

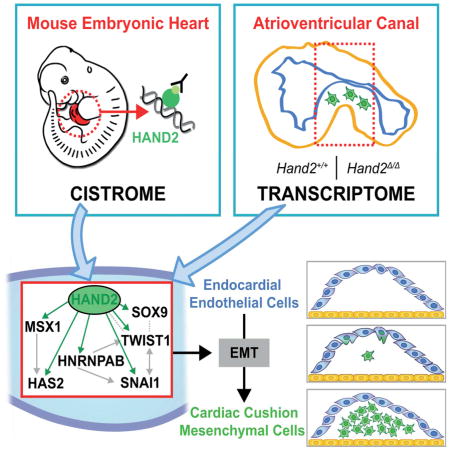
Summary
The HAND2 transcriptional regulator controls cardiac development and we uncover additional essential functions in the endothelial to mesenchymal transition (EMT) underlying cardiac cushion development in the atrioventricular canal (AVC). In Hand2-deficient mouse embryos, the EMT underlying AVC cardiac cushion formation is disrupted and we combined ChIP-Seq of embryonic hearts with transcriptome analysis of wild-type and mutants AVCs to identify the functionally relevant HAND2 target genes. The HAND2 target gene regulatory network (GRN) includes most genes with known functions in EMT processes and AVC cardiac cushion formation. One of these is Snai1, an EMT master regulator whose expression is lost from Hand2-deficient AVCs. Re-expression of Snai1 in mutant AVC explants partially restores this EMT and mesenchymal cell migration. Furthermore, the HAND2-interacting enhancers in the Snai1 genomic landscape are active in embryonic hearts and other Snai1-expressing tissues. These results show that HAND2 directly regulates the molecular cascades initiating AVC cardiac valve development.
eTOC Blurb
Laurent et al. combine ChIP-Seq with transcriptome analysis to identify the HAND2 target gene network that controls the EMT and mesenchymal cell migration during cardiac cushion formation in the atrioventricular canal (AVC). The HAND2 transcriptional targets include Snai1, whose re-expression in Hand2-deficient AVC explants partially restores mesenchymal cell migration.
Introduction
Perturbations affecting cardiac progenitors result in embryonic lethality and severe congenital heart defects, which are a major cause of infant and even adult mortality (Bruneau, 2008). In particular, different types of congenital heart defects are caused by alterations in the progenitors of the second heart field (SHF; reviewed by Kelly, 2012). SHF progenitors migrate into the developing heart tube, where they contribute to the most developing structures including the inflow pole, both atria and ventricles and the outflow tract (OFT). The four heart chambers are formed by rapid proliferative expansion, while the cardiomyocytes in the OFT and atrioventricular canal (AVC) proliferate less and remain undifferentiated (Christoffels et al., 2010; Greulich et al., 2011). The AVC connects the left ventricle to the forming atria, is required for chamber septation and gives rise to the atrioventricular node and mitral and tricuspid valves (Christoffels et al., 2010; Lin et al., 2012). In mouse embryos, development of the AVC valves begins at embryonic day E9.5, when endocardial cells undergo an endothelial to mesenchymal transition (EndMT or EMT) in response to signals from the myocardium. The delaminating endocardial cells migrate into the cardiac jelly and give rise to the cardiac cushion mesenchyme, which are then remodeled into the mature valve structures (MacGrogan et al., 2014). The EMT in the AVC is controlled by BMP2 signaling from the myocardium, which synergizes with myocardial TGFβ2 and endocardial NOTCH signaling to activate downstream effectors that include the Snai1 transcriptional regulator (Luna-Zurita et al., 2010; Ma et al., 2005; Niessen et al., 2008; Timmerman et al., 2004). SNAI1 is a key EMT regulator in embryos and various diseases such as tumor metastasis (reviewed by Nieto, 2011). Its inactivation in the endothelial compartment disrupts the EMT underlying AVC cardiac cushion formation (Wu et al., 2014).
Another transcription factor essential for heart development is HAND2, which also functions in developing branchial arches and limb buds (Srivastava et al., 1997). In the developing heart, HAND2 is expressed in the myocardial compartment of the right ventricle and OFT, the epicardium and valve progenitors in both OFT and AVC (VanDusen and Firulli, 2012; VanDusen et al., 2014b). Consistent with its complex expression pattern, genetic inactivation of Hand2 in mice disrupts development of limb buds, branchial arches, aortic arch arteries and the right ventricle, which causes embryonic lethality (Srivastava et al., 1997). Specific inactivation in developing heart tissues has revealed essential Hand2 functions in the cardiac neural crest cells that contribute to cardiac cushions in the OFT, survival of SHF progenitors, heart chamber trabeculation and epicardial cell differentiation (Barnes et al., 2011; Holler et al., 2010; Tsuchihashi et al., 2011; VanDusen et al., 2014a). Previous studies had also pointed to HAND2 functions in cardiac cushion formation, but the potential essential functions have not been identified (Holler et al., 2010; Liu et al., 2009; VanDusen et al., 2014a). In humans, mutations in HAND2 have been linked to congenital heart malformations that include ventricular septal defects (Shen et al., 2010; Sun et al., 2016).
We have used ChIP-Seq to define the genome-wide interaction profile of endogenous HAND2 chromatin complexes in mouse embryonic hearts. This analysis shows that HAND2 interacts with non-coding regions associated to large number of genes functioning during heart development. Most importantly, our analysis revealed essential HAND2 functions in the EMT underlying AVC cardiac cushion formation, which is disrupted in Hand2-deficient embryos. Combining transcriptome analysis of wild-type and Hand2-deficient AVCs with the HAND2 ChIP-Seq dataset identified the HAND2 target genes that function in AVC cardiac cushion development. In particular, this analysis revealed that the EMT regulator Snai1 is a transcriptional target of HAND2. The failure of endocardial cells to invade the cardiac jelly in Hand2-deficient AVCs is partially restored by re-expressing SNAI1 in mutant explants. Last but not least, we show that the two Snai1-associated enhancers interacting with HAND2 chromatin complexes recapitulate major aspects of Snai1 expression in mouse embryos.
