

Abstract
Background
MicroRNA (miRNA) is a type of noncoding RNA that can repress the expression of target genes through posttranscriptional regulation. In addition to numerous physiologic roles for miRNAs, they play an important role in pathophysiologic processes affecting cardiovascular health. Previously, we reported that nuclear encoded microRNA (miR‐181c) is present in heart mitochondria, and importantly, its overexpression affects mitochondrial function by regulating mitochondrial gene expression.
Methods and Results
To investigate further how the miR‐181 family affects the heart, we suppressed miR‐181 using a miR‐181‐sponge containing 10 repeated complementary miR‐181 “seed” sequences and generated a set of H9c2 cells, a cell line derived from rat myoblast, by stably expressing either a scrambled or miR‐181‐sponge sequence. Sponge‐H9c2 cells showed a decrease in reactive oxygen species production and reduced basal mitochondrial respiration and protection against doxorubicin‐induced oxidative stress. We also found that miR‐181a/b targets phosphatase and tensin homolog (PTEN), and the sponge‐expressing stable cells had increased PTEN activity and decreased PI3K signaling. In addition, we have used miR‐181a/b−/− and miR‐181c/d−/− knockout mice and subjected them to ischemia‐reperfusion injury. Our results suggest divergent effects of different miR‐181 family members: miR‐181a/b targets PTEN in the cytosol, resulting in an increase in infarct size in miR‐181a/b−/− mice due to increased PTEN signaling, whereas miR‐181c targets mt‐COX1 in the mitochondria, resulting in decreased infarct size in miR‐181c/d−/− mice.
Conclusions
The miR‐181 family alters the myocardial response to oxidative stress, notably with detrimental effects by targeting mt‐COX1 (miR‐181c) or with protection by targeting PTEN (miR‐181a/b).
Keywords: microRNA, miR‐181, mitochondria, mitochondrial miRNA, mitochondrial respiratory complex IV, mt‐COX1, oxidative stress, phosphatase and tensin homolog, PI3 kinase, reperfusion injury
Subject Categories: Ischemia, Oxidant Stress, Myocardial Infarction, Genetically Altered and Transgenic Models, Cell Signalling/Signal Transduction
Introduction
Mitochondria, which possess their own DNA, mRNA, tRNA, and ribosomes,1 are semiautonomous organelles, but they import many critical proteins that are encoded by nuclear genes.1, 2 Critical to mitochondrial function are 5 respiratory chain complexes in the inner mitochondrial membrane that generate a proton gradient across the membrane, which produces ATP.1, 2, 3, 4 Most of the respiratory chain complex subunits are encoded by nuclear genes except for some subunits of complexes I, III, and IV, which are encoded by mitochondrial DNA and synthesized on mitochondrial ribosomes.1 Therefore, coordination of nuclear gene expression and mitochondrial gene expression is essential.
MicroRNAs (miRNAs) are small (~19‐25 nt), noncoding RNAs that regulate posttranscriptional gene expression by binding to the 3′‐UTR of target mRNAs,5 which blocks translation and/or causes mRNA degradation.5 Several groups have suggested that miRNAs exist in mitochondria,6, 7, 8, 9, 10 and supporting this idea, recent work from our group demonstrated that miRNAs exist in heart mitochondria and are functionally important.11, 12 Previously, we showed that miRNA (miR)‐181c, derived from the nuclear genome, translocates to the mitochondria and, importantly, regulates mitochondrial gene expression and alters mitochondrial function. Specifically, we have demonstrated that miR‐181c regulates the mitochondrial gene mt‐COX1, a subunit of complex IV of the respiratory chain, through binding to its 3′‐end. In addition, we found that overexpression of miR‐181c results in decreased mt‐COX1 protein, complex IV remodeling, and increased production of reactive oxygen species (ROS).12
The goal of the present study was to determine whether loss of function of miR‐181 family members in the heart alters cardiac function by altering mitochondrial function. Here, we created a cardiac‐targeted miR‐181 sponge consisting of an antisense sequence of the miR‐181 “seed” sequence to sequester and thereby lower the activity of all endogenous miR‐181 family members. We have found that miR‐181a/b functions to regulate phosphatase and tensin homolog (PTEN) in the cytosol, whereas miR‐181c regulates mt‐COX1 in the mitochondria. In addition, we have validated these in vitro findings in an in vivo mouse model using miR‐181a/b−/− (containing the miR‐181a1 and miR‐181b1 cluster) and miR‐181c/d−/− (containing the miR‐181c and miR‐181d cluster) knockout mice. Our data suggest divergent roles for different miR‐181 family members, which may be important under conditions associated with increased incidence of heart disease.
