

Abstract
PDZ domains play crucial roles in cell signaling processes and are therefore attractive targets for the development of therapeutic inhibitors. In many cases, C-terminal peptides are the physiological binding partners of PDZ domains. To identify both native ligands and potential inhibitors we thus have screened arrays synthesized by the process of inverted peptides (PIPE), a variant of SPOT synthesis that generates peptides with free C-termini. Here, we present the development of a new functionalized cellulose membrane as solid support along with the optimized PIPEPLUS technology. Improved resolution and accuracy of the synthesis were shown with peptide arrays containing both natural and non-natural amino acids. These new screening possibilities will advance the development of active, selective and metabolically stable PDZ interactors.
Keywords: SPOT synthesis, C-terminus, PDZ domain, Peptide array, Cellulose functionalization
Graphical Abstract
While small molecules have traditionally dominated medicinal chemistry1, peptides have gained interest and are now broadly used as therapeutic or diagnostic tools2. Compared to small molecules, peptides display high selectivity and potency; they often exhibit good tolerability and predictable metabolism3. In particular, peptides show promise as inhibitors of protein-protein interactions (PPIs) that regulate a variety of cell signaling processes4. A common class of PPIs is mediated by PDZ domains, which are named after the proteins in which they were first discovered: post-synaptic density 95 (PSD-95), disc large tumor suppressor (DlgA), and zonula occludens 1 (ZO-1)5. These domains are found in many human proteins. They are composed of 80–90 amino acids and most commonly recognize the C-terminal carboxylate and four to seven residues of peptide ligands6,7. PDZ domain-mediated interactions (PDMIs) are involved in the transient assembly and localization of macromolecular complexes associated with fundamental cellular functions such as polarization, protein trafficking, tight-junction formation, and intercellular communication8–10. In addition, individual PDZ domains have been implicated in many human diseases (e.g. cystic fibrosis11, glioblastoma12) and are attractive therapeutic targets for the development of peptidyl inhibitors.
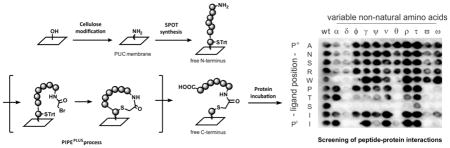
Yeast two-hybrid13, phage-display14, and SPOT synthesis15 approaches have been used to screen PPIs and develop putative peptidyl inhibitors. SPOT synthesis, which has progressively gained interest due to its accuracy and versatility, allows for the rapid synthesis of arrays of peptides grafted onto a cellulose support (Figure 1A)16. A synthesized peptide array can be incubated with any desired protein, followed by immunoblotting to reveal novel PPI sequences.
Figure 1.

SPOT synthesis and PIPE principles. (A): General concept of the SPOT synthesis. (B): Key steps of the process of inverted peptides. (a): coupling of the bromoacetyl moiety (1 M in DMF, 4-time coupling, 15 min reaction each), (b): selective cysteine detritylation with TFA (7% in DCM), (c): macrocycle formation, (d): one-pot acid cleavage and side chain deprotection (60% TFA followed by 90% TFA).
However, in standard SPOT synthesis, the resulting peptides are C-terminally attached to the membrane, making this technique unsuitable for the screening of PDZ domain ligands. Therefore, methods for generating peptides with free carboxyl functions have been developed and improved over the years17,18,19. In the latest version, known as the “process of inverted peptides” (PIPE), peptide orientation is inverted in three steps (Figure 1B): 1) synthesis of the peptide with the incorporation of p hydroxymethylphenoxyacetic acid (HMPA) as an acid-labile linker; 2) cyclization of the peptide chain involving an N-terminal bromoacetyl function; and 3) hydrolysis of the acid-labile bond with concomitant side-chain deprotection.
Within this PIPE protocol, several steps are crucial in determining peptide quality, which in turn governs the signal-to-noise ratio and the probability of false-positive or false-negative results. In this study, we present: 1) the development of a new amino-functionalized cellulose membrane; 2) optimization of the PIPE protocol and 3) proof-of-principle for the identification of PDZ-binding peptides containing non-natural amino acids.
