

Abstract
Pyrroloquinazoline is a privileged chemical scaffold with diverse biological activities. We recently described a series of N-3 acylated 1,3-diaminopyrroloquinazolines with potent anticancer activities. The N-1 primary amino group in 1,3-diaminopyrroloquinazoline is critical for its inhibitory activity against dihydrofolate reductase (DHFR). In order to design out this unnecessary DHFR inhibition activity and further expand the chemical space associated with pyrroloquinazoline, we removed this N-1 primary amino group. In this report, we describe our design and synthesis of a series of N-3 acylated monoaminopyrroloquinazolines. Biological evaluation of these compounds identified a naphthamide 4a as a potent anticancer agent (GI50 = 88-200 nM), suggesting that removing the N-1 primary amino group in 1,3-diaminopyrroloquinazoline is a useful chemical modification that can be introduced to improve the anticancer activity.
Keywords: anticancer, privileged scaffold, pyrroloquinazoline, reduction
Graphical abstract
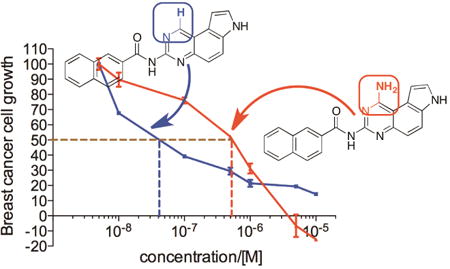
Privileged chemical scaffolds possess specific biological activities against multiple disparate targets depending on the unique combination of substitution pattern on the scaffold.1 These structures are great starting points to discover small molecule ligands to different classes of biomolecules including proteins and nucleic acids.1,2 Perhaps the most prominent member among the privileged scaffolds is benzodiazepine which has been shown to present numerous biological activities including γ-aminobutyric acid (GABA) receptor modulation,3,4 cholecystokinin (CCK) receptor modulation5-7 and apoptosis-inducing activity.8,9 In medicinal chemistry, structure-activity relationship (SAR) studies on privileged structures are generally highly productive with clearly discernible SAR patterns.1,10
We recently became interested in privileged pyrroloquinazoline scaffold to develop potential anticancer agents.2,10 7H-Pyrrolo[3,2-f]quinazoline-1,3-diamine (1, Figure 1) was originally synthesized as a dihydrofolate reductase (DHFR) inhibitor in the 1970s.11 Over the ensuing four decades, derivatives of 1 have been shown to display additional biological activities by inhibiting diverse targets including G-protein coupled protease-activated receptors (PARs),12 protein tyrosine phosphatase 1B (PTP1B)13 and serum paraoxonase (PON).2,14 During this period, the chemical space associated with 1 was heavily investigated, most of which focused on different alkyl groups of varying hydrophobicity at N-7.2 Recently, we further expanded the chemical space associated with 1 through developing a unique suite of methods to regioselectively mono-N-acylate the three nucleophilic nitrogens in 1 (i.e. N-1, N-3 and N-7) (Figure 1).10 From this focused library of mono-N-acylated 1,3-diaminopyrroloquinazolines, we found that compounds 2 with an acyl group at N-3 are generally more potent than other acylated counterparts as antiproliferative agents in breast cancer cells.10 Among these, compound 3 was the most potent inhibitor of MDA-MB-231 and MDA-MB-468 cell growth with sub-micromolar or low micromolar GI50,10 a concentration required to inhibit 50% of the cell growth.
Figure 1.

Chemical structures of 1,3-diaminopyrroloquinazolines 1-3 and acylated monoaminopyrroloquinazolines 4.
In our previous published report, we showed that compound 3 did not inhibit human DHFR although the original compound 1 did so potently.10 This difference was ascribed to the presence of a bulky naphthoyl group in 3, which could not be tolerated in the active site of DHFR.10 Upon examination of reported DHFR inhibitors,15 virtually all of them contain the pharmacophore of 2,4-diaminopyrmidine moiety and the amino group highlighted in 1 (Figure 1) is always a primary amino group. This primary amino group can form two hydrogen bonds with DHFR through the N-H functionality as a hydrogen bond donor.16 Along this line of analysis, replacement of this amino group with a hydrogen atom would be expected to generate compounds devoid of DHFR inhibitory activity. Consistent with this hypothesis, replacement of 4-amino group in 2,4-diamino-6,7-diphenylpteridine with a hydrogen atom resulted in complete loss of DHFR inhibition.17 Furthermore, this structural change is also expected to bring significant changes to the electrostatic potential (ESP) surfaces. As models to test this hypothesis, we optimized the structures of acetamides 2b and 4b (Figure 1, R = Me) at the HF/6-31g(d,p) level of theory in Jaguar in Schrödinger modeling package. Then the ESP surfaces were calculated from their respective electron densities (Figure 2).10 It is obvious that both the sterics and electronics around position 1 of the pyrroloquinazoline nucleus are significantly different between 2b and 4b (Figure 2), suggesting that this structural change will further enhance the diversity of the chemical space belonging to pyrroloquinazoline.2 In this report, we describe the synthesis and anticancer potential of a series of such compounds 4.
