

Abstract
The lung is one of several moderately radiosensitive organs. Radiation-induced lung injury (RILI), including acute radiation pneumonitis and chronic radiation-induced pulmonary fibrosis, occurs most often in radiotherapy of lung cancer, esophageal cancer, and other thoracic cancers. Clinical symptoms of RILI include dry cough, shortness of breath, chest pain, fever, and even severe respiratory failure and death. The occurrence of RILI is a complex process that includes a variety of cellular and molecular interactions which ultimately leads to large fibroblast accumulation, proliferation, and differentiation, resulting in excessive extracellular matrix deposits, causing pulmonary fibrosis. The progress that has been made in recent years in the understanding of cellular and molecular mechanisms of RILI is summarized in this review.
MeSH Keywords: Abnormalities, Radiation-Induced; Fibroblasts; Macrophage Activation; Transforming Growth Factor beta1
Background
There are a series of cellular and molecular changes that occur when the lung tissue suffers ionizing radiation that does not cause immediate clinical symptoms. Pulmonary irradiation can produce a lot of reactive oxygen species and reactive nitrogen which causes oxidative damage of DNA, lipid, and protein. The resulting injury or apoptosis of alveolar epithelial cells and vascular endothelial cells then induce a series of inflammatory reactions and chemotaxis of monocytes, lymphocytes, and granulocytes, which gather at the site of tissue injury. The result is secretion of large amounts of inflammatory cytokines, chemokines, and growth factors, such as TGFβ, IFN-γ, ET-1, IL4, IL-13, which aggregate more inflammatory cells. When this damage becomes chronic, it ultimately leads to pulmonary fibrosis. In summary, a variety of cells and molecules are involved in the complex process of radiation-induced lung injury (RILI) (Figure 1).
Figure 1.
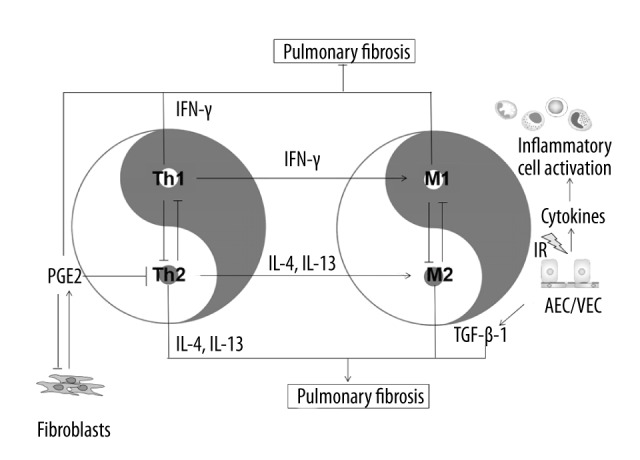
When lung tissue suffers ionizing radiation, alveolar epithelial cells (AEC) and vascular endothelial cells (VEC) are damaged, and then secrete large amounts of cytokines, which induce acute radiation pneumonitis. In this period, Th-1 derived IFN-γ induces activation of M1 macrophages. As a major effector cytokine of Th1 cells, IFN-γ auto-amplifies Th1 responses and cross-inhibits the differentiation and function of Th2 cells and the expression of Th2-derived cytokines IL-4 and IL-13, which induce activation of M2 macrophages that promote fibrosis through the production of TGF-β1. Meanwhile, activated fibroblasts secret great amounts of PGE2, which, in turn, triggers a negative feedback on the production of cytokines, and inhibits the transendothelial migration of T cells and transition to Th2 cells, and downregulates the functions of fibroblasts, including proliferation, collagen synthesis, and myofibroblast transformation.
