

Cardiovascular‐related mortality accounts for >600 000 deaths per year and is the leading cause of death in the United States.1 In addition to factors such as age; a high‐fat, high‐glucose diet; exercise; and certain comorbidities, male sex is independently associated with higher incidence and mortality among multiple subtypes of cardiovascular disease.2 Although sex‐based differences have been observed at the population level, the molecular mechanisms underlying these differences are poorly understood.
Cardiomyopathy can be either acquired (secondary to myocardial infarction or myocarditis) or genetic (caused by diseases such as muscular dystrophy or familial hypertrophic cardiomyopathy). One group of genetic cardiomyopathies, including Friedreich's ataxia and hemochromatosis, is characterized by cardiac iron overload leading to the formation of reactive oxygen species (ROS) and cardiomyocyte cell death.3, 4 Interestingly, more symptomatic hemochromatosis is associated with male sex.5 In addition, cardiac iron accumulation has also been shown to play a causative role in cardiac ischemic injury and acquired cardiomyopathy.6 These findings suggest that the sex‐based differences influencing the severity of certain forms of genetic cardiomyopathy, such as hemochromatosis, may also affect outcomes in patients with acquired cardiomyopathy. Understanding the interconnection between sex and iron handling in the heart might provide for the discovery of novel therapeutic targets in cardiomyopathy.
Iron is an essential mineral used for the synthesis of heme and iron/sulfur clusters, both of which are cofactors for a host of enzymes involved in DNA repair, ATP production, and regulation of gene expression. In excess, however, iron can be detrimental because of its involvement in ROS production via the Fenton reaction.7 Consequently, iron is tightly regulated at both the cellular and systemic levels. At the systemic level, the liver synthesizes and secretes the peptide hormone hepcidin, which regulates the release of iron into circulation from the small intestine and macrophages. At the cellular level, a highly conserved iron response protein (IRP)/iron response element (IRE) system serves as the primary regulator of cellular iron homeostasis. Low cellular iron activates IRP, which binds to IREs in various mRNAs and results in increased iron uptake, mobilization from storage, and decreased iron export. If this mechanism is not sufficient, a parallel pathway involving the proteins mammalian target of rapamycin and tristetraprolin is activated to conserve iron for cell survival.7 Although iron uptake is systemically regulated, there are no mechanisms for mammals to actively shed iron, with the exception of blood loss. Because of the regular blood loss that occurs with menstruation, women are more susceptible to iron deficiency and anemia.8 For the very same reason, however, it is speculated that women may also be protected against iron overload disease, such as hemochromatosis.9 This could be a reason why men carrying genetic mutations for hemochromatosis are found to have greater severity of disease.5 Nevertheless, despite compelling hypotheses, the differences in cellular iron handling due to biological sex are poorly characterized.
It is this gap in knowledge that the study by Das et al addresses in this issue of JAHA.10 Using genetic and pharmacological models of cardiac iron overload, the authors demonstrated a sex difference in the severity of cardiac dysfunction despite similar levels of iron overload in the heart. In addition, oophorectomy in female mice mitigated the cardioprotective effects of female sex, and supplementation of estrogen in male mice conferred them. The former experiment was critical in determining whether the protective effects of sex were due to signaling from the ovaries or an intrinsic property of the cardiomyocyte. The latter experiment identified estrogen, as the central factor secreted from the ovaries, underlying the sex‐based difference in cardiac function. Among all proteins study by the group, ferritin levels appeared to be inversely correlated with both cardiac function and tissue ROS levels (Figure). This fits nicely with previous work showing that ferritin‐bound iron is less capable of forming ROS than “free” iron.11 Taken together, these findings suggest that differences in estrogen signaling may differentially regulate cellular iron‐storage machinery.
Figure 1.
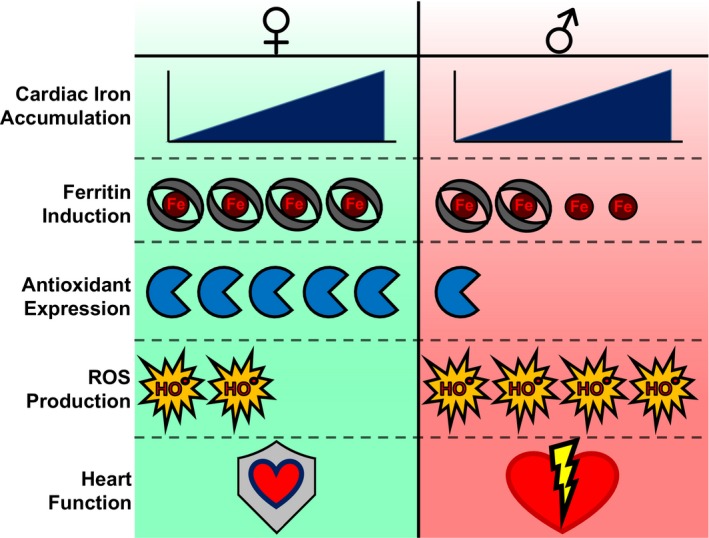
Key effects of sex on the response to iron overload in the murine heart. Male and female mice accumulate roughly equal amounts of cardiac iron in response to iron overload. Female mice express greater levels of ferritin and antioxidant proteins, resulting in reduced reactive oxygen species (ROS) production and preserved cardiac function.