Results
Genomic regions enriched in endogenous HAND2 chromatin complexes identify the range of HAND2 target genes in mouse embryonic hearts
The Hand23xF allele, which encodes a HAND2 protein with a 3xFLAG epitope tag inserted in its N-terminal part (Osterwalder et al., 2014), was used to profile the genomic regions enriched in HAND2-chromatin complexes. Anti-FLAG antibodies were used for chromatin immunoprecipitation, which was followed by massive parallel sequencing (ChIP-Seq). To obtain sufficient chromatin for ChIP-Seq, ~300 hearts per biological replicate were dissected from Hand23xF/3xF mouse embryos at embryonic days E10.25–10.5. Two biological replicates were analyzed and the genome-wide binding profiles using MACS (Zhang et al., 2008) identified 12117 significantly enriched genomic regions. GREAT analysis (McLean et al., 2010) was used to assign these 12117 regions to 7792 neighboring genes, which defines the initial set of putative HAND2 targets (Table S1 and Supplemental Experimental Procedures). Most of the genomic regions enriched in HAND2 chromatin complexes are located ≥10kb away from transcriptional start sites (TSS) and encode evolutionary conserved sequences that overlap the peak summit (Figure 1A, 1B). Functional enrichment was assessed by GREAT using increasingly larger set of peaks (pool of incremental deciles; for details see Supplemental Experimental Procedures). This analysis revealed that terms related to abnormal cardiac morphology and heart development were already enriched when using only the most enriched regions, while terms referring to specific processes such as outflow tract (OFT), right ventricle and atrioventricular canal (AVC) development reached significance using the larger dataset (Figure S1A). In particular, 15 of the 16 most enriched Gene Ontology (GO) terms are relevant to heart development, while the remaining term identifies genes functioning in EMT processes (Figure 1C, see below). De novo motif discovery using HOMER (Heinz et al., 2010) identified the consensus Ebox motif (CATCTG; Dai and Cserjesi, 2002) as the most prevalent among HAND2 peaks (Figure 1D). Other significantly enriched motifs include binding sites for GATA transcription factors (Figure S1B), which are key regulators of heart development (Stefanovic and Christoffels, 2015). Indeed, computational comparison of the HAND2 (Table S1) with a published GATA4 ChIP-Seq dataset (using whole mouse embryonic hearts at E12.5; He et al., 2014) shows that 28.3% of the enriched genomic regions are shared between the two datasets, which is 15-fold higher than expected by chance (data not shown). As development of Hand2-deficient mouse embryos results in lethality by ~E10.5 (Srivastava et al. 1997), we limited our analysis to mutant embryos at E9.0–E9.5. During this early organogenic stage, no aberrant apoptosis occurs in the developing heart in contrast to branchial arches and frontonasal mass (Figure S1C). Therefore, all interactions of HAND2 chromatin complexes with candidate cis-regulatory modules (CRMs) associated to genes of interest were also verified by ChIP-qPCR using embryonic hearts at E9.25–9.5 (Figures 1,4, 6, S3, S6).
Figure 1. ChIP-Seq analysis using the Hand23xF allele identifies the HAND2 cistrome in mouse embryonic hearts.

(A, B) The majority of the genomic regions enriched in HAND2 chromatin complexes from mouse embryonic hearts at E10.5 map ≥10kb away from the closest transcription start site (TSS) and are evolutionarily conserved. (C) The top GO terms associated with HAND2 candidate targets reveal the preferential enrichment of genes functioning in cardiac development. (D) The consensus Ebox motif is most enriched by de novo motif discovery. (E) Selection of VISTA enhancers enriched in HAND2 chromatin complexes. Green intervals indicate the regions with enhancer activity (VISTA enhancer database); blue intervals highlight the regions enriched in HAND2 chromatin complexes (MACS peaks). Distances to the nearest TSS within the TAD are indicated on top. ChIP-Seq profiles of the two biological replicates (E10.5) are shown in light and dark blue, respectively. The H3K27ac ChIP-Seq profile for mouse hearts (E11.5) is shown in grey (Nord et al., 2013). The scheme at the bottom shows the placental mammal conservation (Cons) plot (PhyloP). Representative transgenic LacZ reporter embryos for VISTA enhancers associated to genes functioning in OFT and/or right ventricle development (Gata4, Myocd, Gata6 and Tbx20) are shown below. (F) ChIP-qPCR validation of the ChIP-Seq peaks (panel E) for mouse embryonic hearts at E10.5 (n=3 biological replicates) and E9.25 (n=2; mean ± SD). (G) Expression of the HAND2 targets Gata4, Myocd, Gata6 and Tbx20 in wild-type and Hand2-deficient (Hand2Δ/Δ) embryonic hearts (E9.0–9.5). Scale bars: 100μm. oft: outflow tract, ra: right atrium, rv: right ventricle, lv: left ventricle, avc: atrioventricular canal, la: left atrium. See also Figures S1–S3 and Tables S1–S4.
Figure 4. Transcriptome analysis identifies the transcriptional targets of HAND2 in the AVC.

(A) Heat map of the differentially expressed genes (DEGs) identified by comparing the transcriptomes of wild-type and Hand2-deficient AVCs from mouse embryonic hearts at E9.25–E9.5 (n=4 and n=3 biological replicates were analyzed for mutant and wild-type AVCs, respectively). DEGs are genes whose expression is significantly changed (≥1.5-fold) between wild-type and mutant samples (p<0.05) (B) The box plot shows the number of HAND2 ChIP-Seq peaks in the TADs harboring genes with unchanged, down- or up-regulated expression in mutant AVCs, respectively. The TADs of genes with altered expression encode more HAND2-interacting genomic regions. To account for the different numbers of genes and HAND2 ChIP-Seq peaks per TAD, peak counts were normalized as numbers of peaks per gene for each TAD. (C) GO enrichment analysis for biological processes for the 167 down-regulated and 372 up-regulated genes (in mutant AVCs) that contain HAND2 ChIP-Seq peaks in their TADs. (D) GO analysis to identify the HAND2 target genes with annotated functions in EMT processes and AV cushion morphogenesis. (E) Mouse embryonic hearts isolated at E9.25–E9.5 were used for ChIP-qPCR validation of the most prominent HAND2 ChIP-Seq peaks in the TADs of the genes shown in panel D (n=4 using two biological replicates, p≤0.05). See also Figure S4 and Table S5 and S6.
Figure 6. Re-expression of Snai1 in Hand2-deficient AVC explants partially restores mesenchymal cell migration and regulation of Snai1 expression by enhancers enriched in HAND2 chromatin complexes.