To date, N-modified cellulose-amino-hydroxypropyl ether (N-CAPE) membranes18,19 have been used as supports for PDZ peptide arrays generated by the PIPE protocol19. On N-CAPE membranes, the required amine functionalization is provided by a stable ether bond between the hydroxyl groups of the cellulose and the free amine anchors on which the peptide chain will be elongated. However, the epibromohydrin moiety used to attach the diamine functional group is light-sensitive and very hygroscopic, complicating the preparation. Hence, one of our goals was to develop a more facile synthesis of amino-functionalized membranes that would be compatible both with standard SPOT synthesis and the PIPE variant.
Along with ether formation, cellulose hydroxyl groups can also be functionalized via carbamate synthesis. Based on the work of Stöllner et al.20, we activated the hydroxyl function of the cellulose membrane with 1,1-carbonyldiimidazole (CDI, 45 μmol.cm−2) in acetone, followed by an overnight incubation with 1,3-diaminopropane (DAP, 1 M) in carbonate buffer, leading to the propylamine urethane cellulose (PUC) membrane, as shown in Figure 2. The PUC membrane exhibited an amine density of 900–1,000 nmol.cm−2, a value comparable with the amine density observed for N-CAPE membranes (200–1,200 nmol.cm−2; ref. 16).
Figure 2.

Synthesis and characterization of the PUC membrane. (A) Schematic representation of the PUC membrane functionalization. (a) CDI: 1,1-carbonyldiimidazole (5–40 μmol.cm−2) in acetone, 4 h at room temperature. (b) DAP: 1,3-diaminopropane (5–1,000 mM) pH 9.6, 3–24 h at room temperature. (B) CDI concentration modestly affects amine density (means ± SD, n=3). (C) DAP concentration influences the amine density of the PUC membrane (mean with ranges, n=2). The extent of membrane amino-functionalization was quantified via Fmoc titration (see Materials and Methods).
The stability of the amino functionalization was assessed by coupling Fmoc-βAla-OPfp (0.3 M, 2x coupling) to a PUC membrane (1,000 nmol.cm−2), which was subsequently incubated with the different chemical solvents/conditions used during the PIPE process. Titration of the Fmoc group revealed no significant difference in the stability of the β-alanine linkage compared to untreated membranes (still approximately 1,000 nmol.cm−2, data not shown). As expected, no impairment of the PUC amine functionality is observed upon treatment with the conditions used during the SPOT or PIPE synthesis.
Because peptide density on the cellulose membrane has an impact on the downstream protein screen (e.g., via steric hindrance), it was important to determine and control the parameters that influence the yield of amino-functionalization. Replacing the acetone by a less polar solvent such as 1,4-dioxane substantially reduced efficiency, as no amino-functionalization was observed.
We next studied the influence of the concentrations of CDI and DAP on amine density. Decreasing the CDI concentration to as little as 5 μmol.cm−2 did not substantially reduce amino-functionalization (Figure 2B). However, 20 μmol.cm−2 CDI yielded the most consistent amine densities both within and between membranes (Table S1), so this value was used for all subsequent experiments. In contrast to the experiments with CDI, amine density quantification revealed a clear dose-dependent effect of initial DAP concentration on amine density (Figure 2C). As for the CDI-concentration experiments, we observed that the measured amine densities were highly reproducible at each of the DAP concentrations tested. Finally, we show that shorter DAP incubation periods (≥3h, 20 mM) are enough to obtain an amine density of 200 nmol.cm−2 (S3).
These findings implied that PUC membranes can be synthesized in a reproducible and controlled fashion at amine densities ranging from 100 to 1,000 nmol.cm−2. More importantly, this membrane functionalization does not require the anhydrous conditions that are critical for epibromhydrin coupling during N-CAPE preparation21. For the following experiments designed to optimize the PIPE protocol, we selected a DAP concentration of 20 mM, which results in PUC membranes with a loading capacity of 200 nmol.cm−2. This value is comparable to the loading yields of the commercially available Amino-PEG500-UC540 (400 nmol.cm−2, AIMS Scientific Products GmbH) and to previously used N-CAPE membranes. Additionally, in our experience, starting the synthesis with a membrane loading capacity of 200 nmol.cm−2 has been used successfully for screening a wide range of PDZ domains 18,19,22,23.