Although periodic iron loss has long been considered the protective mechanism against iron overload in women, this study provides the first description of a hormone‐based cardioprotective mechanism that is uncoupled from systemic and cellular iron levels. Nevertheless, the molecular mechanism underlying sex‐based differences in ferritin expression remains to be determined. Although the data suggest that estrogen signaling is essential for the expression of ferritin under iron‐overload conditions, baseline estrogen signaling does not appear to cause any difference in ferritin expression, as demonstrated by the lack of difference in baseline ferritin levels between male and female mice of the same genotype and in female mice with or without oophorectomy. Interestingly, ferritin levels appear to be comparable in male and female mice with genetic hemochromatosis after iron loading, despite significantly lower ferritin mRNA levels in male mice. Consequently, the questions of whether there is an iron‐mediated increase in estrogen receptor translocation to the nucleus and whether there is differential binding of the estrogen receptor to its gene targets under iron‐overload conditions remain to be answered. In addition, the free iron concentration is usually kept low in the cell. Das et al used both Prussian blue staining and inductively coupled plasma mass spectrometry to measure total cellular iron, but those techniques are unable to distinguish different forms of iron (ie, free versus stored iron in ferritin versus iron in heme and iron/sulfur clusters). Whether the difference in ferritin levels reflects difference in free iron levels will require further studies.
Another important issue to consider is that not all intracellular iron accumulation sites are equivalent. Recent studies demonstrated that mitochondrial, rather than cytosolic, iron accumulation contributes significantly to cardiac ischemia–reperfusion injury.6 Therefore, although male and female mice exhibit similar levels of iron accumulation in genetic and pharmacologic models of iron overload, the subcellular distribution of accumulated iron might also explain the sex difference in cardiac dysfunction.
Finally, Das et al implicate the differential expression of antioxidant genes, notably thioredoxin 1, as a mechanism mediating the reduction in ROS production in iron‐overloaded female mice compared with iron‐overloaded male mice. This is consistent with previous reports finding that estradiol or estrogen analogs are capable of upregulating antioxidant gene expression, such as manganese superoxide dismutase, partly through the activation of the nuclear factor κB pathway.12, 13 Nevertheless, this cannot account for the entirety of the sex‐based protective effect because multiple clinical trials investigating the use of antioxidants in the prevention of cardiovascular disease have been unsuccessful.14
The mechanism described in this study may potentially also explain the sex‐based difference in hemochromatosis symptom severity. Initial symptoms of hemochromatosis include abnormal liver function, diabetes mellitus, and electrocardiographic abnormalities, all of which relate to iron overload–associated organ damage.15 Similar to female mice, which have equivalent iron overload but reduced ROS production compared with male mice, female patients also may be protected against ROS damage due to differences in intracellular iron handling and antioxidant expression compared with male patients.
In contrast to the cardioprotective effect of estrogen demonstrated in this study, recent clinical data have suggested a correlation between testosterone supplementation and worse cardiovascular disease outcomes in male patients, leading the U.S. Food and Drug Administration to submit an official statement requiring warnings be added to testosterone product labels.16 Moreover, sex‐based differences in cardiac events could also be explained by the deleterious effects of testosterone on the heart.
Ultimately, more in‐depth studies will be needed to develop novel therapies exploiting these sex‐based differences. Clinical trials investigating the supplementation of estrogen in postmenopausal women with congestive heart failure found a decrease in 12‐month mortality in women with estrogen supplementation.17 Guidelines from the American Academy of Clinical Endocrinologists, however, rated estrogen maintenance therapy as grade D1 because of concerns regarding increased risk of thromboembolism, uterine cancer, and increased cardiovascular events.18 Consequently, future studies investigating targetable components of the estrogen‐mediated iron‐storage pathway will be of great utility.