(A) Snai1 expression is lost from the endocardium of Hand2-deficient hearts. (B) Upper panels: Hand2-deficient AVC explants were infected either with GFP (control) or SNAI1-GFP adenovirus to re-express SNAI1Smaples were analyzed 72 hours after infection. All infected cells that migrated into the matrix are marked by GFP expression (green). Smooth muscle actin (SMA, red) was detected to reveal cellular morphology. Lower panel: Quantitation of cell migration in Hand2-deficient AVC explants. The mean ±SD and all individual data points are shown. The observed increase in mesenchymal cells in SNAI1-GFP infected explants is significant (p= 0.0093, Mann-Whitney test). Scale bars: 100μm. (C) Scheme of the mouse Snai1 TAD (red, HiC-data). The HAND2 ChIP-Seq profiles in the heart (this study), Hand2-expressing tissues (eT) and limb buds (Lb(Osterwalder et al., 2014) are shown below together with the H3K27ac profile in developing hearts (E11.5, Nord et al., 2013). The Snai1 TAD boundaries are marked by CTCF-binding regions in opposite orientation (Gomez-Marin et al., 2015). Green bars indicate the HAND2 target CRMs located +14kb, +45kb and +57kb that were analyzed by LacZ reporter assays in transgenic founder embryos. (D) Snai1 transcript distribution in wild-type mouse embryos (E10.5). (E, F) representative transgenic founder embryos (E10.5) show the activity of LacZ reporter constructs encoding the +45kb and +57kb CRMs, respectively. The upper panels depict whole embryos, dissected hearts and forelimb buds. The lower panels show sagittal sections at the level of the heart. The boxed areas indicate the enlargements shown in the right panels. Asterisks: AVC cardiac cushion mesenchyme; as: aortic sac; avc: atrioventricular canal; ba: branchial arch; la: left atria; lv: left ventricle; oft: outflow tract; ra: right atria; rv: right ventricle. Scale bars: 200μm. See also Figure S5 and S6.
To validate the ChIP-Seq dataset as a resource for identifying HAND2 target genes, we determined which of the genes with previously documented alterations in expression associate with HAND2 ChIP-Seq peaks (Table S2 and references therein). This analysis showed that about half of these genes associate with at least one HAND2 ChIP-Seq peak (n=56/114; Table S2, see also Table S1). This suggests that their altered expression in Hand2-deficient hearts could be a consequence of direct transcription regulation by HAND2. Next, we overlapped the HAND2 binding profiles with enhancers active in mouse embryonic hearts that were identified by a large enhancer screen (VISTA Enhancer Browser: https://enhancer.lbl.gov, Visel et al., 2007). This analysis showed that 71 of the 193 VISTA enhancers active in mouse embryonic hearts overlap with HAND2 ChIP-Seq peaks (Figures 1E, 1F and S2, Table S3). These enhancers include CRMs in the genomic landscapes of the Gata4, Gata6, Myocd and Tbx20 transcriptional regulators, which are essential for OFT and/or right ventricle development. Whole mount in situ hybridization (WISH) showed that the expression of Gata4, Myocd and Gata6 is reduced in Hand2-deficient hearts, while the expression of Tbx20 appears unchanged (E9.25–E9.5, Figure 1G). The requirement of Hand2 for outflow tract (OFT) and right ventricle development is also supported by GO analysis as more than half of the genes with annotated functions in OFT and RV development are associated to HAND2 ChIP-Seq peaks (Figure S3A and Table S4). In particular, several ligands of the signaling pathways required for development of these structures are identified as HAND2 target genes (Figure S3B–C). This includes Wnt11, Wnt5a, Bmp4, Tgfβ2 and Fgf10, whose expression is either reduced or lost from the OFT and/or RV of Hand2-deficient mouse embryos (E8.75–9.25, Figure S3C). Collectively, this first analysis shows that a significant fraction of genes functioning in mouse OFT and RV morphogenesis are likely HAND2 target genes (Fig. 1G and Fig. S3). However, not all candidate target genes analyzed are altered in Hand2-deficient hearts, which points either to cis-regulatory redundancy or raises the possibility that the interaction of HAND2 complexes with the candidate CRMs is not essential for the adjacent genes.
HAND2 is a key regulator of the EMT during AVC cardiac cushion formation
Strikingly, the GO analysis identified EMT as one of the key biological processes associated with the HAND2 cistrome (Figure 1C). An essential process during heart valve development is formation of the mesenchymal compartment of the AVC cardiac cushions as endocardial cells undergo an EMT (see Introduction). In wild-type hearts, HAND2 proteins are expressed by endocardial cells in the AVC and the cells undergoing EMT and the delaminating cells forming the cushion mesenchyme continue to express HAND2 (asterisk, Figure 2A and data not shown). Furthermore, histological analysis reveals the complete absence of mesenchymal cells in the AVC cardiac cushions of Hand2-deficient hearts at E9.0–E9.5, which points to disruption of the EMT process (Figure 2B). As the distribution of PECAM and smooth muscle actin (SMA) positive cells is not altered in Hand2-deficient hearts, the endocardial and myocardial compartments of the AVC appear to have formed normally, which underscores the specific nature of the observed cellular defect (Figure 2C, 2D). TWIST1 forms hetero-dimeric transcriptional complexes with HAND2 and regulates cardiac cushion development (Firulli et al., 2005; VanDusen and Firulli, 2012). Therefore, the distribution of both proteins was comparatively analyzed in developing AVC cardiac cushions (Figure 2E). In wild-type embryos, HAND2 and TWIST1 are co-expressed by the delaminating mesenchymal cells that form the AVC cardiac cushions in wild-type hearts (left panels, Figure 2E). In contrast, these TWIST1-positive cells are absent in Hand2-deficient hearts (right panel, Figure 2E). Together, these results point to complete disruption of AVC cardiac cushion formation in Hand2-deficient mouse embryos.
Figure 2. AVC cardiac cushion agenesis in Hand2-deficient mouse embryos.

(A) Distribution of the endogenous HAND23xF protein (using anti-FLAG antibodies, green fluorescence) during in mouse embryonic hearts at E9.5 and E10.5. Representative sagittal sections are shown. White arrows: endocardium; white arrowheads: myocardium; white asterisks: HAND2 expressing AVC cardiac cushion mesenchymal cells; black arrows: epicardium. Scale bars: 50μm. (B) Hematoxylin/Eosin staining reveals the absence of delaminating endocardial cells with mesenchymal characteristics (white asterisks) in the AVC of Hand2-deficient mouse embryos. White arrow: endocardium. Scale bars: 100μm. (C, D) Detection of the platelet endothelial cell adhesion molecule (PECAM) in the endocardium and smooth muscle actin (SMA) in the AVC myocardium of wild-type and Hand2-deficient embryos. White arrow: endocardium, white arrowhead: myocardium (panels A to D). (E) Colocalization of the HAND23xF (green fluorescence) with TWIST1 transcriptional regulators (red fluorescence) in the AVC of wild-type (Hand23xF/3xF) and Hand2-deficient (Hand2Δ/Δ) embryos at E9.5. Colocalization is detected in delaminating mesenchymal cells (indicated by arrows, left panel), which are missing from the mutant AVC (right panel). Scale bars: 100μm. avc: atrioventricular canal, ba: branchial arches, la: left atrium, lv: left ventricle, oft: outflow tract, peo: proepicardial organ.