Figure 2.
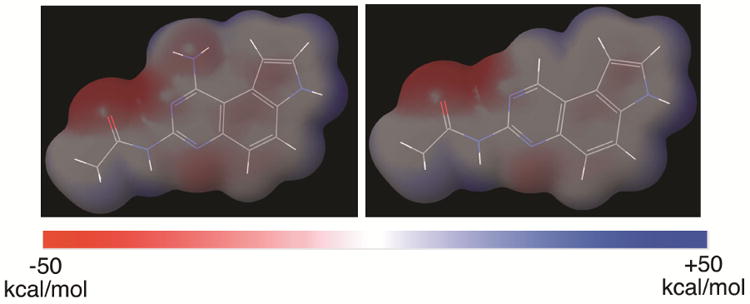
The electrostatic potential (ESP) surface maps of compounds 2b and 4b. The structures were optimized at HF/6-31g(d,p) level of theory implemented in Jaguar (Schrödinger) and the ESP was calculated from mapping the electron density data computed at the HF/6-31g(d,p) level of theory. The surfaces were normalized from −50 kcal/mol to +50 kcal/mol.
Our initial strategy to prepare 4 was to couple mono-amino compound 7 with appropriate anhydrides or NHS esters (Scheme 1). Therefore, the previously reported compound 510 was chlorinated18 with POCl3 to generate 6. Chloride 6 was not stable towards extensive workup and it was thus used directly without further purification for reduction by NaBH4 in the presence of PdCl2(dppf)•DCM19 to provide monoamine 7 in 16% yield. Direct coupling of 7 with NHS ester 8 at an elevated temperature (120 °C) yielded 4a in 11% yield. Attempts to improve this coupling yield at lower or higher temperatures were unfruitful. While this route could provide desired compound 4, the low yields in both the reduction and coupling steps prompted us to investigate an alternative route to prepare 4.
Scheme 1.

Initial synthesis of 4a. Reagents and conditions: a. 1) POCl3, 90°C, overnight; 2) PdCl2(dppf)•DCM, NaBH4, TMEDA, THF, r.t., 16 h; b. DIPEA, DMF, 120 °C.
A revised synthetic route to 4 is presented in Scheme 2. Intermediate 5 was first coupled to an NHS ester (for 9a) or a homoanhydride (for 9b-9f) as described before to give amides 9.10 The hydroxyl group in 9 was then converted into a hydrogen atom by a sequence of chlorination with POCl3 followed by reduction using NaBH4 catalyzed by PdCl2(dppf)•DCM to provide desired compounds 4 in reasonable yields (20-37%).20 While chlorination of the aliphatic amides 9b-9f was carried out successfully at slightly elevated temperatures (70-90 °C), we found that it was necessary to perform chlorination of aromatic amide 9a at lower temperature (room temperature) to obtain 4a.
Scheme 2.

Synthesis of compounds 4 by coupling and reduction. Reagents and conditions: a. anhydrides or NHS ester 8, DMF, 110 °C, 3 h; b. 1) POCl3, DIPEA, 1,4-dioxane, 70-90 °C, overnight (for 4b-4f) or r.t., 4 h (for 4a); 2) PdCl2(dppf)•DCM, NaBH4, TMEDA, THF, r.t., 16-48 h. cLogP values for 4a-4f were calculated from ChemBioDraw Ultra 12.0.
