figure 2A and 2B; Tables 1 and 2), increased exponential time constant of the decay in LV pressure during isovolumic relaxation (τ), and decreased dP/dtmax/dP/dtmin ratio with no change in EF (Figure 2C and 2D; Tables 1 and 2). In contrast, female WT as well as HJVKO mice were protected against iron‐overload‐induced diastolic dysfunction (Figure 2A through 2D; Tables 1 and 2).
Table 1.
Invasive Pressure‐Volume Loop Assessment of Cardiac Function in Iron‐Overloaded WT Male and Female Mice
| WT‐Male Placebo | WT‐Male Iron | WT‐Female Placebo | WT‐Female Iron | WT‐Female OVX‐Placebo | WT‐Female OVX‐Iron | |
|---|---|---|---|---|---|---|
| n | 8 | 12 | 12 | 12 | 8 | 10 |
| HR, bpm | 512±18 | 390±24a | 508±22 | 503±24 | 469±27 | 407±30 |
| LVEDP, mm Hg | 2.9±0.4 | 12.9±0.9a | 3.1±0.4 | 5.3±0.9 | 4.5±0.9 | 12.7±1.3a |
| LVESP, mm Hg | 94±6.2 | 97±2.1 | 96±2.6 | 98±1.9 | 94±4.5 | 84±3.1 |
| LVEDV, μL | 24.8±1.1 | 26.0±3.0 | 26.3±0.9 | 26.8±4.1 | 27.5±2.0 | 34.6±4.5 |
| LVESV, μL | 6.8±0.9 | 7.1±0.9 | 6.9±0.9 | 6.8±0.7 | 4.8±1.3 | 15.2±0.9 |
| SV, μL | 19.9±1.7 | 20.8±0.7 | 20.3±1.5 | 19.2±2.5 | 22.7±2.7 | 19.3±1.8a |
| EF, % | 80.2±3.6 | 80±3.1 | 77.2±3.9 | 71.7±4.9 | 84.8±3.2 | 55.7±5.1b |
| dP/dtmax, mm Hg/s | 8781±86 | 8538±232 | 8612±279 | 8612±279 | 8764±187 | 7923±322 |
| SW, mJ | 0.29±0.038 | 0.28±0.024 | 0.31±0.021 | 0.29±0.059 | 0.25±0.02 | 0.24±0.025 |
| PRSW, mJ/μL | 0.012±0.35 | 0.011±0.008 | 0.012±0.023 | 0.011±0.006 | 0.009±0.005 | 0.006±0.005 |
| dP/dtmax/EDV, mm Hg/s per μL | 358±17 | 351±12 | 349±24 | 353±26 | 328±15 | 253±29b |
| ESPVR, mm Hg/μL | 3.3±0.6 | 3.6±0.5 | 3.4±0.25 | 3.5±0.4 | 3.7±0.3 | 2.4±0.6b |
| τ (Glantz), ms | 8.3±0.4 | 12.0±0.4a | 7.1±1.2 | 8.8±0.5 | 9.8±0.9 | 12.2±0.7b |
| EDPVR, mm Hg/μL | 0.101±0.038 | 0.131±0.090a | 0.105±0.016 | 0.109±0.012 | 0.118±0.01 | 0.141±0.023a |
dP/dt indicates rate of change in LV pressure; dP/dtmax/EDV, Starling contractile index; EDPVR, end‐diastolic pressure‐volume relationship; EF, ejection fraction; ESPVR, end‐systolic pressure‐volume relationship; HR, heart rate; LVEDP, end‐diastolic pressure; LVEDV, end‐diastolic volume; LVESP, end‐systolic pressure; LVESV, end‐systolic volume; OVX, ovariectomized; PRSW, preload‐recruitable stroke work; PV, pressure‐volume; SV, stroke volume; SW, stroke work; WT, wild‐type; τ, LV relaxation time constant.
P<0.05 for effect of iron.
P<0.05 for interaction.
Table 2.