Related Cells
Alveolar epithelial cells and vascular endothelial cells
Ionizing radiation-induced injuries in alveolar epithelial cells and vascular endothelial cells are important contributors to RILI and the resulting chronic progression condition. When alveolar epithelial cells and vascular endothelial cells are injured, the connections between cells is damaged, accompanied by impairment on the regulation of myofibroblast, so that excessive extracellular matrix deposits result in radiation-induced pulmonary fibrosis [1]. Once the barrier of lung tissue is damaged, a large number of blood exudate and inflammatory cells accumulate in the alveolar cavity, which then aggregates numerous fibroblasts and induces their differentiation into myofibroblasts. Activated myofibroblasts then secret angiotensin and hydrogen peroxide, which in turn induces apoptosis of alveolar epithelial cells [2]. The damage to alveolar epithelial cells and vascular endothelial cells results in secretion of a large number of pro-inflammatory and pro-fibrotic cytokines, including TGF-β1, IL-13, ET-1, PGE2 [3].
Th1 and Th2 cells
Th1/Th2 imbalance plays an important role in the development of RILI. When lung tissue is exposed to ionizing radiation, Th1 cells play a role in RILI mainly through the secretion of IFN-γ, whereas Th2 cells play a role mainly through the secretion of IL-4 and IL-13. IFN-γ increases early after irradiation, while Th2-derived IL-4 and IL-13 do not significantly increase in the early stage, but gradually increases over time and are maintained at a high level [4]. IL-4 and IL-13 can co-stimulate with TGF-β1 in collagen synthesis, playing significant roles in tissue remodeling and fibrosis [5,6], while Th1-derived IFN-γ have significant anti-fibrosis and immunomodulatory effects [7]. Excessive Th1 immune response mostly contributes to acute radiation pneumonitis, whereas excessive Th2 immune response mostly contributes to chronic radiation-induced pulmonary fibrosis.
Macrophages and fibroblasts
Interleukins, tumor necrosis factor, transforming growth factor, and platelet-derived growth factor induce activation of macrophages and fibroblasts, which are the major effector cells for synthesis of extracellular matrix [7]. Macrophages can be divided into two types, depending on the induction of cytokines from activated Th1 cells or Th2 cells. Th1-derived cytokine IFN-γ can promote the expression of nitric oxide synthase of macrophages, which is known as classic activated macrophages (M1). And Th2-derived cytokines IL-4 and IL-13 can promote the activity of arginase in macrophages, which is called bypass activated macrophages (M2) [8,9]. In addition, dendritic cells [10] and fibroblasts [11] will have a similar effect when subjected to Th1/Th2-derived cytokines. The synthesis of proline could be eventually promoted by the arginase pathway, and proline is the necessary substance for synthesis of collagen, which is the main component of the extracellular matrix, mainly synthesized and secreted by myofibroblasts. Fibroblasts and myofibroblasts accumulate in three ways: in situ proliferation, epithelial-mesenchymal transformation, and derivation from bone marrow. Accumulation of fibroblasts and myofibroblasts is induced by TGF-β1, PDGF, CXCL12, and other factors, of which the most important is TGF-β1 [12]. A variety of pro-fibrotic cytokines (such as IL-4, IL-13, and TNF-α) and growth factors (such as PDGF and CTGF) connect directly or indirectly with TGF-β1. In the normal repair process, the myofibroblasts shrink the damaged area, and then epithelial cells and endothelial cells divide and migrate to repair the epithelium and endothelium. However, when the tissue suffers sustained injury, chronic inflammation and abnormal repair processes can lead to excessive secretion and deposition of extracellular matrix, and ultimately lead to fibrosis and structural changes.