With the newly synthesized compounds 4a-4f in hand, we evaluated their potential activity in inhibiting breast cancer cell growth and compared them with the corresponding amines 2 (Table 1 and Figure 1). Two different breast cancer cell lines MDA-MB-231 and MDA-MB-468 were used for this purpose. These two cell lines represent triple-negative breast cancer (TNBC) cells and this subtype of breast cancer has the worst prognosis among the different breast cancer subtypes.21 The cells were incubated with different concentrations of the compounds for 72 h. Then the number of remaining viable cells was quantified by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent as reported previously.22-24 The drug concentrations required for 50% growth inhibition (GI50) were calculated from the corresponding dose-response curves and are presented in Table 1. The results shown in Table 1 demonstrated that the aliphatic amides 4b-4f are in general not potent inhibitors of either cell lines with the exception of 4d, which presented single-digit μM activities in both of the cell lines (GI50 = 3.46 ± 1.46 and 2.02 ± 1.10 μM in MDA-MB-231 and MDA-MB-468, respectively) and was ∼10-fold more potent than 2d. The overall weak activity for the aliphatic amides is consistent with the weak activity observed in 2b-2e (Table 1).10 As reported previously,10 compound 2a, being a naphthamide, was the most potent congener in this focused library of compounds. Strikingly, we found that compound 4a was even more potent than 2a in both of the cell lines (Table 1 and Figure 3). The GI50 for 4a was 200 ± 170 nM in MDA-MB-231 cells and 80 ± 26 nM in MDA-MB-468 cells, which represent 5-8-fold improvement over 2a (Figure 3), suggesting that aromatic amides of monoaminopyrroloquinazoline may be more favorable than aromatic amides of 1,3-diaminopyrroloquinazoline in delivering potent antitumor agents. In this series of amides of monoaminopyrroloquinazoline, cLogP or cell-permeability alone is insufficient to determine the antiproliferative activities. For example, while the most potent compound 4a has the highest cLogP value (3.50) (Scheme 2), compound 4f (cLogP = 2.08) was much less potent than 4c (cLogP = 1.37) or 4d (cLogP = 1.90).
Table 1.
Antiproliferative activities (GI50, μM) of compounds 2 and 4 in breast cancer cells.a
| -R | MDA-MB-231 | MDA-MB-468 | ||
|---|---|---|---|---|
|
| ||||
| Series 2b | Series 4 | Series 2b | Series 4 | |
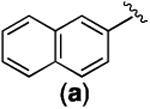
|
1.60 ± 0.51 | 0.20 ± 0.17 | 0.44 ± 0.14 | 0.088 ± 0.026 |
| Me (b) |
27.17 ± 11.40 | >100c | 53.54 ± 29.54 | >100c |
| Et (c) |
21.43 ± 9.86 | 72.84 ± 5.64 | 24.41 ± 3.33 | 28.30 ± 7.05 |
|
n-Pr (d) |
25.52 ± 9.93 | 3.46 ± 1.46 | 27.37 ± 4.52 | 2.02 ± 1.10 |
|
i-Pr (e) |
39.66 ± 22.46 | >100c | 29.80 ± 8.41 | >100c |
|
t-Bu (f) |
N/Ad | >100c | N/Ad | >100c |
The antiproliferative activities of the compounds were assessed using the MTT assay. The cells were incubated with different concentrations of the drugs for 72 h. Then the number of viable cells was quantified by the MTT reagent. The GI50s, presented as mean ± SD (standard deviation), were calculated from the corresponding dose-response curves in Prism 5.0 using non-linear regression analysis. The SD was calculated from at least two independent measurements.
The GI50s for series 2 were from ref10.
The GI50 was not reached at the highest tested concentration (100 μM).
This compound was not assessed.
Figure 3.
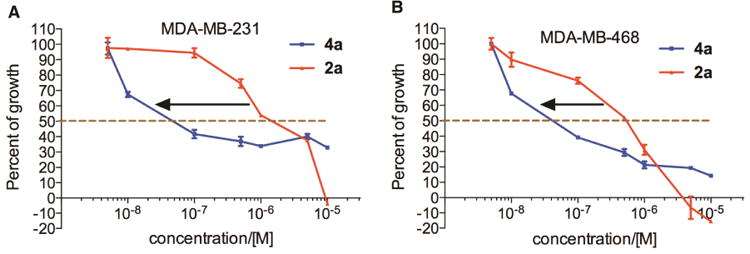
Dose-dependent antiproliferative activity of 2a and 4a in MDA-MB-231 (A) and MDA-MB-468 cells (B). The cells were incubated with increasing concentrations of compounds 2a and 4a for 72 h. Then the number of viable cells was quantified spectrophotometrically by the MTT reagent.