Invasive Pressure‐Volume Loop Assessment of Cardiac Function in Iron‐Overloaded HJVKO Male and Female Mice
| HJVKO Male‐P | HJVKO Male‐Fe | HJVKO Female‐P | HJVKO Female‐Fe | HJVKO Female‐OVX‐P | HJVKO Female‐OVX‐Fe | |
|---|---|---|---|---|---|---|
| n | 8 | 12 | 12 | 8 | 8 | 12 |
| HR, bpm | 431±8 | 426±9a | 471±18 | 467±32a | 409±25 | 372±12 |
| LVEDP, mm Hg | 9.9±1.5 | 17.±2.0a | 8.9±0.8 | 10.1±1.1 | 6.3±0.8 | 10.5±1.8b |
| LVESP, mm Hg | 103±6.1 | 107.±7.4 | 93±2.5 | 91±3.4 | 96±4.3 | 98±4.3 |
| LVEDV, μL | 22.5±2.8 | 24.7±2.6 | 26.7±4.4 | 28.6±3.8 | 44.7±7.0 | 65.5±7.3 |
| LVESV, μL | 4.36±1.72 | 4.7±1.38 | 6.1±1.1 | 5.8±0.9 | 12.2±3.2 | 34.1±1.5 |
| SV, μL | 18.2±1.7 | 20±1.9 | 21±3.7 | 20±2.5 | 32.5±4.4 | 31.4±3.9 |
| EF, % | 80.8±2.9 | 80.9±3.4 | 78.7±3.1 | 70±2.8 | 72.7±4.2 | 51.9±4.3b |
| dP/dtmax, mm Hg/s | 9637±135 | 9201±149 | 8447±145 | 8367±128 | 9263±776 | 7089±477 |
| SW, mJ | 0.341±0.062 | 0.382±0.049 | 0.291±0.009 | 0.303±0.008 | 0.558±0.07 | 0.440±0.016 |
| dP/dtmax/EDV, mm Hg/s per μL | 428±31 | 372±45 | 317±33 | 290±34 | 207±21 | 123±23b |
| ESPVR, mm Hg/μL | 3.14±0.35 | 3.08±0.29 | 3.53±0.39 | 3.55±0.36 | 3.48±1.1 | 2.01±0.34b |
| τ (Glantz), ms | 9.23±0.41 | 13.7±0.32a | 9.29±1.1 | 9.87±0.66 | 10.7±0.72 | 12.73±1.3b |
| EDPVR, mm Hg/μL | 0.103±0.093 | 0.127±0.011a | 0.095±0.009 | 0.091±0.014 | 0.110±0.04 | 0.147±0.004b |
dP/dt indicates rate of change in LV pressure; dP/dtmax/EDV, Starling contractile index; EDPVR, end‐diastolic pressure‐volume relationship; EF, ejection fraction; ESPVR, end‐systolic pressure‐volume relationship; HR, heart rate; HJVKO, hemojuvelin‐null; LVEDP, LV end‐diastolic pressure; LVEDV, LV end‐diastolic volume; LVESP, LV end‐systolic pressure; LVESV, LV end‐systolic volume; PRSW, preload‐recruitable stroke work; SV, stroke volume; SW, stroke work; τ, LV relaxation time constant.
P<0.05 for effect of iron.
P<0.05 for interaction.
We next investigated the mechanism for the sex differences in iron‐overload cardiomyopathy. Estrogen can modulate iron homeostasis by regulating hepcidin and ferroportin expression via an estrogen response element. Iron overload resulted in an equivalent increase in cardiac iron deposition as assessed by Prussian blue staining (Figure 2E and 2F) and quantification of myocardial iron levels using inductively coupled plasma‐resonance mass spectrometry (Figure 2G and 2H) in male and female hearts. Taqman real‐time PCR analysis for ferritin, a ubiquitous intracellular iron‐storage protein, showed that ferritin light‐chain and heavy‐chain mRNA were higher in female than in male WT and HJVKO hearts in response to iron overload (Figure 2I and 2J). Basal mRNA expressions of the iron‐regulatory proteins hepcidin and ferroportin were higher and lower, respectively, in female compared to male WT hearts and were increased in response in iron overload (Figure 2I). Although similar changes were seen in ferroportin expression in HJVKO hearts, hepcidin levels were markedly suppressed and remained unchanged in response to iron overload (Figure 2J). Altered ferritin mRNA levels were reflected in ferritin protein level in WT hearts (Figure 2K), and there was an equivalent increase in ferritin protein in iron‐overloaded HJVKO hearts (Figure 2L). These results clearly document a marked sexual dichotomy in the response to iron overload whereby female mice are clearly protected against iron‐overload cardiomyopathy despite an equivalent degree of myocardial iron deposition.
Female Mice Are Protected Against Iron‐Overload‐Mediated Myocardial Oxidative Stress and Myocardial Fibrosis
Iron‐induced myocardial oxidative stress and lipid peroxidation coupled with reduced myocardial antioxidant reserves are major pathogenic processes in iron‐overload cardiomyopathy.9, 10, 40, 41 Whereas male iron‐overloaded hearts demonstrated marked increase in oxidative stress, iron‐overloaded female WT and HJVKO hearts did not show increased oxidative stress (Figure 3A through 3F). Male WT and HJVKO iron‐overloaded hearts displayed increased dichlorodihydrofluorescein (DCF) and DHE fluorescence and 4‐HNE levels that were markedly suppressed in female hearts (Figure 3A and 3B). The increased oxidative stress in male iron‐overloaded hearts was associated with a severe decrease in GSH levels, a key antioxidant reserve (Figure 3C and 3D), and increased MDA levels, a marker of lipid peroxidation (Figure 3E and 3F). In contrast, female iron‐overloaded hearts demonstrated a marked resistance to oxidative damage reflected in reduced glutathione (Figure 3C and 3D) and unchanged MDA (Figure 3E and 3F) levels. The expression of 2 key antioxidant enzymes, catalase and heme oxygenase 1 (HMOX1), and antioxidants thioredoxin 1 (TRXN1) and metallothionein‐1 (MT1) was evaluated. Baseline catalase expression was greater in female hearts but increased in male hearts (Figure 3G and 3H), whereas HMOX1 levels increased to a greater extent in female WT iron‐overloaded hearts (Figure 3I and 3J). Expression of TRXN1 and MT1 was greater in female compared to male hearts, but although TRXN1 expression did not change in response to iron overload (Figure 3K and 3L), MT1 expression increased equivalently (Figure 3M and 3N).
