

Abstract
Cardiac β-adrenergic receptors (β-AR) and Ca2+-Calmodulin dependent protein kinase (CaMKII) regulate both physiological and pathophysiological Ca2+ signaling. Elevated diastolic Ca2+ leak from the sarcoplasmic reticulum (SR) contributes to contractile dysfunction in heart failure and to arrhythmogenesis. β-AR activation is known to increase SR Ca2+ leak via CaMKII-dependent phosphorylation of the ryanodine receptor. Two independent and reportedly parallel pathways have been implicated in this β-AR-CaMKII cascade, one involving exchange protein directly activated by cAMP (Epac2) and another involving nitric oxide synthase 1 (NOS1). Here we tested whether Epac and NOS function in a single series pathway to increase β-AR induced and CaMKII-dependent SR Ca2+ leak. Leak was measured as both Ca2+ spark frequency and tetracaine-induced shifts in SR Ca2+, in mouse and rabbit ventricular myocytes. Direct Epac activation by 8-CPT (8-(4-chlorophenylthio)-2′-O-methyl-cAMP) mimicked β-AR-induced SR Ca2+ leak, and both were blocked by NOS inhibition. The same was true for myocyte CaMKII activation (assessed via a FRET-based reporter) and ryanodine receptor phosphorylation. Inhibitor and phosphorylation studies also implicated phosphoinositide 3-kinase (PI3K) and protein kinase B (Akt) downstream of Epac and above NOS activation in this pathway. We conclude that these two independently characterized parallel pathways function mainly via a single series arrangement (β-AR-cAMP-Epac-PI3K-Akt-NOS1-CaMKII) to mediate increased SR Ca2+ leak. Thus, for β-AR activation the cAMP-PKA branch effects inotropy and lusitropy (by effects on Ca2+ current and SR Ca2+-ATPase), this cAMP-Epac-NOS pathway increases pathological diastolic SR Ca2+leak. This pathway distinction may allow novel SR Ca2+ leak therapeutic targeting in treatment of arrhythmias in heart failure that spare the inotropic and lusitropic effects of the PKA branch.
Keywords: excitation-contraction coupling, sarcoplasmic reticulum, ryanodine receptor, calcium calmodulin-dependent protein kinase, Epac, nitric oxide synthase, Cardiac myocyte, Calcium transport, adrenergic signaling, CaMKII
Graphical abstract
1. Introduction
β-adrenergic receptor (β-AR) mediated myocyte Ca2+ mishandling is commonly described in heart failure (HF) and arrhythmia, which are increasing rapidly [1]. Therefore understanding fundamental mechanisms of β-AR effects on Ca2+ handling is critical. β-AR activation is an integral part of the cardiac fight-or-flight response, but its chronic activation (e.g. in HF) contributes to pathological hypertrophic remodeling, contractile dysfunction and arrhythmia.
Acute β-AR activation enhances cardiac contraction (ionotropy) and relaxation (lusitropy), in large part by increasing myocyte Ca2+ transients and accelerating [Ca2+]i reuptake by the sarcoplasmic reticulum (SR). These effects are mainly produced by PKA-dependent phosphorylation of L-type Ca2+ channels which increases Ca2+ current (ICa), and phospholamban (PLB) which enhances SR Ca2+ uptake and content [Ca2+]SRT (making more Ca2+ available for release; Fig 1, right). The higher Ca2+ transient causes stronger contraction, functionally offsetting myofilament Ca2+ desensitization by PKA (that otherwise participates in lusitropy) [2]. Studies have also shown that β-AR can also sensitizes ryanodine receptors (RyR) gating and SR Ca2+ leak, but while PKA can phosphorylate RyR, the functional effects on RyR are mediated via CaMKII activation and phosphorylation of RyR [3–18]. Chronic CaMKII activation and consequent increase of SR Ca2+ is now generally accepted as part of the HF syndrome [19–21]. Indeed, SR Ca2+ leak can contribute directly to both diastolic and systolic dysfunction in HF as well as β-AR triggered arrhythmias, via Ca2+ wave-induced inward Na+/Ca2+ exchange current (INCX) that causes delayed afterdepolarizations (DADs), triggered action potentials and premature ventricular contractions (PVCs) [3, 4, 22, 23]. Therefore, inhibition of SR Ca2+ leak is a valid therapeutic strategy in HF [19–21] which could improve systolic and diastolic function and limit β-AR-induced arrhythmias.
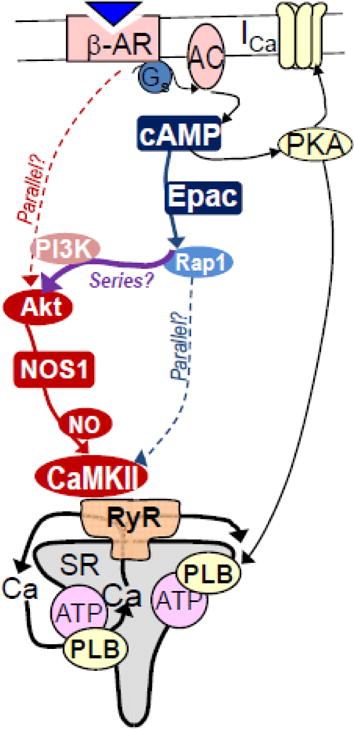
Figure 1. Proposed pathway for β-AR-induced increase of diastolic SR Ca2+ leak.
β-AR activation stimulates G-protein (Gs) dependent activation of adenylyl cyclase causing cAMP production that activates both Epac and PKA. The PKA branch enhances Ca2+ current (ICa) and phospholamban (PLB) sensitive SR Ca2+-ATPase (ATP). The Epac branch activates a cascade leading to NOS- and CaMKII-dependent RyR2 phosphorylation that promotes SR Ca2+ leak. Broken lines indicate a previously held idea that two parallel pathways might mediate Epac and NOS effects on RyR2.
Two pathways have been independently implicated to mediate β-AR activation of CaMKII and SR Ca2+ leak. One pathway involves exchange protein directly activated by cAMP (Epac), a cAMP target parallel to PKA, which may involve some downstream Epac targets leading to CaMKII autophosphorylation and RyR phosphorylation RyR (Fig 1, blue) [10–18]. Indeed, we have shown that this pathway specifically requires β1-AR, Epac2 (which localizes at myocyte T-tubules), CaMKIIδ and RyR2 phosphorylation at S2814 [14, 16]. For this pathway, it is clear that cAMP is involved and that PKA only contributes indirectly (via PLB-dependent increase in SR Ca2+ load), but the details of the pathway from the Epac-Rap1 level to CaMKII are not well resolved (with several mediators implicated, Fig 1) [10–17].
The other β-AR to CaMKII-RyR pathway involving nitric oxide synthase 1 (NOS1), seemed cAMP-independent, but involving protein kinase B (PKB or Akt) as upstream activators of NOS1dependent CaMKII activation via S-nitrosylation (Fig 1, red) [3–8, 24]. But in this case, the steps upstream from the β-AR to Akt were less clearly defined. This pathway was thought to be independent of cAMP and Epac because neither forskolin (direct adenylyl cyclase activator) nor 8-CPT (selective Epac agonist) mimicked β-AR effects [4–6]. This raised the idea of β-arrestin mediated signaling to CaMKII [24], as a parallel pathway from (β-AR to Akt and NOS1; Fig 1). Recent studies that have revealed the molecular mechanism by which S-nitrosylation occurs and mediates CaMKII activation [7, 25], localization of NOS1 at the junctional SR domain,[9] and robust evidence for cardiac CaMKIIδ in regulating RyR2 have solidified our understanding of the bottom part of the NOS1-RyR pathway.