In conclusion, a series of acylated monoaminopyrroloquinazolines was designed to remove a pharmacophore typically present in DHFR inhibitors. These compounds were synthesized in a facile and generally applicable strategy of coupling and reduction. The facile synthesis of these compounds allowed us to have access to further expanded chemical space associated with pyrroloquinazoline.10 Evaluation of the antiproliferative activities of these compounds in two breast cancer cell lines revealed that aliphatic amides possess relative weak antiproliferative potential. On the other hand, the naphthamide 4a represents a potent inhibitor of breast cancer cell growth. These results suggest that aromatic amides of monoaminopyrroloquinazoline can be a new class of potential cancer therapeutics.
Acknowledgments
This work was made possible by the generous financial supports provided by Oregon Health & Science University (OHSU) School of Medicine, OHSU Technology Transfer and Business Development, Oregon Clinical and Translational Research Institute, Oregon Medical Research Foundation, Lloyd Fund and National Institutes of Health (R01GM122820).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Welsch ME, Snyder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chao B, Li BX, Xiao X. MedChemComm. 2015;6:510–520. doi: 10.1039/C4MD00485J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sternbach LH. J Med Chem. 1979;22:1–7. doi: 10.1021/jm00187a001. [DOI] [PubMed] [Google Scholar]
- 4.Sigel E. Curr Top Med Chem. 2002;2:833–839. doi: 10.2174/1568026023393444. [DOI] [PubMed] [Google Scholar]
- 5.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RSL, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J. J Med Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 6.Desai AJ, Lam PC, Orry A, Abagyan R, Christopoulos A, Sexton PM, Miller LJ. J Med Chem. 2015;58:9562–9577. doi: 10.1021/acs.jmedchem.5b01110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang RS, Lotti VJ. Proc Natl Acad Sci U S A. 1986;83:4923–4926. doi: 10.1073/pnas.83.13.4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blatt NB, Bednarski JJ, Warner RE, Leonetti F, Johnson KM, Boitano A, Yung R, Richardson BC, Johnson KJ, Ellman JA, Opipari AW, Jr, Glick GD. J Clin Invest. 2002;110:1123–1132. doi: 10.1172/JCI16029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boitano A, Ellman JA, Glick GD, Opipari AW., Jr Cancer Res. 2003;63:6870–6876. [PubMed] [Google Scholar]
- 10.Chen J, Kassenbrock A, Li BX, Xiao X. Medchemcomm. 2013;4:1275–1282. doi: 10.1039/C3MD00134B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ledig KW. U S., Ed U.S. 1978;US4118561
- 12.Ahn HS, Arik L, Boykow G, Burnett DA, Caplen MA, Czarniecki M, Domalski MS, Foster C, Manna M, Stamford AW, Wu Y. Bioorg Med Chem Lett. 1999;9:2073–2078. doi: 10.1016/s0960-894x(99)00339-x. [DOI] [PubMed] [Google Scholar]
- 13.Cheung AW, Banner B, Bose J, Kim K, Li S, Marcopulos N, Orzechowski L, Sergi JA, Thakkar KC, Wang BB, Yun W, Zwingelstein C, Berthel S, Olivier AR. Bioorg Med Chem Lett. 2012;22:7518–7522. doi: 10.1016/j.bmcl.2012.10.035. [DOI] [PubMed] [Google Scholar]
- 14.Aviram M, Rosenblat M, Bisgaier CL, Newton RS, Primo-Parmo SL, La Du BN. J Clin Invest. 1998;101:1581–1590. doi: 10.1172/JCI1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debnath AK. J Med Chem. 2002;45:41–53. doi: 10.1021/jm010360c. [DOI] [PubMed] [Google Scholar]
- 16.Tosso RD, Andujar SA, Gutierrez L, Angelina E, Rodriguez R, Nogueras M, Baldoni H, Suvire FD, Cobo J, Enriz RD. Journal of chemical information and modeling. 2013;53:2018–2032. doi: 10.1021/ci400178h. [DOI] [PubMed] [Google Scholar]