We next assessed the extent of myocardial hypertrophy, fibrosis, and apoptosis, key pathological events in heart disease. Increased interstitial fibrosis is a characteristic feature of diastolic dysfunction. Iron overload in male WT and HJVKO mice resulted in increased myocardial interstitial fibrosis as seen in the PSR staining and quantification of collagen content (Figure 4A and 4B). Myocardial fibrosis in male iron‐overloaded mice was associated with increased mRNA expression of procollagen I and III (Figure 4C and 4D) and myocardial disease markers, atrial natriuretic factor (ANF), BNP, β‐myosin heavy chain (β‐MHC), and α‐skeletal actin (α‐SkA) (Figure 4E and 4F). Interestingly, female WT and HJVKO mice were protected against iron‐overload‐mediated increase in myocardial fibrosis and the increased expression of collagen genes and disease markers (Figure 4A through 4F). Abnormalities in myocardial Ca2+‐handling proteins are linked to cardiac dysfunction.42 However, we found no alteration in myocardial SERCA2a and NCX1 protein levels in male and female mice in response to iron overload (Figure 4G and 4H). In addition, both female and male mice did not show increased myocardial apoptotic cell death in response to iron overload as shown by TUNEL staining (Figure 5). We next assessed estrogen receptors (ER) in the heart and showed that although basal myocardial ER‐α protein levels were higher in male compared to female WT hearts, ER‐α and ER‐β levels were unaffected by iron overload in WT and HJVKO hearts (Figure 6). Our results collectively demonstrate that whereas male mice showed severe myocardial oxidative stress and fibrosis, female mice were markedly resistant to iron‐overload‐mediated myocardial oxidative damage and fibrosis.
Ovariectomy Results in Loss of Protection and Increased Susceptibility to Iron‐Overload Cardiomyopathy in Female Mice
To determine the effects of estrogen on the observed protection from iron‐overload cardiomyopathy in female mice, we performed bilateral ovariectomies (OVX) in female WT and HJVKO mice (Figure 7). Hematoxylin‐eosin staining was used to confirm the complete removal of both ovaries from every mouse that underwent OVX, and plasma 17β‐estradiol levels were markedly lowered in both female WT and HJVKO mice following OVX and were not affected by iron overload (Figure 8).
Following a 2‐week recovery period, mice were then subjected to iron overload. Importantly, OVX‐WT female mice showed marked worsening of cardiac function in response to iron overload, resulting in systolic and diastolic dysfunction compared with preserved cardiac function in sham‐operated female mice (Figure 9; Tables 1 and 2). Ovariectomy in WT female mice also resulted in diastolic dysfunction based on decreased E′/A′ ratio, prolonged IVRT (Figure 9A), and increased LVEDP, and prolonged LV relaxation time constant (τ) (Figure 9B). Interestingly, loss of ovarian function precipitated systolic dysfunction as shown by the reduced EF, preload‐corrected dP/dtmax, and end‐systolic pressure‐volume relationship (Figure 9C). Representative pressure‐volume loops demonstrated diastolic and systolic dysfunction with LV dilation in OVX iron‐overloaded female WT mice (Figure 9D). Ovariectomy in HJVKO female mice resulted in a similar pattern of cardiac dysfunction characterized by diastolic dysfunction (Figure 9E and 9F) and systolic dysfunction (Figure 9G), as illustrated by representative pressure‐volume loops (Figure 9H).