Here, two labs that helped characterize these two pathways have worked together to further test whether these apparently parallel pathways from β-AR to CaMKII-RyR might be related in series. Much of the work leading to the Epac pathway involved measurement of Ca2+ sparks as an index of SR Ca2+ leak, often used 8-CPT as an Epac agonist and did not explore NOS involvement. The studies leading to NOS1 involvement in β-AR-induced SR Ca2+ leak, more often used the Shannon-Bers method of tetracaine-induced Δ[Ca2+]i and Δ[Ca2+]SRT shifts to measure SR Ca2+ leak. Here, we find similar results regardless of SR Ca2+ leak method, that 8-CPT (when freshly prepared) mimics the β-AR effects on tetracaine-sensitive SR Ca2+ leak, and that NOS mediates Epac-dependent increase of Ca2+ sparks, and that Akt is involved. We conclude that these pathways are largely in series (Fig 1).
2. Materials and Methods
2.1. Myocytes Isolation
Cardiac myocytes were isolated from New Zealand white rabbits and C57BL6 mice using retrograde Langendorff perfusion using Liberase TM (0.075 mg/mL, Roche) and Trypsin (0.0138%, Gibco) (37°C) as previously described [26]. All procedures were approved by the University of California Davis Institutional Animal Care and Use Committee (IACUC) in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
2.2. Ca2+ Spark Measurements
Spontaneous Ca2+ spark were measured in quiescent cardiomyocytes loaded with 5 μM Fluo-4 AM (Molecular Probes) for 30 min [14]. Images were recorded using confocal microscopy in line scan mode (Bio-Rad Radiance 2100, 40× oil immersion objective, 6 ms). Fluo-4 AM was excited at 488 nm using an Argon laser and the emission was collected at 505 nm. All experiments were done in Tyrode’s solution (in mM: 140 NaCl, 4 KCl, 1.1 MgCl2, 10 HEPES, 10 glucose, 1.8 CaCl2; pH7.4 with NaOH). Analysis was made using Firefly, a homemade routine in Python™, which uses analytical criteria similar to other widely used analysis methods [27].
8-CPT is a cAMP analog that is highly selective for Epac vs. PKA activation [28, 29]. The 8-CPT concentration used here (10 μM) has no effect on myocyte Ca2+ handling or Ca2+ sparks in mouse myocytes lacking Epac2 [14] and does not accelerate twitch [Ca2+]i decline [15], in contrast to very potent effects of PKA to accelerate twitch [Ca]i decline [14]. Moreover, direct measurements of PKA activity in intact ventricular myocytes showed no significant increase in PKA activation at 10–100 μM 8-CPT [16]. Thus, as used here 10 μM 8-CPT is selective for Epac-vs. PKA-mediated Ca2+ handling effects.
2.3. SR Ca2+ leak Measurements
The protocol used to measure SR Ca2+ leak in rabbit was as previously described (Fig 2) [5]. Briefly, [Ca2+]i was measured using a fluo-4 (Invitrogen) signal in isolated myocytes in the presence and absence of SR Ca2+ leak. Images were recorded using confocal microscopy in line scan mode (Zeiss LSM DUO in Live Channel mode, 40× water immersion objective, 2 ms/line). Tetracaine was used to rapidly and reversibly block the RyR, thus disrupting the SERCA pump-leak balance (Figure 2). The tetracaine-dependent shift of Ca2+ from the cytosol to the SR (decrease in [Ca2+]i and increase in SR Ca2+ content) is proportional to SR Ca2+ leak.
Figure 2. Exemplar Tetracaine-sensitive SR Ca2+ leak measurement in intact rabbit myocyte.
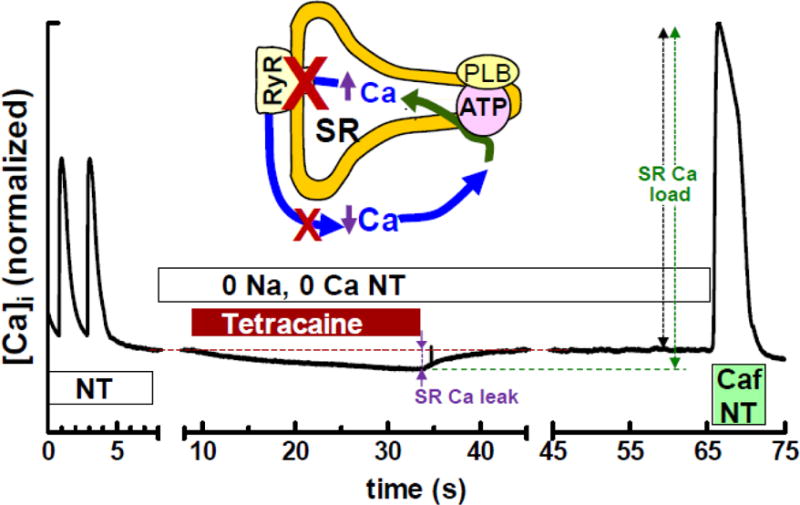
[Ca2+]SRT is varied by stimulation rate. After the last field stimulated twitch in normal Tyrode’s (NT, left) tetracaine blocks RyR-mediated leak in Na+-free, Ca2+-free solution (0 Na, 0 Ca; to prevent sarcolemmal Ca2+ flux). Blocking leak causes net SR Ca2+ uptake such that [Ca2+]i decreases and [Ca2+]SRT increases (inset). Tetracaine wash-off reverses the shift. [Ca2+]SRT is measured by rapid caffeine application and Δ[Ca2+]i. Leak flux is proportional tetracaine-induced Δ[Ca2+] (purple arrows) and Δ[Ca2+]SRT (black vs. green arrows) [32].
Myocytes were subjected to a protocol to load the SR in a graded manner: by emptying the SR with 10 mM caffeine followed either by 30 sec of rest, 30 sec of rest followed by one single stimulation, or field stimulation at 0.25 to 1.0 Hz. Field stimulations at given rates were performed for at least 20 beats to approach steady state cellular and SR Ca2+ content.
After one of the above loading protocols the bath solution was rapidly switched to 0 Na, 0 Ca2+ NT, 1 mM tetracaine. Without Na+ and Ca2+ in the bath, NCX, the primary Ca2+ efflux mechanism at rest, was blocked so that Ca2+ was entrapped in the resting cell [30]. The RyR (and therefore leak) is blocked by tetracaine and the measured resting fluorescence decreases as Ca2+ is taken up into the SR (Figure 2) [5]. Fluo-4 fluorescence was corrected for a 4% quench by tetracaine whenever it was present. Fluorescence was monitored for 30 s followed by another rapid solution switch to 0Na, 0Ca2+ NT with no tetracaine added. With the SR Ca2+ leak restored, diastolic [Ca2+]i rises back to its resting value. Finally, 10mM caffeine in nominally Ca2+ free NT was added to cause SR Ca2+ release. The total SR [Ca2+] ([Ca2+]SRT) was calculated as the difference between the basal and peak total cytosolic [Ca2+] ([Ca2+]T) in the presence of caffeine. The difference in [Ca2+]SRT in the presence and absence of tetracaine (the same as the difference in resting [Ca2+]T) is due to the leak dependent shift of Ca2+ from the cytosol to the SR (i.e. the difference in basal [Ca2+] with and without tetracaine) and the leak rate is proportional to this shift.