To study this processes further, AVCs were dissected from wild-type and mutant hearts at E9.5 and cultured on collagen matrices for 72 hours (Figure 3A, Camenisch et al., 2000). Then, the mesenchymal cells that had migrated from the explant into the matrix were quantitated (Figure 3B): on average 330±40 cells colonize the matrix in wild-type AVC explants (n=10), while ~10-fold fewer mesenchymal cells (36±3) are detected in AVC explants isolated from Hand2-deficient embryos (n=7). In particular, wild-type endocardial cells in proximity of the AVC explant retain their cobblestone-like morphology and a cortical actin ring (Figure 3A, panel i), while cells that migrated further develop actin stress fibers and long filopodia characteristic of mesenchymal cells (Figure 3A, panel ii). In contrast, the few cells invading the matrix in cultures of Hand2-deficient AVCs mostly retain their cobblestone-like morphology (Figure 3A, panels iii and iv). This loss of mesenchymal characteristics shows that the endocardial cells of Hand2-deficient AVCs fail to undergo the EMT giving rise to the mesenchymal cell forming the cardiac cushions.
Figure 3. Hand2-deficient AVC endocardial cells fail to initiate EMT and mesenchymal cell migration.

(A) Smooth muscle actin (red) and F-actin (green) distribution in cells that have migrated into the matrix from AVC explants of wild-type and Hand2-deficient embryos after 72hrs in culture. Scale bars: 100μm. Bottom panels show cells in proximity to the explant (i and iii) and at the far edge of migration (ii and iv). Scale bars: 20μm. (B) Quantification of the numbers of cells that migrated into the matrix from wild-type (n=10) and Hand2-deficient AVC explants (n=7). The mean ± SD (p=0.0001, Mann-Whitney test) and all individual data points are shown.
HAND2 controls the expression of genes that function in the EMT underlying AVC cardiac cushion formation
To identify the gene regulatory networks (GRNs) controlled by HAND2 during AVC cardiac cushion formation, the transcriptomes of dissected wild-type and Hand2-deficient AVCs were analyzed (E9.0–E9.25: 18–23 somites; see Supplemental Experimental Procedures). Statistical analysis showed that 1051 genes are differentially expressed (DEGs: 695 are up-regulated and 365 down-regulated, Figure 4A and Table S5, Figure S4A for GO analysis). Among these genes, the transcriptional targets of HAND2 were identified as those genes harboring one or more HAND2 ChIP-Seq peaks in their Topologically Associating Domains (TADs, Dixon et al., 2012; Figure 4B, 4C and S4B, Table S5). This analysis shows that the TADS of DEGs contain on average a significantly larger number of HAND2-interacting regions (median ~5) than genes whose expression is not altered (median ~1; Figure 4B). GO analysis of these HAND2 transcriptional targets indicates that the 167 DEGs, whose expression is down-regulated in Hand2-deficient AVCs function preferentially in heart and organ development (including cardiac EMT and mesenchyme development), while the 372 up-regulated DEGs function preferentially in cardiovascular and blood vessel development (Figure 4C). In particular, this functional annotation identified a subset of 24 DEGs that function in EMT processes and/or AVC cushion formation (Figure S4C). Combining the transcriptome analysis with HAND2 ChIP-qPCR analysis establishes 19 of these DEGs as direct transcriptional targets of HAND2 in developing hearts at E9.25–9.5 (Figure 4D, 4E). Transcriptome analysis showed that the expression of seven of these HAND2 targets is down-regulated, while 12 are up-regulated in mutant AVCs (Figure 4D, Table S6). These differential effects are not unexpected as HAND2 transcriptional complexes are known to differentially activate or repress gene expression (see Discussion). Together, this analysis uncovers the HAND target GRN functioning in AVC cardiac cushion formation and reveals the differential effects of the Hand2 deficiency on gene expression in the mutant AVC (Figure 4D, 4E).
Next, we used WISH to detect spatial alterations in the AVC of Hand2-deficient hearts at E9.25–9.5 (Figure 5 and S5). To uncover potential global molecular changes in the mutant AVC, we first analyzed the spatial distribution of key regulators whose transcript levels are not changed (Bmp2, Hey2, Notch1, Rbpj, Snai2, Epha3, Tbx20, Table S5, Figure 5A, 5B and data not shown). The spatial distribution of all of these genes is comparable to wild-types in mutant AVCs, as exemplified by the EMT-inducer Bmp2 and the NOTCH transcriptional target Hey2 (Figure 5A, 5B). This indicates that the AVC domain is correctly specified in mutant hearts and corroborates the specificity of the molecular alterations underlying the cardiac cushion agenesis (Figures 2, 3). WISH analysis of HAND2 target genes with significantly changed transcript levels (Figure 4D and Table S6) failed to detect spatial alterations for several of them due to low and/or uniform expression (low: Vasn, Tmem100, Erbb4, Tll1 and Acvrl, uniform: Cyr61 and Hnrnapab, data not shown). Furthermore, the small but significant differences in transcript levels for several additional DEGs could not be reliably detected by WISH (Pitx2, Glipr2, Kdr, Gja5, Figure S5A and data not shown), likely due to the qualitative nature of in situ hybridization that best detects spatial changes. This exemplified by the fact that WISH revealed the ectopic expression of two transcriptionally up-regulated HAND2 target genes in mutant AVCs, namely the NOTCH1 transcriptional mediator Hey1 and the homeobox transcription factor Hhex (Figure 5C, 5D).
Figure 5. Whole mount in situ hybridization reveals spatial changes in some of the differentially expressed HAND2 target genes.

(A, B) Unaltered expression of Bmp2 (panel A) and Hey2 (panel B) indicates that the AVC domain is established correctly in Hand2-deficient hearts. (C, D) Ectopic expression of the Hey1 (panel C) and Hhex transcriptional regulators (panel D) in the mutant AVC corroborated their up-regulation detected by RNA-Seq analysis. (E-F) Loss of Twist1 (panel E), Msx1 (panel F), Sox9 (panel G) and Has2 (panel H) from the mutant AVC is in agreement with their transcriptional down-regulation detected by RNA-Seq analysis. (I) Alcian blue staining of glycosaminoglycans shows the reduced deposition of extra-cellular matrix in the cardiac jelly of Hand2-deficient mouse embryos. Right panels show the enlargements indicated by frames in the left panels. Gene names in black: unaltered, red: increased, blue: reduced transcript levels as determined by RNA-Seq analysis (Figure 4). Scale bars: 100μm. See also Figure S5.