Related Molecules
TGF-β
TGF-β is a potent pro-fibrotic growth factor [13], and its roles are mainly as follows: first it induces proliferation and differentiation of fibroblasts; second it promotes synthesis of collagen by fibroblasts and inhibits synthesis of collagenase and plasminogen activator; and third it aggregates a variety of inflammatory cells and promotes release of PDGF, TNF-α, IL-4, IL-6, IL-13, etc. There are many subtypes of TGF-β, among which TGF-β1, mainly generated by macrophages [14], is mainly associated with fibrosis. In the model of bleomycin-induced pulmonary fibrosis, almost all active TGF-β1 is generated by alveolar macrophages [15]. TGF-β1 is often combined with latency-associated peptide (LAP) in an inactive form [16]. Therefore, the release and activation of TGF-β1 is indispensable for its combination with the receptors TGFBR2 and ALK5, and for the following signal transduction process. Smad2/3, a cytoplasmic effector molecule of TGF-β1, phosphorylates and combines with Smad4, and then translocate into the nucleus to regulate transcription of target genes, including those encoding type I and type III collagen [17]. However, macrophage-derived TGF-β1 often promotes fibrosis [18], while T cell-derived TGF-β1 may inhibit fibrosis [19]. Moreover, a small amount of active TGF-β1 may have an anti-inflammatory effect, and only a relatively large amount of active TGF-β1 has a pro-fibrotic effect [20].
ET-1
Endothelin-1 (ET-1), mainly regulated by TGF-β1 in synthesis and secretion [21], is a potent endothelial-derived 21-amino-acid vasoconstrictor peptide [22]. The expression of ET-1 is induced by TGF-β1 through the ALK5/Smad3 pathway [23]. The activation of ALK5/Smad3/ET-1 pathway inhibits the migration and proliferation of endothelial cells, and increases the expression of fibrosis-associated genes, such as type I collagen and plasminogen activator inhibitor (PAI-1). It has been confirmed that ET-1 is not only a vasoconstrictor, but it is also involved in a number of other physiological processes, such as extracellular matrix deposition [24]. When ET-1 is blocked, the differentiation of fibroblasts into myofibroblasts induced by TGF-β1 will be blocked [25]. In addition, the use of ET-1 antagonist can prolong the survival time of patients with idiopathic pulmonary fibrosis [25].
IL-4
IL-4 is one of the symbolic cytokines of Th2 cells, and it can also be generated by macrophages, fibroblasts and epithelial cells. IL-4 can promote the differentiation of T cells into Th2 cells and the expression of Th2-derived cytokines, and can inhibit the activities of Th1 cells. IL-4 is closely related to radiation-induced pulmonary fibrosis. In serum of patients with idiopathic pulmonary fibrosis [26] and patients with RILI [27], IL-4 is significantly increased. In vitro, when fibroblasts are treated by IL-4, the expression of type I collagen, type III collagen, and fibronectin increases significantly [28–30].
IL-13
IL-13 is secreted mainly by Th2 cells as well as IL-4, and it can also be generated by mast cells, basophils, and macrophages. IL-13 has many similar functions as IL-4 because they the share α chain of IL-4 receptors, which has an effect through the activation of STAT6 [31]. IL-13 is an important pro-fibrotic cytokines and it is closely related with fibrosis of liver [32], lung [33], and skin [34]. Expression of IL-13 is regulated by endogenous transcription factor GATA-3, which can promote the expression of IL-3 through combining with a promoter sequence of IL-13 [35]. Han et al. [36] studied the role of Th2 cells in radiation-induced pulmonary fibrosis, and found that GATA-3, IL-13, and Arg-1 were significantly increased.
IFN-γ
IFN-γ, secreted mainly by Th1 cells, has significant anti-fibrosis and immunomodulatory effects [7], which are associated with inhibition of IL-4, IL-13, and TGF-β1-related pathways [37]. As a major effector cytokine of Th1 cells, IFN-γ auto-amplifies Th1 responses and cross-inhibits the differentiation and function of Th2 cells and the expression of Th2-derived cytokines. In addition, IFN-γ induces the expression of Smad7, which plays an inhibitory effect, such as blocking the activation of Smad3 [38] which can also be directly inhibited when bound with IFN-γ-activated STAT1 [39].