During the PIPE protocol, macrocycle formation is one of the most critical steps. Cyclization is a prerequisite for the inversion of the peptide at the cellulose membrane, and the efficiency of the reaction is controlled by both the reagent and conditions. Typically 2,4-dinitrophenyl bromoacetate (BrAc-DNP) and cesium carbonate are used. However, neither is ideal. BrAc-DNP is not commercially available, and cesium ions can over-activate the cyclizing cysteine (referred to as the cesium effect24). Therefore, we decided to optimize the coupling method with respect to both of these reagents.
To address cyclization conditions, we compared the standard solvent mixture (Cs2CO3/H2O/DMF 0.05/0.5/0.5 [w/v/v]) to several milder conditions such as 2x-concentrated tris-buffered saline (TBS-2X)25 and aqueous sodium bicarbonate (aq. NaHCO3 0.1 M)26 (Table 1). For this purpose, short peptides were synthesized on a PUC membrane modified with HMPA-βAla-Cys(Trt)-βAla (HMPA-BCB), onto which we coupled N-hydroxysuccinimidyl bromoacetate (BrAc-OSu) as a cyclization agent. Coupling efficiency of BrAc-OSu was confirmed by mass spectrometry (Figure S1). After peptide cyclization, residual uncyclized peptide was released by TFA cleavage (Figure S2). The amount of released peptide was determined by UV spectroscopy (A280) (see details in the Supplementary Material). To calibrate the results, Fmoc-βAla-OPfp was coupled to peptides instead of the cyclization agent, such that after TFA cleavage the obtained peptide amount was defined as the 100% value. Cyclization yields (%) for each condition were determined as a fraction of the value obtained with the linear βAla-peptide (see also assay scheme in Figure S2). This indirect quantification method was performed instead of mass spectrometry, as linear soluble peptides spontaneously cyclize in aqueous conditions, preventing accurate calculation of the cyclization ratio (data not shown). The results are shown in Table 1. They confirm a substantial improvement in yield with the bicarbonate-based conditions, which were used in the following experiments.
Table 1.
Yields obtained with different cyclization conditions.
| Peptide length b | Cyclization conditions | Cyclization reagent | Cyclization yield % |
|---|---|---|---|
| 3-mer c | aq. Cs2CO3/DMFa | BrAc-OSu | 25 |
| TBS-2X | BrAc-OSu | Not observed | |
|
| |||
| aq. NaHCO3 0.1 M | BrAc-OSu | 56 | |
| BrAc-DNP | n.d. | ||
| ClAc-OH | 46 | ||
| BrAc-OH | 50 | ||
| MalC3 | n.d. | ||
| MalC4 | n.d. | ||
|
| |||
| 6-mer c | aq. NaHCO3 0.1 M | BrAc-OSu | 50 |
| BrAc-DNP | 24 | ||
| ClAc-OH | n.d. | ||
| BrAc-OH | n.d. | ||
| MalC3 | 20 | ||
| MalC4 | 35 | ||
|
| |||
| 10-mer c | aq. NaHCO3 0.1 M | BrAc-OSu | 44 |
| BrAc-DNP | 33 | ||
| ClAc-OH | n.d. | ||
| BrAc-OH | n.d. | ||
| MalC3 | 17 | ||
| MalC4 | 22 | ||
5% Cs2CO3/H2O/DMF (w/v/v)
Spotted sequences: Z-[AA]n-HMPA-BCB-PUC, with Z = cyclization reagent.
3-mer: WKL; 6-mer: GLSWKL; 10-mer: GALSGLSWKL. n.d. = not determined.