- 17.McCormack JJ, Jaffe JJ. J Med Chem. 1969;12:662–668. doi: 10.1021/jm00304a023. [DOI] [PubMed] [Google Scholar]
- 18.Sirisoma N, Pervin A, Zhang H, Jiang S, Willardsen JA, Anderson MB, Mather G, Pleiman CM, Kasibhatla S, Tseng B, Drewe J, Cai SX. J Med Chem. 2009;52:2341–2351. doi: 10.1021/jm801315b. [DOI] [PubMed] [Google Scholar]
- 19.Chelucci G, Figus S. Journal of Molecular Catalysis A: Chemical. 2014;393:191–209. [Google Scholar]
- 20.The, characterization data are below. 4a: m.p. >250 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1 H), 11.19 (s, 1 H), 9.89 (s, 1 H), 8.70 (s, 1 H), 7.99-8.13 (m, 5 H), 7.59-7.70 (m, 3 H), 7.54 (d, J = 8.9 Hz, 1 H), 7.26-7.32 (m, 1 H); 13C NMR (100 MHz, DMSO-d6) δ 165.64, 156.85, 153.20, 148.01, 134.49, 132.12, 132.05, 131.79, 129.17, 128.79, 127.97, 127.94, 127.66, 126.79, 126.39, 124.74, 122.24, 121.22, 119.54, 116.66, 101.04. 4b: m.p. >250 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.86 (s, 1 H), 10.52 (s, 1 H), 9.77 (s, 1 H), 8.03 (d, J = 8.9 Hz, 1 H), 7.60 (t, J = 2.7 Hz, 1 H), 7.45 (d, J = 8.9 Hz, 1 H), 7.19-7.26 (m, 1 H), 2.25 (s, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 169.06, 156.88, 152.94, 147.83, 131.81, 126.30, 122.19, 121.26, 119.41, 116.16, 100.85, 24.55. 4c: m.p. 229-230 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.87 (s, 1 H), 10.48 (s, 1 H), 9.77 (s, 1 H), 8.03 (d, J = 8.9 Hz, 1 H), 7.59 (t, J = 2.5 Hz, 1 H), 7.44 (d, J = 8.9 Hz, 1 H), 7.22 (s, 1 H), 2.55 (q, J = 7.5 Hz, 2 H), 1.09 (t, J = 7.5 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 172.38, 156.83, 152.94, 147.86, 131.78, 126.26, 122.14, 121.24, 119.39, 116.13, 100.82, 29.57, 9.41. 4d: m.p. 217-218 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.85 (s, 1 H), 10.49 (s, 1 H), 9.77 (s, 1 H), 8.02 (d, J = 9.0 Hz, 1 H), 7.60 (t, J = 2.7 Hz, 1 H), 7.44 (d, J = 8.9 Hz, 1 H), 7.23 (s, 1 H), 2.49-2.53 (m, 2 H), 1.57-1.69 (m, 2 H), 0.94 (t, J = 7.4 Hz, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 171.44, 156.82, 152.91, 147.86, 131.79, 126.27, 122.13, 121.25, 119.41, 116.17, 100.84, 38.22, 18.31, 13.76. 4e: m.p. 221-222 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.85 (s, 1 H), 10.50 (s, 1 H), 9.77 (s, 1 H), 8.03 (d, J = 9.0 Hz, 1 H), 7.60 (t, J = 2.7 Hz, 1 H), 7.45 (d, J = 8.9 Hz, 1 H), 7.23 (s, 1 H), 2.82-2.98 (m, 1 H), 1.12 (d, J = 6.8 Hz, 6 H); 13C NMR (100 MHz, DMSO-d6) δ 175.13, 156.78, 152.96, 147.90, 131.81, 126.25, 122.11, 121.24, 119.42, 116.24, 100.86, 34.38, 19.42. 4f: m.p. 191-192 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.87 (s, 1 H), 10.04 (s, 1 H), 9.80 (s, 1 H), 8.04 (d, J = 8.9 Hz, 1 H), 7.61 (t, J = 2.7 Hz, 1 H), 7.49 (d, J = 8.9 Hz, 1 H), 7.25 (s, 1 H), 1.26 (s, 9 H); 13C NMR (100 MHz, DMSO-d6) δ 176.05, 156.61, 153.14, 147.96, 131.95, 126.28, 122.08, 121.14, 119.49, 116.48, 100.97, 27.05.
- 21.Kang SP, Martel M, Harris LN. Curr Opin Obstet Gynecol. 2008;20:40–46. doi: 10.1097/GCO.0b013e3282f40de9. [DOI] [PubMed] [Google Scholar]
- 22.Xie F, Li BX, Kassenbrock A, Xue C, Wang X, Qian DZ, Sears RC, Xiao X. J Med Chem. 2015;58:5075–5087. doi: 10.1021/acs.jmedchem.5b00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li BX, Yamanaka K, Xiao X. Bioorg Med Chem. 2012;20:6811–6820. doi: 10.1016/j.bmc.2012.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li BX, Xie F, Fan Q, Barnhart KM, Moore CE, Rheingold AL, Xiao X. ACS Med Chem Lett. 2014;5:1104–1109. doi: 10.1021/ml500330n. [DOI] [PMC free article] [PubMed] [Google Scholar]