To determine whether OVX has any effects on iron deposition, we characterized myocardial iron deposition in female mice by Prussian blue staining (Figure 10A and 10B) and inductively coupled plasma‐resonance mass spectrometry (Figure 10C and 10D), which showed that the degree of iron deposition was equivalent in OVX and sham‐operated female mice in response to iron overload. Ovariectomy inhibited iron‐overload‐induced upregulation of myocardial ferritin mRNA (Figure 10E and 10F) and protein (Figure 10G and 10H) levels in WT and HJVKO female mice with the exception of ferritin light‐chain mRNA expression in HJVKO mice (Figure 10F). Ovariectomy in WT and HJVKO females significantly increased the myocardial expression of ferroportin and decreased hepcidin expression in WT hearts in response to iron overload, but the hepcidin expression in HJVKO hearts remained blunted (Figure 10E and 10F). Signaling pathways are known to be sex dependent and may modify the myocardial stress response.21 We then assessed phosphorylation of Akt by Western blot analysis and found that phosphorylation (serine‐473) of Akt remained unchanged in response to iron overload in female mice and was unaffected by OVX (Figure 10I and 10J). Notably, OVX did not increase the susceptibility to apoptotic cell death in response to iron overload, as shown by TUNEL staining (Figure 11), whereas OVX in WT and HJVKO females significantly increased myocardial ER‐α protein levels without affecting the levels of ER‐β (Figure 12). Thus, OVX in female mice resulted in iron‐overload‐induced cardiac dysfunction and lack of upregulation of ferritin levels without a differential effect on myocardial iron deposition.
Loss of Antioxidant Defense Correlated With Iron‐Overload‐Mediated Oxidative Stress and Heart Disease in OVX Female Mice
To understand the contribution of estrogen to the intrinsic antioxidant defense capacity of female mice, we assessed myocardial oxidative stress, hypertrophy, and fibrosis. Interestingly, OVX resulted in a loss of antioxidant defense seen in the WT and HJVKO female hearts and resulted in greatly increased iron overload‐induced myocardial oxidative stress characterized by markedly increased DCF and DHE fluorescence and 4‐HNE levels (Figure 13A and 13B), lowered reduced glutathione levels (Figure 13C and 13D), and increased MDA levels (Figure 13E and 13F). Gene expression analysis revealed decreased expression of catalase (Figure 13G and 13H) and HMOX1 (Figure 13I and 13J) following OVX and a lack of upregulation in response to iron overload. Similarly, baseline expression of TRXN1 (Figure 13K and 13L) and MT1 (Figure 13M and 13N) was lowered in the OVX group, and the iron‐overload associated upregulation of MT1 was blunted (Figure 13M and 13N). Ovariectomy also increased the susceptibility to myocardial interstitial fibrosis in WT and HJVKO female mice, as shown by representative PSR staining and quantification of collagen content (Figure 14A and 14B) coupled with increased mRNA expression of procollagen I and III (Figure 14C and 14D). Pathological myocardial hypertrophy was exacerbated in iron‐overloaded OVX WT and HJVKO females as illustrated by the increased expression of disease markers, ANF, brain natriuretic peptide (BNP), β‐myosin heavy chain (β‐MHC), and α‐SkA (Figure 14E and 14F) and morphometric assessment showing increased LV weights (Figure 14G and 14H). Downregulation of SERCA2a and increased NCX1 levels have been linked to diastolic and systolic dysfunction.42, 43, 44, 45 Importantly, OVX also resulted in decreased SERCA2a (Figure 14I) and increased NCX1 (Figure 14J) protein levels in WT and HJVKO hearts in response to myocardial iron overload. Loss of ovarian function resulted in iron‐overload‐induced oxidative stress linked with increased myocardial fibrosis, pathological hypertrophy, and altered Ca2+ regulatory proteins.
17β‐Estradiol Therapy Rescued Iron‐Overload Cardiomyopathy in Male Iron‐Overloaded Mice
We next determine whether the therapeutic effects of 17β‐estradiol can rescue the iron‐overload cardiomyopathy in male WT mice (Figure 15A). Interestingly, exogenous supplementation with 17β‐estradiol rescued the diastolic dysfunction associated with iron‐overload cardiomyopathy in male mice in the setting of preserved systolic function (Figure 15B). Estradiol therapy resulted in marked normalization of iron‐induced myocardial oxidative stress as determined by DCF and DHE fluorescence (Figure 16A and 16B) and lack of myocardial fibrosis evaluated by PSR staining (Figure 16C) without affecting myocardial iron deposition determined by Prussian blue staining (Figure 16D). These results clearly demonstrate that 17β‐estradiol therapy mediates beneficial effects in a male iron‐overload cardiomyopathy model.