In agreement with the reduced expression detected by RNA-Seq (Figure 4D and Table S6), WISH corroborates the loss of the transcriptional regulators Twist1 (Figure 5E, see also Figure 2E), Msx1 (Figure 5F) and Sox9 (Figure 5G and S5B) from mutant AVCs by E9.5. The down-regulation of Msx1 indicates that BMP signal transduction is disrupted in the mutant AVC (Figure 5F, Table S6). SOX9 regulates the proliferation of the mesenchymal progenitor cells and its loss agrees with the lack of delaminating mesenchymal cells in mutant AVCs (Figure 5G and S5B; Akiyama et al., 2004). Most relevant to the disrupted EMT (Figure 3), the expression of Has2 and Snai1 is significantly down-regulated in Hand2-deficient AVCs (Figure 4D). Has2 encodes the enzyme that produces hyaluronic acid in the cardiac jelly (Camenisch et al., 2000). Genetic inactivation of the mouse Has2 gene disrupts both cardiac jelly deposition and mesenchymal cell migration during cardiac cushion development. In Hand2-deficient hearts, the loss of Has2 is paralleled by reduced extra-cellular matrix/cardiac jelly deposition ((Figure 5H, 5I). Camenisch and co-workers (2000) showed that treatment of Has2-deficient AVC explants with hyaluronic acid restores mesenchymal cell migration. In contrast, culturing Hand2-deficient AVC explants in hyaluronic acid does not suffice to restore migration (data not shown). This is in line with the fact that the genetic inactivation of Hand2 affects the expression of multiple genes required for AVC cushion development (Figures 4, S4).
HAND2 regulates the transcription of Snai1, a key regulator of the EMT and mesenchymal cell migration in the AVC
The transcriptome combined with ChIP-Seq/qPCR analysis (Figure 4D, 4E) and WISH (Figure 6A) establishes Snai1 as a direct transcriptional target of HAND2. As Snai1 is a EMT key regulator (Nieto, 2011), its loss from the mutant AVC (Figure 6A) is likely causally linked to the observed cardiac cushion agenesis (Figure 2). To test this experimentally, Hand2-deficient AVC explants were infected either with adenovirus producing both SNAI1 and GFP proteins (SNAI1-GFP), or control GFP virus (GFP only; Figure 6B; Tao et al., 2011). Quantitative analysis shows that infection of Hand2-deficient AVC explants with SNAI1-producing virus induces migration of a significantly larger fraction of GFP-positive mesenchymal cells into the collagen matrix than GFP alone (Figure 6B, p=0.0093 Mann-Whitney test; see Figure S5C for wild-type controls).
This partial restoration of mesenchymal cell migration reveals the functional importance of the HAND2-Snai1 interactions for the EMT during cardiac cushion formation. Therefore, we analyzed the potential enhancer activities of the three HAND2-interacting CRMs located in the Snai1 TAD (Figure 6C and Figure S6A).
Our previous analysis has shown that these three candidate CRMs are also enriched in HAND2 chromatin complexes isolated from mouse limb buds (Osterwalder et al., 2014) and overlap regions of active chromatins in embryonic hearts (H3K27ac profile in Figure 6C). Their transcription enhancing potential was assessed in transgenic mouse founder embryos using LacZ reporter constructs (Figure 6E–6F). In particular, LacZ activity reminiscent of Snai1 expression (Figure 6D) was detected for reporters encoding the CRMs located +45kb and +57kb downstream of the Snai1 transcription start site (Figure 6E, 6F). In contrast, no LacZ activity was detected using the Snai1 +14kb genomic region, whose sequence is not well conserved in mammals (data not shown). The Snai1 +45kb CRM is active in cells located between the OFT and aortic sac (n=7/10), the posterior (n=10/10) and anterior (n=3/10) limb bud mesenchyme, branchial arches and cranial mesenchyme (n=5/10, Figure 6E). Most relevant with respect to the AVC, the Snai1 +57kb CRM is active in the cardiac cushion mesenchyme of the AVC and OFT (n=7/9, Figure 6F) and in most other embryonic tissues expressing Snai1 (Figure 6B). Together, the activities of these two HAND2-interacting CRMs recapitulate most of the Snai1 expression pattern in mouse embryos (Figure 6D and Figure S6B–S6D). Indeed in Hand2-deficient embryos, Snai1 expression is not only lost from the AVC (Figure 6A), but also significantly reduced in the second branchial arch and forelimb bud mesenchyme (Figure S6E).
Discussion
We show that HAND2 chromatin complexes interact with genomic regions such as enhancers located in the cis-regulatory landscapes of genes functioning in heart morphogenesis. Previous molecular analysis showed that the altered expression of many of these genes correlates well with the defects in right ventricle and OFT morphogenesis observed in Hand2-deficient mouse embryos (Cohen et al., 2012; Tsuchihashi et al., 2011; Zhao et al., 2008). We provide evidence that about half of all genes with altered expression are direct transcriptional targets of HAND2. In addition, our ChIP-Seq analysis reveals that a significant fraction of the genomic regions enriched in HAND2 chromatin complexes are also bound by GATA4 complexes (He et al., 2014). This is interesting in light of previous studies, which showed that HAND2 and GATA4 form transcriptional complexes regulating gene expression in developing hearts (Dai et al., 2002). In addition, it has been shown that AVC enhancers are repressed in the atrial and ventricular myocardium by complexes containing GATA4, HEY1 and/or HEY2 transcriptional repressors (Firulli et al., 2000; Stefanovic et al., 2014). These three repressors plus RUNX2 and TWIST1 are all able to form heterodimers with HAND2 (Firulli et al., 2005; Funato et al., 2009). As the expression of many HAND2 target genes is up-regulated in Hand2-deficient AVCs (this study), HAND2-mediated transcriptional repression is likely functionally relevant to normal AVC development. For example, the HAND2 target Hhex is ectopically expressed in the AVC of Hand2-deficient embryos. Indeed, genetic inactivation of Hhex increases the number of mesenchymal cells in AVC cardiac cushions and causes valve dysplasia (Hallaq et al., 2004).
One key finding of our analysis is that constitutive inactivation of Hand2 disrupts the EMT underlying cardiac cushion formation in the AVC. This disruption of AVC morphogenesis contrasts with the phenotypes resulting from specific inactivation of Hand2 in either the endocardium or mesenchyme. Neither inactivation disrupts AVC cardiac cushion formation, but specifically alters the AVC-derived tricuspid valves (tricuspid atresia; VanDusen et al., 2014a; VanDusen et al., 2014b). This discrepancy is a likely consequence of different Hand2 inactivation kinetics. As genetic inactivation of Hand2 in SHF progenitors also alters AVC development, we cannot formally exclude that recruitment of progenitors to the AVC is compromised in Hand2-deficient embryos (Tsuchihashi et al., 2011), even though the expression of early markers for AVC morphogenesis remains normal (this study).