To determine the choice of cyclization reagent, we next tested a variety of compounds in the synthesis of peptides with different lengths (3-, 6- and 10-mer) and measured cyclization yields for each reagent as described above. First attempts using bromoacetyl bromide (BrAc-Br) or bromoacetic acid (BrAc-OH) activated with N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ) failed (data not shown). Other thiophilic reagents, including BrAc-OSu, chloroacetic acid (ClAc-OH), BrAc-OH, 3-maleimidopropionic acid (MalC3), and 4-maleimidobutyric acid (MalC4), each activated with 0.5 eq. N,N′-diisopropylcarbodiimide (DIC), were evaluated as more promising (Table 1). For all three peptide lengths, BrAc-OSu showed the highest cyclization yield. It was also more efficient than the standard reagent BrAc-DNP when compared directly in the synthesis of 6-mer or 10-mer peptides. Finally, we confirmed the result obtained with BrAc-OSu by mass spectrometry, which revealed the expected mass of the intact macrocyclic compounds (Table S2). These findings indicate that the peptides are not affected by possible side reactions, such as tryptophan bromoacetylation.
Taken together, these experiments allowed us to develop an improved PIPE process, defined as PIPEPLUS, in which BrAc-OSu serves as a cyclization reagent in the presence of aq. NaHCO3. Using this method, we synthesized peptide arrays on a PUC membrane, at a density of 200 nmol.cm−2. To assess the quality of the protocol with a more diverse set of sequences, we synthesized a substitutional analysis (SubAna) array for a peptide (ANSRWPTSII, also called iCAL36) that has been extensively studied by our group27–30. This peptide has been shown to modulate the trafficking of the cystic fibrosis transmembrane conductance regulator (CFTR), by inhibiting its interaction with the PDZ domain of the CFTR-associated ligand (CALP). In brief, each residue of the iCAL36 sequence was individually replaced with each of the 20 naturally occurring L-amino acids to determine the key positions that contribute to CALP binding27. A PIPEPLUS SubAna array was incubated with the CALP domain and washed to remove unbound protein. The interactions between the PDZ domain and the peptides in each spot were revealed using an HRP-based antibody sandwich system (see Supplementary Material).
As shown in Figure 3A, the SubAna signals revealed a homogeneous signal for the wild-type (wt) column. To visualize the amino-acid preferences of iCAL36, signal intensities obtained for each spot were quantified and normalized relatively to the mean values of the wt spots (Figure 3B). To evaluate the effects of single point mutations, the following thresholds were set for sequences with significantly higher, equal, or lower levels of bound CALP than the wt-sequence: >1.6, 1.6 to 0.4, and < 0.4. CALP displayed strong preferences for I/L/V and S/T at position 0 (P0) and position −2 (P−2), respectively, where P0 is defined as the most C-terminal residue. These preferences are in excellent agreement with the assignment of CALP as a bona fide class I PDZ domain31, and with our previous studies27,30,32. At positions P−6 through P−9, the wt amino acids can be replaced by any other, which is typical of the relatively non-specific interactions of N-terminal residues with the PDZ domain14.
Figure 3.

Substitution analysis of the iCAL36 peptide with all natural L-amino acids. Every spot is labeled with the position of the mutation in the iCAL36 sequence (rows) and the identity of the amino-acid replacement (columns). All spots in the left-hand columns are identical and represent the wild-type (wt) peptide. (A) Chemiluminescence signals obtained after antibody incubation of the CAL PDZ incubated peptide array. (B) Quantification of the spot intensities was performed with ImageJ. Mean values of the wild-type (wt) sequences [first column and within the SubAna (written in red)] were used to normalize all values. Squares are color-coded to reflect normalized binding: green, values > 1.6; white, values between 1.6 and 0.4; and orange, values < 0.4.