Sources of Funding
The authors are supported in part by the National Institutes of Health R01 HL127646 and P30 AI117943 to Ardehali and F30 DK109608 to Shapiro.
Disclosures
Ardehali is a consultant to Gerson‐Lehrman Group. The other authors declares no conflicts of interest.
J Am Heart Assoc. 2017;6:e005459. DOI: 10.1161/JAHA.116.005459.
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
References
- 1. Kochanek KD, Murphy SL, Xu J, Tejada‐Vera B. Deaths: final data for 2014. National Vital Statistics Reports. 2016;65:1–121. [PubMed] [Google Scholar]
- 2. Mosca L, Barrett‐Connor E, Wenger NK. Sex/gender differences in cardiovascular disease prevention: what a difference a decade makes. Circulation. 2011;124:2145–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheng C‐F, Lian W‐S. Prooxidant mechanisms in iron overload cardiomyopathy. Biomed Res Int. 2013;2013:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pandolfo M. Molecular pathogenesis of Friedreich ataxia. Arch Neurol. 1999;56:1201–1208. [DOI] [PubMed] [Google Scholar]
- 5. Beaton MD, Adams PC. The myths and realities of hemochromatosis. Can J Gastroenterol. 2007;21:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang H‐C, Wu R, Shang M, Sato T, Chen C, Shapiro JS, Liu T, Thakur A, Sawicki KT, Prasad SVN, Ardehali H. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol Med. 2016;8:247–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang H‐C, Shapiro JS, Ardehali H. Getting to the “Heart” of cardiac disease by decreasing mitochondrial iron. Circ Res. 2016;119:1164–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zimmermann MB, Hurrell RF. Nutritional iron deficiency. Lancet. 2007;370:511–520. [DOI] [PubMed] [Google Scholar]
- 9. Cash WJ, O'Neill S, O'Donnell ME, McCance DR, Young IS, McEneny J, Cadden IS, McDougall NI, Callender ME. Disordered vascular compliance in haemochromatosis. Ir J Med Sci. 2014;183:303–309. [DOI] [PubMed] [Google Scholar]
- 10. Das SK, Patel VB, Basu R, Wang W, DesAulniers J, Kassiri Z, Oudit G. Females are protected from iron‐overload cardiomyopathy independent of iron metabolism: key role of oxidative stress. J Am Heart Assoc. 2017;6:e003456 DOI: 10.1161/JAHA.116.003456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Darshan D, Vanoaica L, Richman L, Beermann F, Kuhn LC. Conditional deletion of ferritin H in mice induces loss of iron storage and liver damage. Hepatology. 2009;50:852–860. [DOI] [PubMed] [Google Scholar]
- 12. La Colla A, Vasconsuelo A, Boland R. Estradiol exerts antiapoptotic effects in skeletal myoblasts via mitochondrial PTP and MnSOD. J Endocrinol. 2013;216:331–341. [DOI] [PubMed] [Google Scholar]
- 13. Borras C, Gambini J, Gomez‐Cabrera MC, Sastre J, Pallardo FV, Mann GE, Vina J. Genistein, a soy isoflavone, up‐regulates expression of antioxidant genes: involvement of estrogen receptors, ERK1/2, and NFkappaB. FASEB J. 2006;20:2136–2138. [DOI] [PubMed] [Google Scholar]
- 14. Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK, Park BJ. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta‐analysis of randomised controlled trials. BMJ. 2013;346:f10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Niederau C, Strohmeyer G, Stremmel W. Epidemiology, clinical spectrum and prognosis of hemochromatosis In: Hershko C, Konijn AM, Aisen P, eds. Progress in Iron Research. Boston, MA: Springer US; 1994:293–302. [DOI] [PubMed] [Google Scholar]
- 16. U.S. Food and Drug Administration . FDA Drug Safety Communication: FDA cautions about using testosterone products for low testosterone due to aging; requires labeling change to inform of possible increased risk of heart attack and stroke with use. Washington, DC; 2015. [DOI] [PubMed]
- 17. Reis SE, Holubkov R, Young JB, White BG, Cohn JN, Feldman AM. Estrogen is associated with improved survival in aging women with congestive heart failure: analysis of the vesnarinone studies. J Am Coll Cardiol. 2000;36:529–533. [DOI] [PubMed] [Google Scholar]
- 18. Goodman NF, Cobin RH, Ginzburg SB, Katz IA, Woode DE. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of menopause. Endocr Pract. 2011;17(suppl 6):1–25. [DOI] [PubMed] [Google Scholar]