Data were selected such that the average [Ca2+]SRT was the same in all groups to ensure that the measured tetracaine-dependent SR Ca2+ leak was independent of SR [Ca2+].
2.4. Fluorescence Resonance Energy Transfer (FRET)
Adenovirus encoding Camui [31] was transfected in cultured cardiomyocytes for 2–4h (M.O.I 10–100)(5% CO2, 37 °C) in PC-1 medium. Experiments were performed 24h after transfection. FRET was measured using confocal microscopy in frame scan mode (Zeiss LSM5 Pascal, ×40 water immersion objective). FRET signal was measured as an increase of the FCFP/FYFP ratio with λex set at 458 nm (Ar laser) for CFP (donor) and λex set at 510 nm for YFP (acceptor) [31]. Donor fluorescence emission was detected at wavelengths of 470–500 CFP and acceptor fluorescence measured at ≥530 nm. Fluorescence images were analyzed using ImageJ software (NIH).
2.5. Western Blotting
Freshly isolated rabbit ventricular myocytes were plated on to laminin (Sigma-Aldrich; 1 mg/ml) coating the wells of 6-well tissue culture plates for further treatment in culture medium containing 2.0 mmol/L CaCl2. 8-(4-Chlorophenylthio)-2″-O-methyladenosine 3′,5′-cyclic monophosphate-sodium salt (8-CPT) and Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME) were obtained from Sigma-Aldrich. LY924002 was from EMD Millipore.
Adhered ventricular myocytes were either pretreated with L-NAME (100 μM), LY924002 (10 μM) or dimethyl sulfoxide (DMSO) for 15 min followed by the addition of 8-CPT (10–25 μM) with gentle swirling to achieve the final desired dilution and incubated for 10–15 min.
Upon completion of experimental treatments, the overlying solution was aspirated and ventricular myocytes were lysed with the immediate addition of hot 1-X Laemmli sample buffer; without β-mercaptoethanol (β-ME) or bromophenol blue dye. Lysates were recovered to a microcentrifuge tube, heated to 95°C for 5 min and stored at −20°C. Sample protein determinations were made using a BCA protein assay kit (Pierce) followed by the addition of β-ME and dye to the samples in order to obtain an appropriate 1-X Laemmli buffer final-volume concentration for each and were again heated as before.
Utilizing standard electrophoresis protocols for SDS-PAGE and western blotting, cell lysates were separated using either pre-cast 4 % or 4–20 %-gradient tris-glycine mini-gels (Novex™, Invitrogen) and transferred to nitrocellulose. Equivalent sample loading of 20–60 μg of protein per well was required. Primary antibodies used for western blotting were directed against phospho-AKT (Ser473; #4690), and pan-AKT (#4690) from Cell Signaling Technology and anti-ryanodine receptor 2 (human RyR2; Alomone labs), or phospho-RyR2 (human, pSer2808 or pSer2814; Badrilla). Purified monoclonal anti-α-actinin immunoglobulin (EA-53; Sigma-Aldrich) was also used to evaluate sample loading. Species specific horseradish peroxidase-conjugated secondary antibodies (Jackson Immunoresearch Labs) were used and visualization was accomplished with Western Lighting™ chemiluminescence reagents (PerkinElmer) and directly imaged using a charge-coupled device (CCD) sensor with controlling software (Marconi Applied Technologies).
For the evaluation of Epac-mediated changes in phospho-RyR, blotted panels were first probed with either anti-pSer2808 or pSer2814 and then stripped completely using 2% SDS-buffer. The efficacy of antibody complex removal was verified by incubation of secondary antibody only for the typical time and antibody dilution with imaging. Next, each panel was re-probed with anti-RyR2 for evaluation of uniform protein loading. Protein bands were quantified (arbitrary units; a.u.) using ImageJ software (NIH) and phospho-RyR was normalized to the corresponding RyR2 band for each group. Statistical significance (P <0.05) was determined using unpaired Student’s t test to compare Control group values to 8-CPT-treated values (n=3) for each experimental group.
3. Results
3.1. Interdependence of Epac and NOS upon β-AR-induced SR Ca2+ Leak
Previous work studying the role of Epac in β-AR-induced SR Ca2+ leak via Ca2+ sparks had not tested nitric oxide or NOS involvement (although NOS1 inhibition was shown to prevent ISO-stimulated increase in total SR Ca2+ leak [6]). Figure 3A and 3C show that 100 nM ISO increased Ca2+ spark frequency by approximately 100% (0.22±0.048 in CTL vs. 0.46±0.12 in ISO, p<0.05). The NOS inhibitor, L-NPA (5 μM) did not significantly alter basal Ca2+ spark frequency, but prevented the ISO-induced increase in Ca2+ sparks (0.19±0.05 vs. 0.46±0.12). This is consistent with prior studies of total leak (using the tetracaine method in Fig 2) [6] and supports the idea that β-AR activation increased SR Ca2+ leak via NOS activation.
Figure 3. β-AR- and Epac-induced Ca2+ sparks.
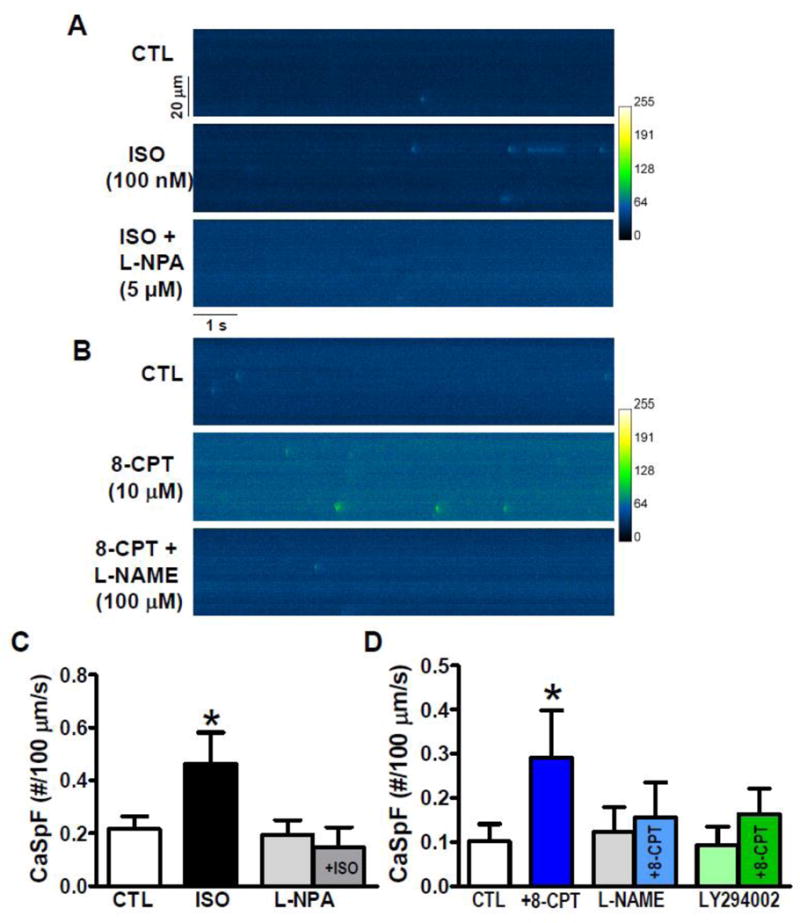
A, B. Raw Ca2+ sparks in intact mouse ventricular myocytes. C. Ca2+ spark frequency (CaSpF) was increased by ISO (100 nM), but not by NOS-inhibitor L-NPA (5 mM, 10 min). However, L-NPA blocked ISO-induced increase in CaSpF (n=9, 6, 5 and 5 myocytes). D. 8-CPT (10 mM, 2 min) induced increased CaSpF, while neither NOS inhibition (100 μM L-NAME, 5 min) nor PI3K inhibition (10 μM LY294002) altered baseline CaSpF. Both L-NAME and LY294002 prevented 8-CPT-induced CaSpF (n=21, 10, 13, 13, 7 and 7 myocytes, respectively; * p<0.05). Resting fluorescence was not significantly altered by L-NPA, 8-CPT, L-NAME or LY94002, but was slightly increased (15%) by ISO, which may be secondary to higher CaSpF.