However, our analysis shows that most genes with known functions in the EMT underlying AVC cardiac cushion formation are direct transcriptional targets of HAND2. Together with the cellular analysis, these results point to specific disruption of the EMT rather than a general arrest of AVC development and suggest that HAND2 is a very up-stream regulator of AVC cardiac cushion morphogenesis. In agreement, re-expression of the HAND2 target Snai1 in Hand2-deficient AVCs explants only partially restores mesenchymal cell migration, which indicates that other HAND2 target DEGs have essential functions in the EMT and/or mesenchymal cell migration during cardiac cushion development. Ingenuity pathway analysis of HAND2 target genes shows that HAND2 enhances the expression of genes such as Msx1 and Hnrnapab, which in turn reinforce the expression of the HAND2 targets Has2, Twist1 and Snai1 (Figure 7). This type of dual transcriptional reinforcement likely increases the robustness of the expression of HAND2 target genes with key functions in AVC morphogenesis. In fact, it is reminiscent of the dual transcriptional reinforcement seen for key genes during limb bud development. In early limb buds, HAND2 reinforces the expression of the Shh morphogen by directly regulating its transcription and indirectly via up-regulating ETS transcription factors, which also positively regulate Shh expression (Osterwalder et al., 2014).
Figure 7. Scheme depicting the interactions among positively regulated HAND2 targets genes.

The HAND2 GRN was constructed using Ingenuity pathway analysis in combination with manual annotation (Chen et al., 2008). Arrows indicate transcriptional up-regulation and direct up-regulation by HAND2 transcriptional complexes is indicated in green. Broken grey lines direct protein-protein interactions. This graph represents the simplest possible scheme to illustrate the relevant direct interactions.
Remarkably, this study identifies HAND2 as key regulator of most genes with known functions in EMT processes and cardiac cushion formation in the developing AVC including Snai1 (Garside et al., 2013). Our analysis also provides evidence that HAND2 directly regulates Snai1 transcription in other embryonic tissues. The notion that the direct transcriptional regulation of Snai1 by HAND2 maybe of more general importance is supported by genetic analysis as Hand2 and Snai1 are both essential for the EMT of epicardial cells and morphogenesis of craniofacial structures such as the palate (Barnes et al., 2011; Murray et al., 2007; Tao et al., 2013; Xiong et al., 2009). In summary, our study identifies the HAND2 target GRN that controls the initiation of cardiac valve formation and provides evidence for its general role in regulating the expression of Snai1 during mouse embryogenesis. Last but not least, the identification of HAND2 as key regulator of AVC cardiac cushion morphogenesis may have important implications for regenerative medicine (see e.g. review by Levine et al., 2015).
Experimental Procedures
Ethics statement, mouse strains and embryos
All experiments conducted with mice and embryos of both sexes at the developmental ages indicated (see Results section and below) were performed in strict accordance with Swiss law. All animal studies were evaluated and approved by the Regional Commission on Animal Experimentation (license 1951). The 3Rs were taken into account in designing the animal studies. The procedures for generating transgenic mice at the Lawrence Berkeley National Laboratory (LBNL) were reviewed and approved by the LBNL Animal Welfare and Research Committee. The Hand2Δ and Hand23xF alleles (Galli et al., 2010; Osterwalder et al., 2014) were outbred into an NMRI background as this prolongs survival of Hand2-deficient embryos in comparison to the previously used 129SvJ/C57BL6 background.
ChIP-Seq analysis
To obtain sufficient material for ChIP-Seq analysis about 600 hearts had to be dissected from Hand23xF/3xF mouse embryos at E10.5. After collection, these were split in two batches and processed as completely independent biological replicates for ChIP-Seq analysis using the M2 anti-FLAG antibody (F1804; Sigma; (Osterwalder et al., 2014). Library construction and sequencing were performed by the Genome Technology Access Center (St. Louis, USA) using an Illumina HiSeq 2500 system. More details are included in the Supplemental Experimental Procedures.
Transcriptome analysis
AVCs dissected from wild-type and Hand2 mutant embryos at E9.0–E9.25 were flash frozen in RLT buffer (Qiagen). Four AVCs were pooled per replicate, keeping the same gender ratio for all replicates (AVCs of 2 male and female embryos). RNA was extracted using the QIAGEN RNeasy mini kit. The quality of total RNA (30–60ng) was analyzed using the Agilent 2100 Bioanalyzer and both wild-type and mutant samples had an RNA integrity number (RIN) of 8.8–9.6. Libraries were prepared using the Clontech SMARTer kit and sequenced on a HiSeq3000 using a single-read 50 cycle protocol. More details on the computational analysis are provided in the Supplemental Experimental Procedures.
Accession Numbers
The primary ChIP-Seq and transcriptome datasets have been deposited in the GEO database under accession numbers GSE73368 and GSE94246, respectively.
AVC explant cultures
AVC explant cultures were set on matrixes of rat-tail collagen-type I (Luna-Zurita et al., 2010, see Supplemental Information for more details). Only wild-type and mutant AVC explants that were still beating, i.e. alive after 72hrs in culture were analyzed. Supplementation with hyaluronic acid (HA): both the collagen matrix and serum-free culture medium were supplemented with 0.75mg/ml HA (Camenisch et al., 2000). Adenoviral infections: the titers of the SNAI1-GFP or GFP adenoviruses (Tao et al., 2011 and Vector Biolabs) were determined in mitomycin-treated mouse embryonic fibroblasts and adjusted such that equal numbers of active virus particles were used. Following attachment to the matrix, mutant and wild-type AVC explants were incubated with 6x106 PFU of either SNAI1-GFP or GFP virus for 12hrs in serum-free medium. Then, the AVC explants were cultured for 60hrs in fresh serum-free medium. After fixation in 4% PFA (30 min room temperature), antigens were detected using anti-SMA-Cy3 antibodies (1:250, Sigma) and Phalloidin-Alexa 488 (1:250, Life Technologies) and nuclei counterstained with Hoechst-33258 and analyzed using a Leica SP5 confocal microscope. The analysis of wild-type controls shows that GFP-virus tends to infect AVC cells more efficiently than SNAI1-GFP virus (Figure S5C). Therefore, the restoration of cell migration following infection of mutant AVCs with SNAI1-GFP virus is rather underestimated (Figure 6B).
Statistical analysis
ChIP-Seq: following initial alignment of sequences, the genome-wide pattern of binding of HAND2 was determined using MACS (version 1.4.2) with a p-value threshold of 1e-5. ChIP-qPCR: mean ± SD were calculated using the Prism (GraphPad Software) Student-t test. Transcriptome: following initial sequence alignment, edgeR was used to normalize the datasets (TMM normalization) and to identify the differentially expressed genes (DEGs). Only genes expressed in all samples were considered (RPM ≥1). DEGs are defined as genes with a q-value ≤0.05 and a linear fold-change ≥1.5. AVC explant cultures: the Mann-Whitney test was used to determine significant differences in numbers of migrating mesenchymal cells. More details on statistical validation of the ChIP-Seq and transcriptome analyses are included in the Supplemental Online Experimental Procedures.
Histology and immunofluorescence analysis
Embryos were collected and fixed overnight in 4% PFA at 4°C and embedded in paraffin wax. Standard protocols were used for histological staining (Hematoxylin and eosin; alcian blue) of 7μm paraffin sections. Minimally 3 biological replicates were analyzed for each stage, genotype and antigen shown. Antibodies are listed the Supplemental Experimental Procedures.