Discussion
Iron‐overload cardiomyopathy remains an important cause of morbidity and mortality in patients with secondary iron‐overload and primary hemochromatosis.5, 6, 7, 8 In this study we investigated sex‐based differences in iron‐overload cardiomyopathy in male and female WT and HJVKO mice. Our chronic acquired and genetic murine models of iron overload recapitulate essential features of clinical iron overload and its associated heart disease.9, 10, 30, 32 Although WT mice with a C57BL6 background can be resistant to iron‐overload cardiomyopathy,46 our murine models had prolonged iron overload, and we used appropriate sex‐, age‐, and strain‐matched mice as appropriate controls. We showed that female acquired and genetic murine models were markedly resistant to iron‐mediated cardiac injury based on functional, histological, and molecular determinants of heart disease. Importantly, we evaluated cardiac function using no‐invasive echocardiography and invasive pressure‐volume loops, and whereas iron‐overloaded females showed normal cardiac function, iron‐overloaded males exhibited diastolic dysfunction. In contrast, iron overload in OVX females resulted in advanced iron‐overload cardiomyopathy characterized by both diastolic and systolic dysfunction. The phenotypic features and response to OVX were similar in both models, highlighting a conserved mechanism for the sex‐dependent cardiac remodeling in response to iron overload. Ferritin mRNA was concordant with ferritin protein levels in WT hearts, but there was some degree of discordance between ferritin mRNA/protein levels in HJVKO hearts, which may be related to differences in the type of iron overload, the genetic strain of the mice, and our assessment of total ferritin protein levels.
Sexual dimorphisms in terms of cardiovascular diseases and iron metabolism exist in humans as well as in experimental animals.16, 21, 47 Iron‐overload‐mediated injury and clinical outcomes in female patients are better compared to male patients with β‐thalassemia,48 sickle cell anemia,49 and primary hemochromatosis.50 Several lines of evidence showed that estrogen exerts pleiotropic cardioprotective effects in well‐established models of heart failure such as pressure overload14 and myocardial ischemia‐reperfusion injury.16, 18, 19, 20, 21, 25 Our study extends these observations, and we demonstrated sex‐specific differences in iron‐overload‐mediated heart disease. In humans sex differences are thought to be related to menstrual blood loss and lowered iron stores and serum ferritin in females compared to males.51 In addition, regulation of iron metabolism is tightly controlled at a systemic level through the hepcidin/ferroportin axis,4, 52 and gonadal steroids can modulate iron metabolism such as estrogen‐mediated suppression of hepcidin and ferroportin expression.47, 52, 53 Rodents have estrous cycles in which there is no menstrual blood loss, and therefore, this cannot be evoked as the mechanism for the sex differences in iron‐overload cardiomyopathy. Our assessment of iron deposition using both quantitative measures and histological staining showed equivalent degrees of myocardial iron overload in females compared to males and in response to OVX.
Instead, our data strongly suggest that the intrinsic response of the myocardial tissue to iron‐induced injury is strongly modulated by sex. Excess iron promotes oxidative stress via the Fenton reaction and is the major pathogenic process in iron‐overload cardiomyopathy.9, 12, 13, 41 Interestingly, we found that preserved ovarian function was associated with a potent antioxidant response in the heart and upregulation of myocardial ferritin levels. Female sex was clearly associated with blunting of iron‐induced myocardial oxidative stress, lipid peroxidation product, and increased intrinsic antioxidant capacity. Conversely, OVX markedly exacerbated iron‐overload‐induced myocardial oxidative stress. In male iron‐overloaded mice, 17β‐estradiol, which mainly acts through its classical receptors ER‐α and/or ER‐β, which are both present in the heart,14 rescued iron‐overload‐induced diastolic dysfunction with normalization of the increased myocardial oxidative stress and fibrosis. These results further support the concept that 17β‐estradiol can mediate therapeutic effects in males as seen in the pressure‐overload heart failure model.14
The ability to counteract iron‐mediated oxidative stress is a key protective mechanism,9, 31, 54 and our results clearly demonstrate that estrogen‐mediated differences in reactive oxygen species (ROS) production could account for the male‐female differences in cardiovascular function and disease.21 Several studies have suggested that female mitochondria generate less ROS,21, 55 and iron‐induced mitochondrial ROS production may also have been curtailed in female hearts. Diastolic dysfunction correlated with increased myocardial fibrosis in the setting of increased myocardial oxidative stress, possibly by activating the myocardial transforming growth factor‐β signaling cascade.56, 57 Importantly, systolic dysfunction in iron‐overloaded OVX female mice was associated with marked reduction in SERCA2a and increased NCX1 levels, which represent a pivotal pathophysiological change in systolic heart failure.42, 45 Our results are consistent with the effects of female reproductive hormones on myocardial excitation‐contraction coupling and clearly support a role of directed antioxidant therapy for iron‐overload cardiomyopathy.31, 58 We demonstrated decreased SERCA2 protein levels, but oxidative posttranslational modification of SERCA2a may also have directly contributed to the myocardial dysfunction.59, 60
Sources of Funding
We acknowledge the funding support from the Canadian Institutes of Health Research (Kassiri and Oudit) and Alberta Innovates‐Health Solutions (AI‐HS) (Kassiri and Oudit). Das is supported by an AI‐HS Graduate Studentship, and Patel is supported by AI‐HS and Heart and Stroke Foundation of Canada Post‐Doctoral Fellowships.
Disclosures
None.
(J Am Heart Assoc. 2017;6:e003456. DOI: 10.1161/JAHA.116.003456.)