We next tested whether direct specific Epac activation of Ca2+ sparks (by 10 μM 8-CPT) [14, 15] would be sensitive to NOS inhibition. Figures 3B and D show that the robust Ca2+ spark activation by 8-CPT was prevented by 100 μM L–NAME (which inhibits NOS). Again, L-NAME alone did not alter basal Ca2+ sparks, indicating (as in Fig 3C) that this NOS signaling to Ca2+ sparks is minimal prior to activation by ISO or 8-CPT (CTL: 0.10±0.04, 8-CPT:0.29±0.11 vs. L-NAME; 0.12±0.06). These results indicate that NOS is, in fact activated downstream of Epac and mediates the Epac-induced activation of Ca2+ sparks, and may mediate the β-AR-induced and Epac-dependent activation of Ca2+ sparks.
Our previous work measuring tetracaine-sensitive SR Ca2+ leak also showed that ISO-induced increase was reliant upon NOS activity, but was not mimicked by 8-CPT [6]. We revisited this issue since full 8-CPT efficacy requires fresh solution preparation for each experiment, which we were unaware of during that study. Figure 4 shows the results of these experiments in rabbit ventricular myocytes stimulated to steady-state at varying frequencies to obtain a range of [Ca2+]SRT (Figure 2). A rapid switch to Ca2+-and Na+-free Tyrode’s solution blocks the Na+/Ca2+ exchanger, keeping the total cellular [Ca2+] constant. Tetracaine (1 mM) blocks the RyR causing a shift of Ca2+ from the cytosol into the SR (lowering [Ca2+]i and raising [Ca2+]SRT, proportional to the SR Ca2+ leak) [32, 33]. We found that Epac activation with 8-CPT increased SR Ca2+ leak (Figure 4B 19.2±2.4 vs. 12.3±1.6 μmol/l cytosol ∆[Ca2+]SRT, n=11,18, p<0.05) when considering only data for the same average [Ca2+]SRT (Figure 4A, 120.9 vs. 120.6 μmol/l cytosol), consistent with the spark data above. Furthermore, 100 μM of the NOS inhibitor L-NAME completely prevented this increase (10.1±1.3 μmol/l cytosol ∆[Ca2+]SRT, n=11), confirming the involvement of NOS is the pathway. We conclude that Epac can mimic the β-AR-induced SR Ca2+ leak, as seen with Ca2+ spark measurements, and that the prior study [6] missed this because of a technical limitation (see corrigendum link to be added in proof). Here we used freshly prepared perfusate, to which 8-CPT was added from frozen stock, just prior to the experiment.
Figure 4. 8-CPT and ISO-induced SR Ca2+ leak in rabbit myocytes.
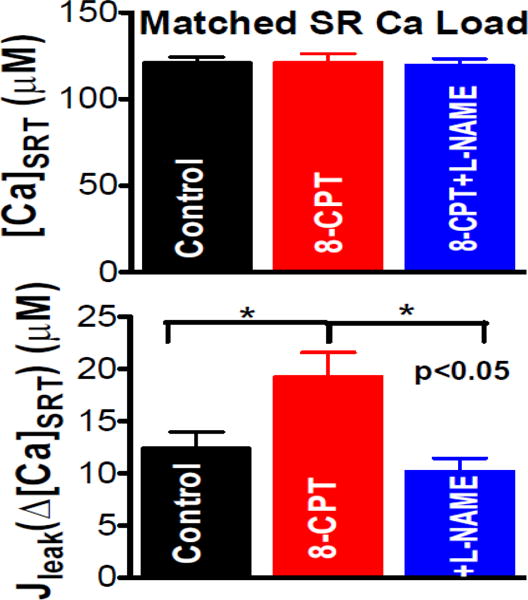
The tetracaine-induced approach (Fig 2) was used to assess SR Ca2+ leak (Δ[Ca2+]SRT, bottom) in myocyte records with matched SR Ca2+ content ([Ca2+]SRT, top). SR Ca2+ leak induced by 10 μM 8-CPT was prevented by NOS inhibition (100 μM L-NAME; n= 18, 11, 11 myocytes; * p<0.05).
3.2. β-AR-dependent CaMKII activation is by a Epac/NOS-dependent pathway
In order to test that each pathway operates via CaMKII activation downstream of both Epac and NOS, we used the FRET-based activity reporter Camui [31] (Figure 5A) to measure the response of CaMKII to both ISO and 8-CPT treatment. Figures 5B & C show that ISO and 8-CPT significantly activate CaMKII, as measured by a reduction of FRET (shown as an increase in FCFP/FYFP). Furthermore, consistent with the SR Ca2+ leak data above, the NOS inhibitor L-NAME (100 μM) prevented the Epac activation of CaMKII (Figure 5B) and the NO-donor SNAP mimicked the CaMKII activation by both ISO and 8-CPT. This also shows that SNAP prevented further CaMKII activation by 8-CPT (Figure 5C), consistent with no further 8-CPT activation of already S-nitrosylated CaMKII [6, 7, 25]. Note also that PKA inhibition (H89) did not significantly reduce ISO-induced CaMKII activation (Figure 5C).
Figure 5. CaMKII activation via Epac.