Generation and analysis of LacZ transgenic founder embryos
Genomic regions were amplified by PCR from mouse genomic DNA (Snai1 +45kb, Snai1 +14kb) or recovered as restriction fragment (Snai1 +57kb, BAC clone RP23-193B17) and cloned into the Hsp68-promoter-LacZ reporter vector (Osterwalder et al., 2014). Transgenic founder embryos were generated by pronuclear injection, analyzed by LacZ staining and transgenic embryos identified by genotyping.
Supplementary Material
Highlights.
HAND2 controls development of the AVC cardiac cushions forming mitral/tricuspid valves
HAND2 is a key regulator of the EMT underlying cardiac cushion mesenchyme formation
Identification of the HAND2 target gene networks that control EMT and AVC development
HAND2 acts upstream of the EMT key regulator Snai1 in AVC and other embryonic tissues
Acknowledgments
The Genome Technology Access Center (Dept. of Genetics at Washington University School of Medicine) is acknowledged for deep sequencing and R. Ivanek (DBM Bioinformatics Core) performed the bioinformatics analysis of the ChIP-Seq datasets. All calculations were performed using the sciCORE (http://scicore.unibas.ch/) scientific computing core facility at University of Basel. We thank D. Speziale for technical assistance, A. Offinger’s team for excellent animal care and P. Lorentz (DBM Bio-Optics Core Facility) for imaging support. V. Afzal, B. Mannion and I. Plajzer-Frick provided assistance with transgenics and L. Bazzani the GFP adenovirus preparations. T. Papoutsi, C. Rolando and O. Pertz are acknowledged for advice on AVC explants and infections. We are grateful to V. Christoffels for helpful input and anonymous reviewers for critical input that resulted in a significantly improved study. This research was mostly supported by SNF grants 31003A_146248 and 310030B_166685 to AZ and RZ and by funds from the University of both Basel. The stay of MO in the group of AV was supported by an SNSF early mobility postdoctoral fellowship. IB, JA and AV were supported by NIH grants R24HL123879, UM1HL098166, R01HG003988 and U54HG006997 and research at LBNL was performed under Dept. of Energy Contract DE-AC02-05CH11231 to University of California. JL was supported by NIH/NHLBI grant 1R01HL127033.
Footnotes
Author Contributions
FL performed the ChIP-Seq and follow-up functional analysis, AG the transcriptome and WISH analysis for revision, JG participated in different aspects of the experimental studies and IB performed the bioinformatics analysis. MO was involved in initiating this study, JL generated the SNAI1-GFP virus, JA the LacZ reporters and AV provided the resources for the transgenic and bioinformatics analysis. The experimental study design was done and the manuscript written by FL, JLR, AZ and RZ with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama H, Chaboissier MC, Behringer RR, Rowitch DH, Schedl A, Epstein JA, de Crombrugghe B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc Natl Acad Sci USA. 2004;101:6502–6507. doi: 10.1073/pnas.0401711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes RM, Firulli BA, VanDusen NJ, Morikawa Y, Conway SJ, Cserjesi P, Vincentz JW, Firulli AB. Hand2 loss-of-function in Hand1-expressing cells reveals distinct roles in epicardial and coronary vessel development. Circ Res. 2011;108:940–949. doi: 10.1161/CIRCRESAHA.110.233171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–948. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- Camenisch T, Spicer A, Brehm-Gibson T, Biesterfeldt J, Augustine M, Calabro AJ, Kubalak S, Klewer S, McDonald J. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106:349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Ishii M, Sucov HM, Maxson RE., Jr Msx1 and Msx2 are required for endothelial-mesenchymal transformation of the atrioventricular cushions and patterning of the atrioventricular myocardium. BMC Dev Biol. 2008;8:75. doi: 10.1186/1471-213X-8-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels VM, Smits GJ, Kispert A, Moorman AF. Development of the pacemaker tissues of the heart. Circ Res. 2010;106:240–254. doi: 10.1161/CIRCRESAHA.109.205419. [DOI] [PubMed] [Google Scholar]
- Cohen ED, Miller MF, Wang Z, Moon RT, Morrisey EE. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development. 2012;139:1931–1940. doi: 10.1242/dev.069377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai YS, Cserjesi P. The basic helix-loop-helix factor, HAND2, functions as a transcriptional activator by binding to E-boxes as a heterodimer. J Biol Chem. 2002;277:12604–12612. doi: 10.1074/jbc.M200283200. [DOI] [PubMed] [Google Scholar]
- Dai YS, Cserjesi P, Markham BE, Molkentin JD. The transcription factors GATA4 and dHAND physically interact to synergistically activate cardiac gene expression through a p300-dependent mechanism. J Biol Chem. 2002;277:24390–24398. doi: 10.1074/jbc.M202490200. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli BA, Hadzic DB, McDaid JR, Firulli AB. The basic helix-loop-helix transcription factors dHAND and eHAND exhibit dimerization characteristics that suggest complex regulation of function. J Biol Chem. 2000;275:33567–33573. doi: 10.1074/jbc.M005888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli BA, Krawchuk D, Centonze VE, Vargesson N, Virshup DM, Conway SJ, Cserjesi P, Laufer E, Firulli AB. Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat Genet. 2005;37:373–381. doi: 10.1038/ng1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato N, Chapman SL, McKee MD, Funato H, Morris JA, Shelton JM, Richardson JA, Yanagisawa H. Hand2 controls osteoblast differentiation in the branchial arch by inhibiting DNA binding of Runx2. Development. 2009;136:615–625. doi: 10.1242/dev.029355. [DOI] [PubMed] [Google Scholar]
- Galli A, Robay D, Osterwalder M, Bao X, Benazet JD, Tariq M, Paro R, Mackem S, Zeller R. Distinct roles of Hand2 in initiating polarity and posterior Shh expression during the onset of mouse limb bud development. PLoS Genet. 2010;6:e1000901. doi: 10.1371/journal.pgen.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garside VC, Chang AC, Karsan A, Hoodless PA. Co-ordinating Notch, BMP, and TGF-beta signaling during heart valve development. Cell Mol Life Sci. 2013;70:2899–2917. doi: 10.1007/s00018-012-1197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Marin C, Tena JJ, Acemel RD, Lopez-Mayorga M, Naranjo S, de la Calle-Mustienes E, Maeso I, Beccari L, Aneas I, Vielmas E, et al. Evolutionary comparison reveals that diverging CTCF sites are signatures of ancestral topological associating domains borders. Proc Natl Acad Sci USA. 2015;112:7542–7547. doi: 10.1073/pnas.1505463112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich F, Rudat C, Kispert A. Mechanisms of T-box gene function in the developing heart. Cardiovasc Res. 2011;91:212–222. doi: 10.1093/cvr/cvr112. [DOI] [PubMed] [Google Scholar]