PGE2
Prostaglandin E2 (PGE2), derived from arachidonic acid by the catalytic action of cyclooxygenase (COX), plays the role of pro-inflammatory mediator in a variety of diseases. However, in lung tissue, PGE2 plays a unique role in limiting the inflammatory response and the process of tissue repair [40]. PGE2 can inhibit the secretion of TGF-β, the migration of T cells and their differentiation into Th2 cells, and the differentiation of fibroblast into myofibroblast. There are a variety of cytokines produced in the early stage of pulmonary irradiation resulting in large amounts of PGE2 produced by stimulated fibroblasts. In turn, PGE2 triggers a negative feedback on the production of cytokines, and downregulates the function of fibroblasts, including proliferation, collagen synthesis, and capacity of differentiation into myofibroblast [40]. In the late stage of pulmonary irradiation, with the increased differentiation of fibroblast into myofibroblast and continued damage of epithelial cells, the generation of PGE2 decreases [41,42]; this leads to sustained activation of the immune response.
In summary, when the lung tissue suffers RILI, an abnormal repair process can ultimately lead to pulmonary fibrosis, in which a variety of pro-fibrotic cytokines, such as TGF-β, ET-1, IL-4, and IL-13, are involved. Otherwise, IFF-γ and PGE-2 T play a role in the inhibition of pulmonary fibrosis.
Conclusions
When lung tissue suffers ionizing radiation, alveolar epithelial cells and vascular endothelial cells are damaged, and inflammatory mediators are released. Blood vessel dilation and increased permeability allows for efficient accumulation of blood exudate and inflammatory cells at the site of tissue injury. In the early stage of pulmonary irradiation, Th-1 derived IFN-γ induces activation of M1 macrophages. As a major effector cytokine of Th1 cells, IFN-γ auto-amplifies Th1 responses and cross-inhibits the differentiation and function of Th2 cells and the expression of Th2-derived cytokines. If the tissue-damaging irritant persists, the exacerbated inflammatory response leads to substantial lung tissue damage, after which the Th2-derived cytokines IL-4 and IL-13 drive the conversion of the immune response into an abnormal wound healing response, which is characterized by the accumulation of M2 macrophages that promote fibrosis through the production of TGF-β1. Meanwhile, cytokines stimulate the release of great amounts of PGE2 through activated fibroblasts. PGE2, in turn, triggers a negative feedback on the production of cytokines, and inhibits the transendothelial migration of T cells and the transition to Th2 cells, and downregulates the functions of fibroblasts, including proliferation, collagen synthesis, and myofibroblast transformation.
A few achievements have been made on the cellular and molecular mechanisms of RILI, while there is remarkably little progress in the development of safe and effective therapeutic strategies. Therefore, in-depth studies on the prevention and treatment of RILI should be continued.
Footnotes
Competing interests
The authors have no competing interests to disclose.
Source of support: This work was supported in part by grants from National Natural Science Foundation of China (Grants nos. 31670861, 81472911 and 31270900) and Shanghai Municipal Commission of Health and Family Planning (no. 20164Y0038)
References
- 1.Du Bois RM. Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov. 2010;9(2):129–40. doi: 10.1038/nrd2958. [DOI] [PubMed] [Google Scholar]
- 2.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–61. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 3.Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc Am Thorac Soc. 2006;3(4):364–72. doi: 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- 4.Huang Y, et al. Grape seed pro-anthocyanidins ameliorates radiation-induced lung injury. J Cell Mol Med. 2014;18(7):1267–77. doi: 10.1111/jcmm.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang Y, Liu W, Liu H, et al. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(−/−) mice. Am J Pathol. 2003;163(4):1261–73. doi: 10.1016/s0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaviratne M, Hesse M, Leusink M, et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-beta independent. J Immunol. 2004;173(6):4020–29. doi: 10.4049/jimmunol.173.6.4020. [DOI] [PubMed] [Google Scholar]