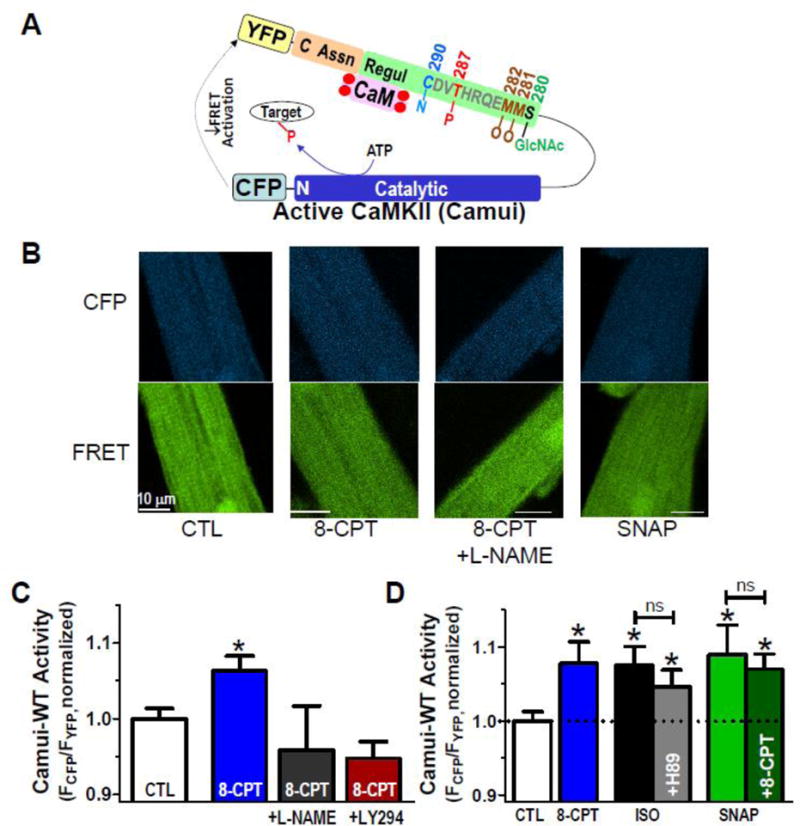
A. Schematic of the FRET-based activity reporter Camui, where activation (opening of regulatory (Regul) and catalytic domains causes reduced FRET between cyan and yellow fluorescent proteins (CFP and YFP). Sites of autophosphorylation (P), S-nitrosylation (N), oxidation (O) and O-GlcNAcylation (GlcNAc) are indicated. B. Rabbit myocyte images of CFP and FRET (YFP emission) under indicated treatments. C. Mean Camui signals at baseline (CTL), with 10 μM 8-CPT alone (30 min) or after pretreatment with 100 μM L-NAME or 10 μM LY294002 (n=41, 44, 13 and 10, respectively). D. Mean Camui signals in control rabbit cardiomyocytes at baseline (CTL), ± 10 μM 8-CPT (30 min), ISO (100 nM) ± H89 (1 μM) or SNAP ± 8-CPT (n =33, 24, 13, 17, 18 and 14 for each bar, respectively).
We also tested whether the phosphorylation of RyR in response to Epac activation was dependent on NOS, using Western blots with site-specific antibodies against pRyR-S2809 and pRyR-S2815. Rabbit cardiomyocytes were stimulated ±8-CPT with and without L-NAME. Figure 6 shows that 8-CPT promoted RyR phosphorylation at both the site known to be a CaMKII target (S2815) as well as S2808 (best known as a PKA target, but can also be a CaMKII target [34]).
Figure 6. Epac-mediated increase in RyR phosphorylation is NOS-dependent.
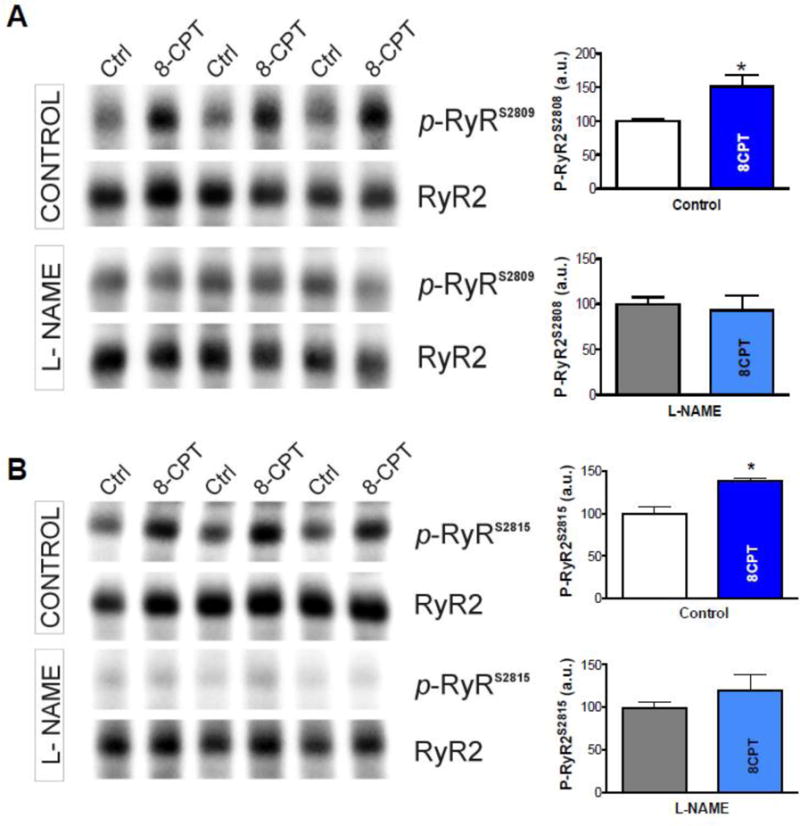
Western blots for total RyR and RyR phosphorylated at S2809 (A) and S2815 (B). Right panels show an Epac-dependent increase in phosphorylation at both residues in response to 8-CPT treatment, which was prevented by 100 μM L-NAME (*p<0.05 vs. without 8-CPT, t-test, n=3).
All together these data (Figures 3–6) demonstrate that β-AR activation activates CaMKII-dependent increase SR Ca2+ leak via Epac-mediated NOS1 activation. The fact that Epac-dependent SR Ca2+ leak is also NOS-dependent clearly shows that these two independently developed pathways are, indeed, parts of the same pathway with NOS1 downstream of Epac and with CaMKII downstream of both. The use of the tetracaine protocol also confirms that these effects on the SR Ca2+ leak are not simply the result of an increase of SR Ca2+ load. Finally, the effect is not species-dependent, as it is present in both mouse and rabbit, and it is observable both in Ca2+ spark and tetracaine-sensitive SR Ca2+ leak measurements.
3.3. Epac/NOS-dependent SR Ca2+ leak is mediated by PI3K and Akt
The data above are consistent with our proposed unified series pathway for β-AR activation of SR Ca2+ leak in two distinct areas (Figure 1). This work indicates that β-AR activation and subsequent generation of cAMP lead to activation of Epac. Furthermore, farther down the pathway, activation of NOS1 results in nitrosylation and activation of CaMKII and generation of increased SR Ca2+ leak via RyR phosphorylation. But the pathway between Epac and NOS1 is still less clear. The remaining experiments aim to clarify this further.
Previous data suggested that ISO-induced Akt activation might be involved in the NOS1 activity that leads to increased SR Ca2+ leak [6]. In order to confirm and extend this idea, we performed Western blots with antibodies against activated pAkt-S473 in myocytes stimulated ± 8-CPT and flash frozen as above for the RyR. To our surprise, initial results revealed no difference in phosphorylation levels ± 8-CPT (Figure 7A). Noticing that the baseline Akt phosphorylation was rather high in control, we pretreated the myocytes with the phosphoinositide 3-kinase (PI3K) inhibitor LY924002. This reduced baseline Akt phosphorylation levels, and revealed an enhancement of Akt phosphorylation upon application of 8-CPT (Figure 7B & C, 100.0±7.6 vs. 373.2±63.4, n=3, p<0.05, t-test).