- Hallaq H, Pinter E, Enciso J, McGrath J, Zeiss C, Brueckner M, Madri J, Jacobs HC, Wilson CM, Vasavada H, et al. A null mutation of Hhex results in abnormal cardiac development, defective vasculogenesis and elevated Vegfa levels. Development. 2004;131:5197–5209. doi: 10.1242/dev.01393. [DOI] [PubMed] [Google Scholar]
- He A, Gu F, Hu Y, Ma Q, Ye LY, Akiyama JA, Visel A, Pennacchio LA, Pu WT. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat Commun. 2014;5:4907. doi: 10.1038/ncomms5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler KL, Hendershot TJ, Troy SE, Vincentz JW, Firulli AB, Howard MJ. Targeted deletion of Hand2 in cardiac neural crest-derived cells influences cardiac gene expression and outflow tract development. Dev Biol. 2010;341:291–304. doi: 10.1016/j.ydbio.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RG. The second heart field. Curr Top Dev Biol. 2012;100:33–65. doi: 10.1016/B978-0-12-387786-4.00002-6. [DOI] [PubMed] [Google Scholar]
- Levine RA, Hagege AA, Judge DP, Padala M, Dal-Bianco JP, Aikawa E, Beaudoin J, Bischoff J, Bouatia-Naji N, Bruneval P, et al. Mitral valve disease--morphology and mechanisms. Nat Rev Cardiol. 2015;12:689–710. doi: 10.1038/nrcardio.2015.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CJ, Lin CY, Chen CH, Zhou B, Chang CP. Partitioning the heart: mechanisms of cardiac septation and valve development. Development. 2012;139:3277–3299. doi: 10.1242/dev.063495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Barbosa AC, Chapman SL, Bezprozvannaya S, Qi X, Richardson JA, Yanagisawa H, Olson EN. DNA binding-dependent and -independent functions of the Hand2 transcription factor during mouse embryogenesis. Development. 2009;136:933–942. doi: 10.1242/dev.034025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Zurita L, Prados B, Grego-Bessa J, Luxan G, del Monte G, Benguria A, Adams RH, Perez-Pomares JM, de la Pompa JL. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest. 2010;120:3493–3507. doi: 10.1172/JCI42666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601–5611. doi: 10.1242/dev.02156. [DOI] [PubMed] [Google Scholar]
- MacGrogan D, Luxan G, Driessen-Mol A, Bouten C, Baaijens F, de la Pompa JL. How to make a heart valve: from embryonic development to bioengineering of living valve substitutes. Cold Spring Harb Perspect Med. 2014;4:a013912. doi: 10.1101/cshperspect.a013912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray SA, Oram KF, Gridley T. Multiple functions of Snail family genes during palate development in mice. Development. 2007;134:1789–1797. doi: 10.1242/dev.02837. [DOI] [PubMed] [Google Scholar]
- Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D, Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182:315–325. doi: 10.1083/jcb.200710067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer-Frick I, Shoukry M, Afzal V, et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell. 2013;155:1521–1531. doi: 10.1016/j.cell.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder M, Speziale D, Shoukry M, Mohan R, Ivanek R, Kohler M, Beisel C, Wen X, Scales SJ, Christoffels VM, et al. HAND2 targets define a network of transcriptional regulators that compartmentalize the early limb bud mesenchyme. Dev Cell. 2014;31:345–357. doi: 10.1016/j.devcel.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Li X, Shen A, Wang Q, Liu C, Guo Y, Song Z, Li Z. Transcription factor HAND2 mutations in sporadic Chinese patients with congenital heart disease. Chin Med J. 2010;123:1623–1627. [PubMed] [Google Scholar]
- Srivastava D, Thomas T, Lin Q, Kirby M, Brown D, Olson E. Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nat Genet. 1997;16:154–160. doi: 10.1038/ng0697-154. [DOI] [PubMed] [Google Scholar]
- Stefanovic S, Barnett P, van Duijvenboden K, Weber D, Gessler M, Christoffels VM. GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development. Nat Commun. 2014;5:3680. doi: 10.1038/ncomms4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic S, Christoffels VM. GATA-dependent transcriptional and epigenetic control of cardiac lineage specification and differentiation. Cell Mol Life Sci. 2015;72:3871–3881. doi: 10.1007/s00018-015-1974-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YM, Wang J, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Shi HY, Hou XM, Huang RT, et al. A HAND2 Loss-of-Function Mutation Causes Familial Ventricular Septal Defect and Pulmonary Stenosis. G3 (Bethesda) 2016;6:987–992. doi: 10.1534/g3.115.026518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao G, Levay AK, Gridley T, Lincoln J. Mmp15 is a direct target of Snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Dev Biol. 2011;359:209–221. doi: 10.1016/j.ydbio.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao G, Miller LJ, Lincoln J. Snai1 is important for avian epicardial cell transformation and motility. Dev Dyn. 2013;242:699–708. doi: 10.1002/dvdy.23967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes & Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchihashi T, Maeda J, Shin CH, Ivey KN, Black BL, Olson EN, Yamagishi H, Srivastava D. Hand2 function in second heart field progenitors is essential for cardiogenesis. Dev Biol. 2011;351:62–69. doi: 10.1016/j.ydbio.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDusen NJ, Firulli AB. Twist factor regulation of non-cardiomyocyte cell lineages in the developing heart. Differentiation. 2012;84:79–88. doi: 10.1016/j.diff.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDusen NJ, Casanovas J, Vincentz JW, Firulli BA, Osterwalder M, Lopez-Rios J, Zeller R, Zhou B, Grego-Bessa J, De La Pompa JL, et al. Hand2 is an essential regulator for two Notch-dependent functions within the embryonic endocardium. Cell Rep. 2014a;9:2071–2083. doi: 10.1016/j.celrep.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDusen NJ, Vincentz JW, Firulli BA, Howard MJ, Rubart M, Firulli AB. Loss of Hand2 in a population of Periostin lineage cells results in pronounced bradycardia and neonatal death. Dev Biol. 2014b;388:149–158. doi: 10.1016/j.ydbio.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35:D88–92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZQ, Rowe RG, Lim KC, Lin Y, Willis A, Tang Y, Li XY, Nor JE, Maillard I, Weiss SJ. A Snail1/Notch1 signalling axis controls embryonic vascular development. Nat Commun. 2014;5:3998. doi: 10.1038/ncomms4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, He F, Morikawa Y, Yu X, Zhang Z, Lan Y, Jiang R, Cserjesi P, Chen Y. Hand2 is required in the epithelium for palatogenesis in mice. Dev Biol. 2009;330:131–141. doi: 10.1016/j.ydbio.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol. 2008;317:614–619. doi: 10.1016/j.ydbio.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.