- 7.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4(8):583–94. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 9.Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: Competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. 1998;160(11):5347–54. [PubMed] [Google Scholar]
- 10.Munder M, Eichmann K, Morán JM, et al. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163(7):3771–77. [PubMed] [Google Scholar]
- 11.Witte MB, Barbul A, Schick MA, et al. Upregulation of arginase expression in wound-derived fibroblasts. J Surg Res. 2002;105(1):35–42. doi: 10.1006/jsre.2002.6443. [DOI] [PubMed] [Google Scholar]
- 12.Araya J, Nishimura SL. Fibrogenic reactions in lung disease. Annu Rev Pathol. 2010;5:77–98. doi: 10.1146/annurev.pathol.4.110807.092217. [DOI] [PubMed] [Google Scholar]
- 13.Tomcik M, Zerr P, Pitkowski J, et al. Heat shock protein 90 (Hsp90) inhibition targets canonical TGF-beta signalling to prevent fibrosis. Ann Rheum Dis. 2014;73(6):1215–22. doi: 10.1136/annrheumdis-2012-203095. [DOI] [PubMed] [Google Scholar]
- 14.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–61. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 15.Khalil N, Corne S, Whitman C, Yacyshyn H. Plasmin regulates the activation of cell-associated latent TGF-beta 1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am J Respir Cell Mol Biol. 1996;15(2):252–59. doi: 10.1165/ajrcmb.15.2.8703482. [DOI] [PubMed] [Google Scholar]
- 16.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116(Pt 2):217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 17.Roberts AB, Russo A, Felici A, Flanders KC. Smad3: A key player in pathogenetic mechanisms dependent on TGF-beta. Ann NY Acad Sci. 2003;995:1–10. doi: 10.1111/j.1749-6632.2003.tb03205.x. [DOI] [PubMed] [Google Scholar]
- 18.Lee CG, Homer RJ, Zhu Z, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1) J Exp Med. 2001;194(6):809–21. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitani A, Fuss I, Nakamura K, et al. Transforming growth factor (TGF)-beta1-producing regulatory T cells induce Smad-mediated interleukin 10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-beta1-mediated fibrosis. J Exp Med. 2003;198(8):1179–88. doi: 10.1084/jem.20030917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puthawala K, Hadjiangelis N, Jacoby SC, et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008;177(1):82–90. doi: 10.1164/rccm.200706-806OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SD, Lee DS, Chun YG, et al. Transforming growth factor-beta1 induces endothelin-1 in a bovine pulmonary artery endothelial cell line and rat lungs via cAMP. Pulm Pharmacol Ther. 2000;13(6):257–65. doi: 10.1006/pupt.2000.0252. [DOI] [PubMed] [Google Scholar]
- 22.Miyauchi T, Masaki T. Pathophysiology of endothelin in the cardiovascular system. Annu Rev Physiol. 1999;61:391–415. doi: 10.1146/annurev.physiol.61.1.391. [DOI] [PubMed] [Google Scholar]
- 23.Castañares C, Redondo-Horcajo M, Magán-Marchal N, et al. Signaling by ALK5 mediates TGF-beta-induced ET-1 expression in endothelial cells: A role for migration and proliferation. J Cell Sci. 2007;120(Pt 7):1256–66. doi: 10.1242/jcs.03419. [DOI] [PubMed] [Google Scholar]
- 24.Xu SW, Howat SL, Renzoni EA, et al. Endothelin-1 induces expression of matrix-associated genes in lung fibroblasts through MEK/ERK. J Biol Chem. 2004;279(22):23098–103. doi: 10.1074/jbc.M311430200. [DOI] [PubMed] [Google Scholar]
- 25.Shi-wen X, Kennedy L, Renzoni EA, et al. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum. 2007;56(12):4189–94. doi: 10.1002/art.23134. [DOI] [PubMed] [Google Scholar]