As a reference, we synthesized three iCAL36 SubAna arrays using standard PIPE synthesis27. These were independently incubated with CALP and analyzed in the same way as described above (Figure S3). Comparison of the measured values showed overall good agreement with the PIPEPLUS results. In particular, there is excellent concordance at positions P−1 and P−2. However, while the patterns are broadly consistent, there are some differences. In particular, the PIPEPLUS method reveals more stringent amino-acid preferences at P0 (I/L/V). Furthermore, significantly stronger binding signals could be observed for replacements of Trp with a number of side chains at P−5, while those did not appear as strong in previous analyses. However, the signal intensities measured on cellulose membrane only allow relative comparison, and binding constants should be measured by other methods such as surface plasmon resonance (SPR), fluorescence polarization (FP) or isothermal titration calorimetry (ITC).
As a further refinement, we tested the compatibility of the PIPEPLUS protocol with non-natural amino acids (NNAA). Such residues expand the range of chemical space available for peptide screening. They can also modulate properties, such as peptide affinity or stability that are relevant for the development of therapeutic peptides. First of all, we analyzed the coupling efficiency of 11 Fmoc-NNAA-OH activated with CDI on the HMPA linker. Results of the Fmoc titration assays showed coupling efficiencies between 650 nmol.cm−2 for 2-aminobutyric acid (t) and 140 nmol.cm−2 for tert-butylglycine (a) (Figure 4A). All tested Fmoc-NNAA-OH reagents thus exceeded a coupling efficiency of 100 nmol.cm−2, which is the minimum concentration required for PIPE-related SPOT synthesis33. After this preliminary coupling of each NNAA, the sequence WKL was synthetically appended. The resulting spots were punched out, transferred into Eppendorf tubes, and treated with TFA for peptide release and side-chain deprotection. Crude product analyses of the precipitated peptides by RP-HPLC revealed excellent purity, with values ranging from 70% to 95% (Figure S4).
Figure 4.

Implementation of non-natural amino acids in the PIPEPLUS protocol. (A) Determination of the coupling efficiency for the 11 C-terminal non-natural amino acids. Fmoc-NNAA-OH (0.2 M) were activated in DMF with CDI and coupled 4-times on a HMPA-BCB functionalized membrane. Graphical representation of the coupling yields (means with ranges) achieved by titration of the Fmoc-piperidine adduct cleaved from one spot (n = 2). The minimal coupling efficiency for SPOT synthesis (100 nmol.cm−2) is shown as a horizontal line as reported in 33. (B) Substitution analysis of the iCAL36 peptide using the 11 NNAAs. Chemiluminescence signals obtained after antibody incubation of the CAL PDZ incubated peptide array. (C) Quantification of the spot intensities with ImageJ. All values were normalized relative to the mean signal intensities of the wild-type (wt) spots (written in red). Green: values > 1.6, white: values between 1.6 and 0.4 and orange: values < 0.4. α: tert-butylglycine, δ: 3-nitrotyrosine, φ: ornithine, γ: norleucine, ψ: β-thienylalanine, ν: cyclohexylglycine, θ: piperidine-4-carboxylic acid, ρ: norvaline, τ: 2-aminobutyric acid, ϖ: p-biphenylalanine, ω: 1-naphthylalanine.
Finally, we incorporated these NNAA in a substitution analysis of the iCAL36 peptide using the PIPEPLUS method. After CALP incubation and antibody detection, we observed robust binding at several spots, and wt signals with similar baseline intensities in each row in the array (0.7 – 1.2) (Figure 4B and 4C). Using the same signal thresholds as described for Figure 3B, we found that the key residue serine at P−2 could not be replaced by any of the NNAA that were tested. Interestingly, the P0 isoleucine appeared to be successfully replaced by hydrophobic amino acids such as norleucine (γ), cyclohexylglycine (ν), norvaline (ρ), or aminobutyric acid (τ). These results are in agreement with the structure of the CAL PDZ domain, which bears a hydrophobic pocket for the C-terminal amino acids29. Quantification and comparison of the spot intensities allowed also the identification of peptide sequences with higher or equal binding signals compared to the wt-sequence. Interesting sequences could be selected such as ANSρWPTSII [norvaline (1.7)] to inhibit arginine cleavage by endopeptidases or ANSRWPTSIτ [2-aminobutyric acid (3.1)] to inhibit exopeptidase-mediated degradation. Crystallographic evidence shows that CAL PDZ has additional space for binding at P−5 that is not fully occupied by tryptophan30. Thus, replacement with the voluminous 1-naphthylalanine (which has the highest binding signal: 4.5) could generate a ligand with more potency for CAL and higher specificity versus other PDZ domains.