References
- 1. Sawicki KT, Chang HC, Ardehali H. Role of heme in cardiovascular physiology and disease. J Am Heart Assoc. 2015;4:e001138 doi: 10.1161/JAHA.114.001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Conrad ME, Umbreit JN. Disorders of iron metabolism. N Engl J Med. 2000;342:1293–1294. [PubMed] [Google Scholar]
- 3. Pietrangelo A. Hereditary hemochromatosis—a new look at an old disease. N Engl J Med. 2004;350:2383–2397. [DOI] [PubMed] [Google Scholar]
- 4. Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. [DOI] [PubMed] [Google Scholar]
- 5. Olivieri NF. The beta‐thalassemias. N Engl J Med. 1999;341:99–109. [DOI] [PubMed] [Google Scholar]
- 6. Murphy CJ, Oudit GY. Iron‐overload cardiomyopathy: pathophysiology, diagnosis, and treatment. J Cardiac Fail. 2010;16:888–900. [DOI] [PubMed] [Google Scholar]
- 7. Pennell DJ, Udelson JE, Arai AE, Bozkurt B, Cohen AR, Galanello R, Hoffman TM, Kiernan MS, Lerakis S, Piga A, Porter JB, Walker JM, Wood J; American Heart Association Committee on Heart Failure and Transplantation of the Council on Clinical Cardiology and Council on Cardiovascular Radiology and Imaging . Cardiovascular function and treatment in β‐thalassemia major: a consensus statement from the American Heart Association. Circulation. 2013;128:281–308. [DOI] [PubMed] [Google Scholar]
- 8. Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, McLaren CE, Bahlo M, Nisselle AE, Vulpe CD, Anderson GJ, Southey MC, Giles GG, English DR, Hopper JL, Olynyk JK, Powell LW, Gertig DM. Iron‐overload‐related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221–230. [DOI] [PubMed] [Google Scholar]
- 9. Oudit GY, Trivieri MG, Khaper N, Husain T, Wilson GJ, Liu P, Sole MJ, Backx PH. Taurine supplementation reduces oxidative stress and improves cardiovascular function in an iron‐overload murine model. Circulation. 2004;109:1877–1885. [DOI] [PubMed] [Google Scholar]
- 10. Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, Yazdanpanah M, Wilson GJ, Schwartz A, Liu PP, Backx PH. L‐type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron‐overload cardiomyopathy. Nat Med. 2003;9:1187–1194. [DOI] [PubMed] [Google Scholar]
- 11. Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: part II: animal and human studies. Circulation. 2003;108:2034–2040. [DOI] [PubMed] [Google Scholar]
- 12. Sawicki KT, Shang M, Wu R, Chang HC, Khechaduri A, Sato T, Kamide C, Liu T, Naga Prasad SV, Ardehali H. Increased heme levels in the heart lead to exacerbated ischemic injury. J Am Heart Assoc. 2015;4:e002272 doi: 10.1161/JAHA.115.002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Munzel T, Gori T, Keaney JF Jr, Maack C, Daiber A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J. 2015;36:2555–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iorga A, Li J, Sharma S, Umar S, Bopassa JC, Nadadur RD, Centala A, Ren S, Saito T, Toro L, Wang Y, Stefani E, Eghbali M. Rescue of pressure overload‐induced heart failure by estrogen therapy. J Am Heart Assoc. 2016;5:e002482 doi: 10.1161/JAHA.115.002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Witt H, Schubert C, Jaekel J, Fliegner D, Penkalla A, Tiemann K, Stypmann J, Roepcke S, Brokat S, Mahmoodzadeh S, Brozova E, Davidson MM, Ruiz Noppinger P, Grohe C, Regitz‐Zagrosek V. Sex‐specific pathways in early cardiac response to pressure overload in mice. J Mol Med. 2008;86:1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer S, van der Meer P, van Tintelen JP, van den Berg MP. Sex differences in cardiomyopathies. Eur J Heart Fail. 2014;16:238–247. [DOI] [PubMed] [Google Scholar]
- 17. Konhilas JP, Leinwand LA. The effects of biological sex and diet on the development of heart failure. Circulation. 2007;116:2747–2759. [DOI] [PubMed] [Google Scholar]
- 18. Miller VM. Why are sex and gender important to basic physiology and translational and individualized medicine? Am J Physiol Heart Circ Physiol. 2014;306:H781–H788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Satoh M, Matter CM, Ogita H, Takeshita K, Wang CY, Dorn GW II, Liao JK. Inhibition of apoptosis‐regulated signaling kinase‐1 and prevention of congestive heart failure by estrogen. Circulation. 2007;115:3197–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. [DOI] [PubMed] [Google Scholar]