Figure 7. Epac activation by 8-CPT and Akt phosphorylation.
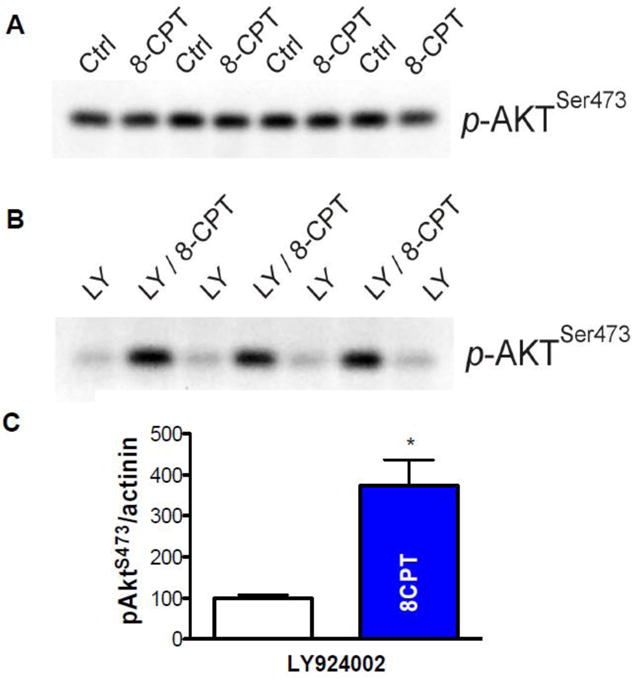
A. 8-CPT does not appear to increase phospho-473 Akt (p-AktSer473). B. After 10 μM LY924002 pretreatment to decrease background phosphorylation, 8-CPT causes demonstrative phosphorylation of Akt. C. Mean data from B (P<0.05, t-test, n=3).
It is evident from the experiments above that inhibition of PI3K strongly influenced global Akt phosphorylation. However, the requirement for local PI3K stimulation in the Epac-dependent activation of SR Ca2+ leak is unknown. To test this, we performed Ca2+ spark experiments with the PI3K inhibitor LY294002 (Figure 3D). LY294002 inhibited the ability of 8-CPT to promote Ca2+ spark frequency. In contrast LY294002 alone had no effect on Ca2+ sparks. The data suggest that the activation of Akt, leading to increased SR Ca2+ leak, is dependent upon PI3K activity. Considering these Ca2+ spark results (Fig 3D) together with the need to inhibit global PI3K activity to detect the 8-CPT-induced effect on Akt phosphorylation might mean that local Akt near NOS1, CaMKII and RyR may be a more important readout, than is global Akt. In conclusion, Epac2 activation of NOS1 upon β-AR appears to be mediated by Akt activation via PI3K (Fig 1).
4. Discussion
Prior work had demonstrated two apparently independent parallel pathways by which cardiac β-ARs activate CaMKII-dependent diastolic SR Ca2+ leak involving Epac2 (Fig 1, blue) [10–18] and NOS1 (Fig 1, red) [4–9]. Here, we have demonstrated that these are part of a single series signaling cascade involving both Epac and NOS (Fig 1). Key novel findings here demonstrate such a link. Indeed, direct Epac activation by 8-CPT: 1) activates Ca2+ sparks, an effect prevented by inhibition of NOS or PI3K (as seen for ISO), 2) increases tetracaine-sensitive SR Ca2+ leak in rabbit (overturning prior conclusions [6]) and again this was NOS-dependent, 3) activates myocyte CaMKII in a NOS- and PI3K-dependent manner (as seen for ISO [6]), and 4) promotes NOS-dependent RyR S2815 phosphorylation. We conclude that β1-AR activation triggers a series cascade via cAMP-Epac2-PI3K-Akt-NOS1-CaMKIIδ to cause phosphorylation of RyR at S2815 to increase pathological diastolic SR Ca2+ leak.
4.1. Epac activation mimics β-AR-induced SR Ca2+ Leak in rabbit and mouse
We had previously reported that direct Epac activation by 8-CPT failed to activate SR Ca2+ leak in rabbit ventricular myocytes using the tetracaine-sensitive SR Ca2+ leak method, and inferred that the Epac2- and NOS1-dependent pathways were independent in signaling from β-AR to RyR2 [6]. However, this was at odds with much of our prior mouse and rat work, where 8-CPT reliably increased SR Ca2+ leak assessed as Ca2+ sparks [14–17]. That included a study in which mice with genetic knockout of Epac1, Epac2 and CaMKIIδ, as well as knock-in of mutant RyR2 (S2814A and S2814D) were used for molecular dissection of these pathway components [14]. This left several possible explanations. First, the Epac-dependence seen in mouse and rat might be absent in rabbit. Second, the Epac-dependence might only be evident when SR Ca2+ leak is measured using Ca2+ sparks and not by total tetracaine-sensitive SR Ca2+ leak measurements. That is, Epac might promote more spark-mediated leak, but equally reduce spark-independent leak [35–37]). Third, there might be a technical problem. Working together we realized that 8-CPT storage conditions are important for efficacy, which sharply declines in aqueous solution at 23°C during an experimental day [14–17]. Here, we repeated the prior rabbit ventricular myocyte 8-CPT experiments [6], and found that freshly prepared 8-CPT induced a robust increase in SR Ca2+ leak that was prevented by NOS inhibition (Fig 4). These new rabbit results mesh well with our mouse data where the 8-CPT-induced Ca2+ spark enhancement was prevented by NOS inhibition (Fig 3). So the prior negative 8-CPT result [6] was likely a now-resolved technical issue, and we infer that this Epac-NOS-CaMKII-RyR pathway is likely universal in mammals (at least rabbit, mouse and rat).
Our novel data with the direct CaMKII activity reporter in rabbit myocytes shows that both ISO and 8-CPT similarly activate CaMKII and that that depends on both NOS and PI3K activity. This agrees with recent work demonstrating that CaMKII can be activated by direct S-nitrosylation of Cys-290 in the CaMKIIδ regulatory domain [7, 25]. This S-nitrosylation causes CaMKII to be trapped in the autonomous open (active) conformation, which can also expose the neighboring autophosphorylation site (T287). This may explain the previously reported 8-CPT-induced CaMKII autophosphorylation [15]. That is, the data in Fig 5B suggests that without the 8-CPT-induced S-nitrosylation of CaMKII, autophosphorylation does not occur (because that itself would strongly promote the active open conformation [31]). Thus S-nitrosylation may promote autophosphorylation which could further prolong CaMKII activation. Taken together, the data support the idea that Epac and NOS are in the same pathway, and that NOS is downstream of Epac2 (Fig 1).