Finally, the purity of the peptides synthesized by PIPEPLUS was estimated by HPLC to be between 60% and 90% as exemplified in Figure S5.
In conclusion, the optimized process of inverted peptides (PIPEPLUS) is a powerful SPOT synthesis variant allowing the preparation of rationally-designed peptide arrays for the screening of PDMIs as exemplified here for the CAL PDZ domain. Three aspects of the synthesis were optimized with respect to yield and synthetic accessibility: the amino functionalization of the cellulose membrane, the cyclization conditions, and the cyclization reagent. The PIPEPLUS protocol is now more widely accessible to the scientific community. All required compounds are commercially available, and the method is user-friendly and easily adaptable to any research laboratory using SPOT synthesis. PIPEPLUS arrays show an improved sensitivity, reproducibility and signal-to-noise ratio compared to previous described methods. As exemplified by the substitutional analysis of the iCAL36 sequence, both natural and non-natural amino acids can be included in the synthesis, expanding the scope of peptide sequence optimization for any biological application. Looking forward, the improved chemistry of our protocol means that peptide substitution screening can potentially be extended to a wide variety of amino acid-like building blocks, including β-, γ- or ω-amino acids. These improvements and modifications will likely accelerate the speed and fidelity by which potent and selective peptide inhibitors of PDMIs are identified and developed.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01DK101541 and T32GM008704). We thank Dr. Bernhard Ay for technical support and fruitful discussions.
Footnotes
Supplementary data associated with this article can be found in the online version.
References and notes
- 1.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 2.Antosova Z, Mackova M, Kral V, Macek T. Trends Biotechnol. 2009;27:628–635. doi: 10.1016/j.tibtech.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Fosgerau K, Hoffmann T. Drug Discov Today. 2015;20:122–128. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Pawson T, Nash P. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy MB. Trends Biochem Sci. 1995;20:350. doi: 10.1016/s0968-0004(00)89074-x. [DOI] [PubMed] [Google Scholar]
- 6.Sheng M, Sala C. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Harris BZ, Lim WA. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 8.Nourry C, Grant SGN, Borg JP. Sci STKE Signal Transduct Knowl Environ. 2003;2003:RE7. doi: 10.1126/stke.2003.179.re7. [DOI] [PubMed] [Google Scholar]
- 9.Dev KK. Nat Rev Drug Discov. 2004;3:1047–1056. doi: 10.1038/nrd1578. [DOI] [PubMed] [Google Scholar]
- 10.Grillo-Bosch D, Choquet D, Sainlos M. Drug Discov Today Technol. 2013;10:e531–540. doi: 10.1016/j.ddtec.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Cheng J, Moyer BD, Milewski M, Loffing J, Ikeda M, Mickle JE, Cutting GR, Li M, Stanton BA, Guggino WB. J Biol Chem. 2002;277:3520–3529. doi: 10.1074/jbc.M110177200. [DOI] [PubMed] [Google Scholar]
- 12.Kegelman TP, Wu B, Das SK, Talukdar S, Beckta JM, Hu B, Emdad L, Valerie K, Sarkar D, Furnari FB, Cavenee WK, Wei J, Purves A, De SK, Pellecchia M, Fisher PB. Proc Natl Acad Sci U S A. 2017;114:370–375. doi: 10.1073/pnas.1616100114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sidhu SS, Bader GD, Boone C. Curr Opin Chem Biol. 2003;7:97–102. doi: 10.1016/s1367-5931(02)00011-x. [DOI] [PubMed] [Google Scholar]