- 21. Murphy E. Estrogen signaling and cardiovascular disease. Circ Res. 2011;109:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grohe C, Kahlert S, Lobbert K, Stimpel M, Karas RH, Vetter H, Neyses L. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. 1997;416:107–112. [DOI] [PubMed] [Google Scholar]
- 23. Bendale DS, Karpe PA, Chhabra R, Shete SP, Shah H, Tikoo K. 17‐β Oestradiol prevents cardiovascular dysfunction in post‐menopausal metabolic syndrome by affecting SIRT1/AMPK/H3 acetylation. Br J Pharmacol. 2013;170:779–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Eickels M, Grohe C, Cleutjens JP, Janssen BJ, Wellens HJ, Doevendans PA. 17beta‐Estradiol attenuates the development of pressure‐overload hypertrophy. Circulation. 2001;104:1419–1423. [DOI] [PubMed] [Google Scholar]
- 25. Sharkey LC, Holycross BJ, Park S, Shiry LJ, Hoepf TM, McCune SA, Radin MJ. Effect of ovariectomy and estrogen replacement on cardiovascular disease in heart failure‐prone SHHF/Mcc‐ fa cp rats. J Mol Cell Cardiol. 1999;31:1527–1537. [DOI] [PubMed] [Google Scholar]
- 26. Krijt J, Cmejla R, Sykora V, Vokurka M, Vyoral D, Necas E. Different expression pattern of hepcidin genes in the liver and pancreas of C57BL/6N and DBA/2N mice. J Hepatol. 2004;40:891–896. [DOI] [PubMed] [Google Scholar]
- 27. Widdowson EM, McCance RA. Sexual differences in the storage and metabolism of iron. Biochem J. 1948;42:577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramakrishnan U, Kuklina E, Stein AD. Iron stores and cardiovascular disease risk factors in women of reproductive age in the United States. Am J Clin Nutr. 2002;76:1256–1260. [DOI] [PubMed] [Google Scholar]
- 29. Sullivan JL. Iron and the genetics of cardiovascular disease. Circulation. 1999;100:1260–1263. [DOI] [PubMed] [Google Scholar]
- 30. Das SK, DesAulniers J, Dyck JR, Kassiri Z, Oudit GY. Resveratrol mediates therapeutic hepatic effects in acquired and genetic murine models of iron‐overload. Liver Int. 2016;36:246–257. [DOI] [PubMed] [Google Scholar]
- 31. Das SK, Wang W, Zhabyeyev P, Basu R, McLean B, Fan D, Parajuli N, DesAulniers J, Patel VB, Hajjar RJ, Dyck JR, Kassiri Z, Oudit GY. Iron‐overload injury and cardiomyopathy in acquired and genetic models is attenuated by resveratrol therapy. Sci Rep. 2015;5:18132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115:2187–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Idris AI. Ovariectomy/orchidectomy in rodents. Methods Mol Biol. 2012;816:545–551. [DOI] [PubMed] [Google Scholar]
- 34. Wang W, McKinnie SM, Patel VB, Haddad G, Wang Z, Zhabyeyev P, Das SK, Basu R, McLean B, Kandalam V, Penninger JM, Kassiri Z, Vederas JC, Murray AG, Oudit GY. Loss of Apelin exacerbates myocardial infarction adverse remodeling and ischemia‐reperfusion injury: therapeutic potential of synthetic Apelin analogues. J Am Heart Assoc. 2013;2:e000249 doi: 10.1161/JAHA.113.000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z, Oudit GY. Angiotensin‐converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. 2010;122:717–728, 18 p following 728. [DOI] [PubMed] [Google Scholar]
- 36. Mori J, Patel VB, Abo Alrob O, Basu R, Altamimi T, Desaulniers J, Wagg CS, Kassiri Z, Lopaschuk GD, Oudit GY. Angiotensin 1‐7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ Heart Fail. 2014;7:327–339. [DOI] [PubMed] [Google Scholar]
- 37. McLean BA, Zhabyeyev P, Patel VB, Basu R, Parajuli N, DesAulniers J, Murray AG, Kassiri Z, Vanhaesebroeck B, Oudit GY. PI3Kα is essential for the recovery from Cre/tamoxifen cardiotoxicity and in myocardial insulin signalling but is not required for normal myocardial contractility in the adult heart. Cardiovasc Res. 2015;105:292–303. [DOI] [PubMed] [Google Scholar]
- 38. Patel VB, Wang Z, Fan D, Zhabyeyev P, Basu R, Das SK, Wang W, Desaulniers J, Holland SM, Kassiri Z, Oudit GY. Loss of p47phox subunit enhances susceptibility to biomechanical stress and heart failure because of dysregulation of cortactin and actin filaments. Circ Res. 2013;112:1542–1556. [DOI] [PubMed] [Google Scholar]
- 39. Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. [DOI] [PubMed] [Google Scholar]
- 40. Jansova H, Machacek M, Wang Q, Haskova P, Jirkovska A, Potuckova E, Kielar F, Franz KJ, Simunek T. Comparison of various iron chelators and prochelators as protective agents against cardiomyocyte oxidative injury. Free Radic Biol Med. 2014;74:210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berdoukas V, Coates TD, Cabantchik ZI. Iron and oxidative stress in cardiomyopathy in thalassemia. Free Radic Biol Med. 2015;88:3–9. [DOI] [PubMed] [Google Scholar]