4.2. Local Nature of the β -AR-induced SR Ca2+ Leak
Our prior work leading to NOS involvement in this pathway [4–6], indicated that bulk activation of adenylyl cyclase by forskolin did not mimic the ISO-induced SR Ca2+ leak. We interpreted this as evidence for a potentially cAMP-independent pathway to CaMKII activation [24]. The demonstrable involvement of Epac (which is cAMP-dependent) has caused us to re-interpret this observation. Forskolin certainly increases global [cAMP], but increasing evidence shows that β-AR can induce discrete local cAMP microenvironments relatively independent of global cAMP concentration [38]. Our findings that Epac2, which mediates the 8-CPT effect [14], is highly localized, along with RyR, along T-tubules [16], is consistent with this interpretation. That is, we hypothesize very local β-AR, Epac, NOS1 and CaMKIIδ signaling to RyR2 (where all 5 proteins are known to be concentrated). We think this is a plausible hypothesis, but details may require further testing (e.g. with targeted fluorescence reporters).
Another aspect of our present results that is consistent with highly localized signaling, is the PI3K-Akt results. Prior data [6] indicated that β-AR activation of SR Ca2+ leak was Akt-dependent. So, we expected that Epac activation would stimulate Akt. While we did find that 8-CPT increased Akt phosphorylation at S473 this was demonstrable only after inhibiting basal global Akt phosphorylation (Figure 7). This might be because the local pool of Akt participating in this pathway is a small (local) fraction of overall myocyte Akt. While precise details of the PI3K- and Akt-dependence of NOS1 activation merit further study, both kinases seem to be involved [6].
4.3. Remaining pathway gaps in the series from β-AR to RyR
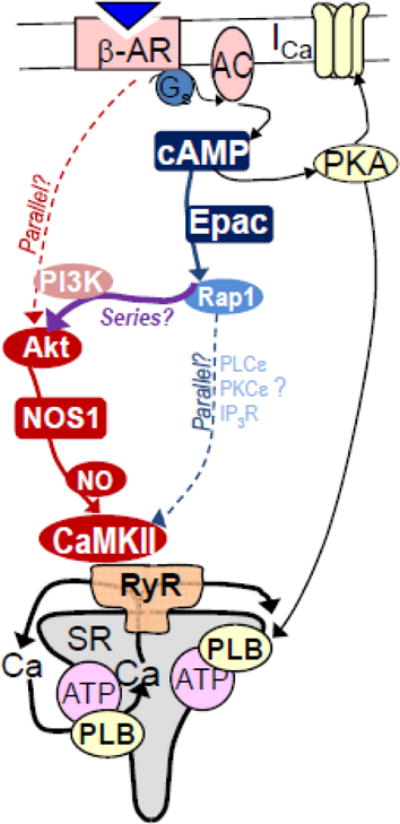
This series arrangement is now molecularly well-defined at the top (β1-AR, cAMP, Epac2) and bottom (Akt-NOS1-CaMKII-RyR) ends (Fig 1). However, some of the implicated intermediaries connecting Epac2 to Akt are less completely resolved molecularly. While Rap1 and PI3K seem critical at exactly this point, the involvement of other previously implicated components downstream of Epac (PLCε, PKCε and maybe InsP3 receptors [12, 13, 17]) is less clear. There is also precedent for Epac-induced activation of PI3K in the regulation of Kv potassium channels in pancreatic β-cells Epac [39].
4.4. cAMP signaling via PKA to inotropy/lusitropy vs. via CaMKII to SR Ca2+ leak
The effects of β-AR to enhance Ca2+ transient amplitude and decline kinetics is mediated largely by PKA-dependent phosphorylation of L-type Ca2+ channels and phospholamban. This is undisputed and causes increased SR Ca2+ load, release and faster relaxation. PKA-dependent effects on myofilament proteins (troponin I, myosin-binding protein C and titin) the Na+/K+-ATPase and other ion channels also modulate the integrated β-AR effects on contractility and diastolic function [2]. An important distinction is that these well characterized cAMP effects via PKA operate in parallel to the cAMP-Epac-CaMKII pathway that we describe here. CaMKII phosphorylates many of the same protein targets as PKA (often at different sites). While cAMP-PKA has stronger functional effects than CaMKII on Ca2+ current and SR Ca-ATPase, the opposite is true for RyR2 (where CaMKII effects are much more functionally robust [40, 41]).
4.5 SR Ca2+ Leak as a Potential Therapeutic Target
Since elevated diastolic SR Ca2+ leak in HF (and other pathologies) can contribute to reduced systolic and diastolic function and also arrhythmogenesis, it is a potential therapeutic target [42]. We have shown that the enhanced SR Ca2+ leak in HF can be reversed by acute CaMKII inhibition but not PKA inhibition [3]. So inhibition of this Epac-dependent pathway to CaMKII-RyR2 may allow inhibition of β1-AR-induced SR Ca2+ leak, while preserving the positive inotropic and lusitropic effects of PKA-dependent phosphorylation [43]. In principle, any of the identified components from Epac2 to RyR2 itself could be targets to suppress pathological SR Ca2+ leak. Critically, because this pathway is distinct from the PKA branch leading to increased inotropy and lusitropy, this might be done without reducing contractility, diastolic function or stroke volume that may limit β-AR blocker efficacy.
Epac activation has also been reported to increase myofilament Ca2+ sensitivity via CaMKII-dependent phosphorylation of myofilament proteins, resulting in stronger contraction for a given Ca2+ transient [18, 44]. To the extent that this Epac myofilament effect is beneficial in HF, it would need assessment when targeting the Epac induced SR Ca2+ leak therapeutically.
Highlights.
Beta-adrenergic receptors (B-AR) induce arrhythmogenic SR Ca leak in adult rabbit and mouse ventricular myocytes.
Previously proposed parallel pathways involving Epac or NOS signals are actually in series.
This SR Ca leak is via a B1AR-cAMP-Epac2-PI3K-Akt-NOS1-CaMKII -RyR2 phosphorylation cascade.
This parallels B-AR/cAMP inotropic and lusitropic PKA effects on Ca current and SR Ca uptake.