- 26.Emura M, Nagai S, Takeuchi M, et al. In vitro production of B cell growth factor and B cell differentiation factor by peripheral blood mononuclear cells and bronchoalveolar lavage T lymphocytes from patients with idiopathic pulmonary fibrosis. Clin Exp Immunol. 1990;82(1):133–39. doi: 10.1111/j.1365-2249.1990.tb05416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Büttner C, Skupin A, Reimann T, et al. Local production of interleukin-4 during radiation-induced pneumonitis and pulmonary fibrosis in rats: Macrophages as a prominent source of interleukin-4. Am J Respir Cell Mol Biol. 1997;17(3):315–25. doi: 10.1165/ajrcmb.17.3.2279. [DOI] [PubMed] [Google Scholar]
- 28.Fertin C, Nicolas JF, Gillery P, et al. Interleukin-4 stimulates collagen synthesis by normal and scleroderma fibroblasts in dermal equivalents. Cell Mol Biol. 1991;37(8):823–29. [PubMed] [Google Scholar]
- 29.Doucet C, Brouty-Boyé D, Pottin-Clémenceau C, et al. Interleukin (IL) 4 and IL-13 act on human lung fibroblasts. Implication in asthma. J Clin Invest. 1998;101(10):2129–39. doi: 10.1172/JCI741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tiggelman AM, Boers W, Linthorst C, et al. Collagen synthesis by human liver (myo)fibroblasts in culture: evidence for a regulatory role of IL-1 beta, IL-4, TGF beta and IFN gamma. J Hepatol. 1995;23(3):307–17. [PubMed] [Google Scholar]
- 31.Zurawski SM, Vega F, Jr, Huyghe B, Zurawski G. Receptors for interleukin-13 and interleukin-4 are complex and share a novel component that functions in signal transduction. EMBO J. 1993;12(7):2663–70. doi: 10.1002/j.1460-2075.1993.tb05927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reiman RM, Thompson RW, Feng CG, et al. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect Immun. 2006;74(3):1471–79. doi: 10.1128/IAI.74.3.1471-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park SW, Ahn MH, Jang HK, et al. Interleukin-13 and its receptors in idiopathic interstitial pneumonia: Clinical implications for lung function. J Korean Med Sci. 2009;24(4):614–20. doi: 10.3346/jkms.2009.24.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JW, Zoumalan RA, Valenzuela CD, et al. Regulators and mediators of radiation-induced fibrosis: Gene expression profiles and a rationale for Smad3 inhibition. Otolaryngol Head Neck Surg. 2010;143(4):525–30. doi: 10.1016/j.otohns.2010.06.912. [DOI] [PubMed] [Google Scholar]
- 35.Kishikawa H, Sun J, Choi A, et al. The cell type-specific expression of the murine IL-13 gene is regulated by GATA-3. J Immunol. 2001;167(8):4414–20. doi: 10.4049/jimmunol.167.8.4414. [DOI] [PubMed] [Google Scholar]
- 36.Han G, Zhang H, Xie CH, Zhou YF. Th2-like immune response in radiation-induced lung fibrosis. Oncol Rep. 2011;26(2):383–88. doi: 10.3892/or.2011.1300. [DOI] [PubMed] [Google Scholar]
- 37.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: Implications for immune responses and autoimmune diseases. Immunity. 2009;31(4):539–50. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397(6721):710–13. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 39.Ghosh AK, Yuan W, Mori Y, et al. Antagonistic regulation of type I collagen gene expression by interferon-gamma and transforming growth factor-beta. Integration at the level of p300/CBP transcriptional coactivators. J Biol Chem. 2001;276(14):11041–48. doi: 10.1074/jbc.M004709200. [DOI] [PubMed] [Google Scholar]
- 40.Vancheri C, Mastruzzo C, Sortino MA, Crimi N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004;25(1):40–46. doi: 10.1016/j.it.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 41.Gauldie J, Kolb M, Sime PJ. A new direction in the pathogenesis of idiopathic pulmonary fibrosis? Respir Res. 2002;3:1. doi: 10.1186/rr158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olman MA. Epithelial cell modulation of airway fibrosis in asthma. Am J Respir Cell Mol Biol. 2003;28(2):125–28. doi: 10.1165/rcmb.F257. [DOI] [PubMed] [Google Scholar]