- 14.Tonikian R, Zhang Y, Sazinsky SL, Currell B, Yeh JH, Reva B, Held HA, Appleton BA, Evangelista M, Wu Y, Xin X, Chan AC, Seshagiri S, Lasky LA, Sander C, Boone C, Bader GD, Sidhu SS. PLoS Biol. 2008;6:e239. doi: 10.1371/journal.pbio.0060239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank R. Tetrahedron. 1992;48:9217–9232. [Google Scholar]
- 16.Volkmer R. Chem Bio Chem. 2009;10:1431–1442. doi: 10.1002/cbic.200900078. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmüller U, Russwurm M, Kleinjung F, Ashurst J, Oschkinat H, Volkmer-Engert R, Koesling D, Schneider-Mergener J. Angew Chem. 1999;111:2180–2184. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<2000::AID-ANIE2000>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 18.Boisguerin P, Leben R, Ay B, Radziwill G, Moelling K, Dong L, Volkmer-Engert R. Chem Biol. 2004;11:449–459. doi: 10.1016/j.chembiol.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Boisguerin P, Ay B, Radziwill G, Fritz RD, Moelling K, Volkmer R. Chembiochem Eur J Chem Biol. 2007;8:2302–2307. doi: 10.1002/cbic.200700518. [DOI] [PubMed] [Google Scholar]
- 20.Stöllner D, Scheller FW, Warsinke A. Anal Biochem. 2002;304:157–165. doi: 10.1006/abio.2001.5603. [DOI] [PubMed] [Google Scholar]
- 21.Bhargava S, Licha K, Knaute T, Ebert B, Becker A, Grötzinger C, Hessenius C, Wiedenmann B, Schneider-Mergener J, Volkmer-Engert R. J Mol Recognit JMR. 2002;15:145–153. doi: 10.1002/jmr.565. [DOI] [PubMed] [Google Scholar]
- 22.Mastny M, Heuck A, Kurzbauer R, Heiduk A, Boisguerin P, Volkmer R, Ehrmann M, Rodrigues CDA, Rudner DZ, Clausen T. Cell. 2013;155:647–658. doi: 10.1016/j.cell.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weski J, Meltzer M, Spaan L, Mönig T, Oeljeklaus J, Hauske P, Vouilleme L, Volkmer R, Boisguerin P, Boyd D, Huber R, Kaiser M, Ehrmann M. Chembiochem Eur J Chem Biol. 2012;13:402–408. doi: 10.1002/cbic.201100643. [DOI] [PubMed] [Google Scholar]
- 24.Dijkstra G, Kruizinga WH, Kellogg RM. J Org Chem. 1987;52:4230–4234. [Google Scholar]
- 25.Jakab A, Schlosser G, Feijlbrief M, Welling-Wester S, Manea M, Vila-Perello M, Andreu D, Hudecz F, Mezo G. Bioconjug Chem. 2009;20:683–692. doi: 10.1021/bc800324g. [DOI] [PubMed] [Google Scholar]
- 26.Robey FA, Fields RL. Anal Biochem. 1989;177:373–377. doi: 10.1016/0003-2697(89)90068-7. [DOI] [PubMed] [Google Scholar]
- 27.Vouilleme L, Cushing PR, Volkmer R, Madden DR, Boisguerin P. Angew Chem Int Ed Engl. 2010;49:9912–9916. doi: 10.1002/anie.201005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cushing PR, Vouilleme L, Pellegrini M, Boisguerin P, Madden DR. Angew Chem Int Ed Engl. 2010;49:9907–9911. doi: 10.1002/anie.201005585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amacher JF, Cushing PR, Weiner JA, Madden DR. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2011;67:600–603. doi: 10.1107/S1744309111009985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amacher JF, Cushing PR, Brooks L, Boisguerin P, Madden DR. Structure. 2014;22:82–93. doi: 10.1016/j.str.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 32.Amacher JF, Cushing PR, Bahl CD, Beck T, Madden DR. J Biol Chem. 2013;288:5114–5126. doi: 10.1074/jbc.M112.401588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ay B, Landgraf K, Streitz M, Fuhrmann S, Volkmer R, Boisguerin P. Bioorg Med Chem Lett. 2008;18:4038–4043. doi: 10.1016/j.bmcl.2008.05.116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.