- 42. Hajjar RJ. Potential of gene therapy as a treatment for heart failure. J Clin Invest. 2013;123:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Basu R, Oudit GY, Wang X, Zhang L, Ussher JR, Lopaschuk GD, Kassiri Z. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am J Physiol Heart Circ Physiol. 2009;297:H2096–H2108. [DOI] [PubMed] [Google Scholar]
- 44. Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1‐dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sipido KR, Volders PG, Vos MA, Verdonck F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc Res. 2002;53:782–805. [DOI] [PubMed] [Google Scholar]
- 46. Musumeci M, Maccari S, Sestili P, Massimi A, Corritore E, Marano G, Catalano L. The C57BL/6 genetic background confers cardioprotection in iron‐overloaded mice. Blood Transfus. 2013;11:88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hou Y, Zhang S, Wang L, Li J, Qu G, He J, Rong H, Ji H, Liu S. Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element. Gene. 2012;511:398–403. [DOI] [PubMed] [Google Scholar]
- 48. Borgna‐Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, Romeo MA, Forni GL, Gamberini MR, Ghilardi R, Piga A, Cnaan A. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89:1187–1193. [PubMed] [Google Scholar]
- 49. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. [DOI] [PubMed] [Google Scholar]
- 50. Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, Dawkins FW, Acton RT, Harris EL, Gordeuk VR, Leiendecker‐Foster C, Speechley M, Snively BM, Holup JL, Thomson E, Sholinsky P; Hemochromatosis and Iron Overload Screening (HEIRS) Study Research Investigators . Hemochromatosis and iron‐overload screening in a racially diverse population. N Engl J Med. 2005;352:1769–1778. [DOI] [PubMed] [Google Scholar]
- 51. Zacharski LR, Ornstein DL, Woloshin S, Schwartz LM. Association of age, sex, and race with body iron stores in adults: analysis of NHANES III data. Am Heart J. 2000;140:98–104. [DOI] [PubMed] [Google Scholar]
- 52. Brewer C, Otto‐Duessel M, Wood RI, Wood JC. Sex differences and steroid modulation of cardiac iron in a mouse model of iron overload. Transl Res. 2014;163:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qian Y, Yin C, Chen Y, Zhang S, Jiang L, Wang F, Zhao M, Liu S. Estrogen contributes to regulating iron metabolism through governing ferroportin signaling via an estrogen response element. Cell Signal. 2015;27:934–942. [DOI] [PubMed] [Google Scholar]
- 54. Berndt C, Lillig CH, Holmgren A. Thiol‐based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292:H1227–H1236. [DOI] [PubMed] [Google Scholar]
- 55. Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res. 2010;106:1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Richter K, Konzack A, Pihlajaniemi T, Heljasvaara R, Kietzmann T. Redox‐fibrosis: impact of TGFβ1 on ROS generators, mediators and functional consequences. Redox Biol. 2015;6:344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Saito K, Ishizaka N, Aizawa T, Sata M, Iso‐o N, Noiri E, Mori I, Ohno M, Nagai R. Iron chelation and a free radical scavenger suppress angiotensin II‐induced upregulation of TGF‐β1 in the heart. Am J Physiol Heart Circ Physiol. 2005;288:H1836–H1843. [DOI] [PubMed] [Google Scholar]
- 58. MacDonald JK, Pyle WG, Reitz CJ, Howlett SE. Cardiac contraction, calcium transients, and myofilament calcium sensitivity fluctuate with the estrous cycle in young adult female mice. Am J Physiol Heart Circ Physiol. 2014;306:H938–H953. [DOI] [PubMed] [Google Scholar]
- 59. Balderas‐Villalobos J, Molina‐Munoz T, Mailloux‐Salinas P, Bravo G, Carvajal K, Gomez‐Viquez NL. Oxidative stress in cardiomyocytes contributes to decreased SERCA2a activity in rats with metabolic syndrome. Am J Physiol Heart Circ Physiol. 2013;305:H1344–H1353. [DOI] [PubMed] [Google Scholar]
- 60. Lancel S, Qin F, Lennon SL, Zhang J, Tong X, Mazzini MJ, Kang YJ, Siwik DA, Cohen RA, Colucci WS. Oxidative posttranslational modifications mediate decreased SERCA activity and myocyte dysfunction in Galphaq‐overexpressing mice. Circ Res. 2010;107:228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]