Acknowledgments
We thank Kayvon Jabbari for his technical assistance. This work was supported by grants from the National Institutes of Health (R01-HL030077 and P01-HL080101 to DMB) and American Heart Association (14GRNT20380907 to TRS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.M, Writing Group. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. American Heart Association Statistics, S Stroke Statistics, Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Negroni JA, Morotti S, Lascano EC, Gomes AV, Grandi E, Puglisi JL, Bers DM. Beta-Adrenergic Effects on Cardiac Myofilaments and Contraction in an Integrated Rabbit Ventricular Myocyte Model. J Mol Cell Cardiol. 2015;81:162–175. doi: 10.1016/j.yjmcc.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97(12):1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 4.Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J Mol Cell Cardiol. 2010;49(1):25–32. doi: 10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curran J, Hinton MJ, RÌos E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100(3):391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 6.Curran J, Tang L, Roof SR, Velmurugan S, Millard A, Shonts S, Wang H, Santiago D, Ahmad U, Perryman M, Bers DM, Mohler PJ, Ziolo MT, Shannon TR. Nitric oxide-dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PLoS One. 2014;9(2):e87495. doi: 10.1371/journal.pone.0087495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erickson JR, Nichols CB, Uchinoumi H, Stein ML, Bossuyt J, Bers DM. S-Nitrosylation induces both autonomous activation and inhibition of calciumcalmodulin dependent protein kinase II delta. J Biol Chem. 2015 doi: 10.1074/jbc.M115.650234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gutierrez DA, Fernandez-Tenorio M, Ogrodnik J, Niggli E. NO-dependent CaMKII activation during beta-adrenergic stimulation of cardiac muscle. Cardiovasc Res. 2013;100(3):392–401. doi: 10.1093/cvr/cvt201. [DOI] [PubMed] [Google Scholar]
- 9.Jian Z, Han H, Zhang T, Puglisi J, Izu LT, Shaw JA, Onofiok E, Erickson JR, Chen YJ, Horvath B, Shimkunas R, Xiao W, Li Y, Pan T, Chan J, Banyasz T, Tardiff JC, Chiamvimonvat N, Bers DM, Lam KS, Chen-Izu Y. Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Science Signal. 2014;7(317):ra27. doi: 10.1126/scisignal.2005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hothi SS, Gurung IS, Heathcote JC, Zhang Y, Booth SW, Skepper JN, Grace AA, Huang CL. Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflugers Archiv: Euro J Physiol. 2008;457(2):253–270. doi: 10.1007/s00424-008-0508-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metrich M, Lucas A, Gastineau M, Samuel JL, Heymes C, Morel E, Lezoualc’h F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ Res. 2008;102(8):959–965. doi: 10.1161/CIRCRESAHA.107.164947. [DOI] [PubMed] [Google Scholar]
- 12.Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and Phospholipase C{epsilon} Regulate Ca2+ Release in the Heart by Activation of Protein Kinase C{epsilon} and Calcium-Calmodulin Kinase II. J Biol Chem. 2009;284(3):1514–22. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac-mediated activation of phospholipase C(epsilon) plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem. 2007;282(8):5488–5495. doi: 10.1074/jbc.M608495200. [DOI] [PubMed] [Google Scholar]
- 14.Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J, Bers DM. Epac2 mediates cardiac beta1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation. 2013;127(8):913–22. doi: 10.1161/CIRCULATIONAHA.12.148619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pereira L, Métrich M, Fernández-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Bénitah J-P, Lezoualc’h F, Gómez AM. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol. 2007;583(Pt 2):685–694. doi: 10.1113/jphysiol.2007.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pereira L, Rehmann H, Lao DH, Erickson JR, Bossuyt J, Chen J, Bers DM. Novel Epac fluorescent ligand reveals distinct Epac1 vs. Epac2 distribution and function in cardiomyocytes. Proc Natl Acad Sci U S A. 2015;112(13):3991–3996. doi: 10.1073/pnas.1416163112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pereira L, Ruiz-Hurtado G, Morel E, Laurent AC, Metrich M, Dominguez-Rodriguez A, Lauton-Santos S, Lucas A, Benitah JP, Bers DM, Lezoualc’h F, Gomez AM. Epac enhances excitation-transcription coupling in cardiac myocytes. J Mol Cell Cardiol. 2012;52(1):283–291. doi: 10.1016/j.yjmcc.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz-Hurtado G, Dominguez-Rodriguez A, Pereira L, Fernandez-Velasco M, Cassan C, Lezoualc’h F, Benitah JP, Gomez AM. Sustained Epac activation induces calmodulin dependent positive inotropic effect in adult cardiomyocytes. J Mol Cell Cardiol. 2012;53(5):617–625. doi: 10.1016/j.yjmcc.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51(4):468–73. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mustroph J, Neef S, Maier LS. CaMKII as a target for arrhythmia suppression. Pharmacol Ther. 2016 doi: 10.1016/j.pharmthera.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120(12):4375–4387. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88(11):1159–67. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 23.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local Œ ≤-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res. 2012;110(11):1454–1464. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mangmool S, Shukla AK, Rockman HA. beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J Cell Biol. 2010;189(3):573–587. doi: 10.1083/jcb.200911047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coultrap SJ, Bayer KU. Nitric oxide induces Ca2+-independent activity of the Ca2+/calmodulin-dependent protein kinase II (CaMKII) J Biol Chem. 2014;289(28):19458–19465. doi: 10.1074/jbc.M114.558254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uchinoumi H, Yang Y, Oda T, Li N, Alsina KM, Puglisi JL, Chen-Izu Y, Cornea RL, Wehrens XH, Bers DM. CaMKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J Mol Cell Cardiol. 2016;98:62–72. doi: 10.1016/j.yjmcc.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Ce ll Physiol. 2007;293(3):C1073–81. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- 28.Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4(11):901–6. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- 29.Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E. Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods. 2008;5(4):277–8. doi: 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- 30.Bers DM, Bassani JW, Bassani RA. Competition and redistribution among calcium transport systems in rabbit cardiac myocytes. Cardiovasc Res. 1993;27(10):1772–1777. doi: 10.1093/cvr/27.10.1772. [DOI] [PubMed] [Google Scholar]
- 31.Erickson JR, Patel R, Ferguson A, Bossuyt J, Bers DM. Fluorescence resonance energy transfer -based sensor Camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ Res. 2011;109(7):729–38. doi: 10.1161/CIRCRESAHA.111.247148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91(7):594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 33.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93(7):592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 34.Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266(17):11144–52. [PubMed] [Google Scholar]
- 35.Zima AV, Bovo E, Bers DM, Blatter LA. Ca(2)+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588(Pt 23):4743–57. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santiago DJ, Curran JW, Bers DM, Lederer WJ, Stern MD, Rios E, Shannon TR. Ca sparks do not explain all ryanodine receptor-mediated SR Ca leak in mouse ventricular myocytes. Biophys J. 2010;98(10):2111–20. doi: 10.1016/j.bpj.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol. 2014;76:107–27. doi: 10.1146/annurev-physiol-020911-153308. [DOI] [PubMed] [Google Scholar]
- 38.Liu S, Li Y, Kim S, Fu Q, Parikh D, Sridhar B, Shi Q, Zhang X, Guan Y, Chen X, Xiang YK. Phosphodiesterases coordinate cAMP propagation induced by two stimulatory G protein-coupled receptors in hearts. Proc Natl Acad Sci U S A. 2012;109(17):6578–83. doi: 10.1073/pnas.1117862109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Wang H, Guo Q, Li X, Gao J, Liu Y, Yang C, Niu L, Yang J. PI3K is involved in P2Y receptor-regulated cAMP/Epac/Kv channel signaling pathway in pancreatic beta cells. Biochem Biophys Res Commun. 2015;465(4):714–8. doi: 10.1016/j.bbrc.2015.08.057. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90(3):309–16. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 41.Guo T, Zhang T, Mestril R, Bers DM. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99(4):398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 42.Bers DM. Stabilizing ryanodine receptor gating quiets arrhythmogenic events in human heart failure and atrial fibrillation. Heart Rhythm. 2016 doi: 10.1016/j.hrthm.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Bers DM. Beyond beta blockers. Nat Med. 2005;11(4):379–80. doi: 10.1038/nm0405-379. [DOI] [PubMed] [Google Scholar]
- 44.Kaur S, Kong CH, Cannell MB, Ward ML. Depotentiation of intact rat cardiac muscle unmasks an Epac-dependent increase in myofilament Ca(2+) sensitivity. Clin Exp Pharmacol Physiol. 2016;43(1):88–94. doi: 10.1111/1440-1681.12504. [DOI] [PubMed] [Google Scholar]