Methods
Animals
Male C57BL6/J (Jackson Laboratories, Bar Harbor, Maine) were used as WT control for both miR‐181a/b−/− and miR‐181c/d−/− mice. We used 12‐week‐old male mice for all of our studies. They were provided with food and water ad libitum. Mice were treated humanely, and all experimental procedures were approved by the Institutional Animal Care and Use Committee of Johns Hopkins University.
miR‐181 Cardiac‐Specific Sponge Expression Vector
We used plasmid Enhanced Green Fluorescent Protein as a backbone to clone the miR‐181 sponge sequence (sponge group), or a scramble sequence (control group) was inserted between the XhoI and KpnI sites. We also cloned α‐MHC (cardiospecific promoter, generously provided by Dr Jeffery D. Molkentin at Cincinnati Children's Hospital, Cincinnati, OH) into the AscI and NheI sites. This vector also contains an EGFP sequence, which not only helped us to generate stable H9c2 cells but also will help us to confirm its expression level and specificity in the heart of our animal models.
Generation of Stable miR‐181‐Sponge‐H9c2 and Scramble‐H9c2 Cell Line
Using an electroporator (Nucleofector Amaxa, Lonza, Gaithersburg, MD) and following the protocol for rat myoblasts (H9c2), we transfected either the scramble (284‐nt‐long scramble sequence, which replaced the same length of miR‐181‐sponge sequence) or the miR‐181‐sponge construct. After 48 hours of transfection we flow‐sorted the GFP‐positive cells, taking advantage of the plasmid Enhanced Green Fluorescent Protein plasmid. Our backbone has neomycin resistance. So, 24 hours after the flow sorting, we cultured our cells with geneticin (G418 sulfate) (500 μmol/L) in our culture medium for 16 more days following the guidelines from Lonza (Gaithersburg, MD). We calculated the 16 days based on our “kill curve” data from G418 treatment with different concentrations on untransfected H9c2 cells. We seeded 5 to 10 cells in each well of a 96‐well plate by flow‐sorting the G418‐treated cells. We followed the single cloned cells from each well and grew them in a single well of a 12‐well plate followed by a single well in 24‐well plate. From each well of the 24‐well plate we froze the cells (passage 3, p3) and performed all our experiments with the cells between p5 to p10.
Mitochondrial Respiration and Metabolic Studies
Analysis of the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of stable H9c2 cells was performed with a Seahorse XF96 Extracellular Flux Analyzer (Cellular Dynamics, Madison, WI) instrument. In brief, scramble‐H9c2 or miR‐181‐sponge‐H9c2 cells were seeded in the medium at a density of 8000 cells/well on V3‐PET TC‐coated plates, and the OCR and ECAR were measured in real time.
RNA Isolation
Total RNA was isolated from whole hearts, mitochondrial fraction of the hearts, and from the stable H9c2 cell line using a miRNeasy kit (Qiagen, Valencia, CA) and RNase free DNase kit (Qiagen, Valencia, CA).12 To characterize the integrity of the isolated RNA, spectrophotometric evaluation was performed using Nanodrop (Thermo Scientific, Wilmington, DE). All the samples whose A260 (absorbance at 260 nm) value was more than 0.15 were used for further experiments; an A260<0.15 suggests poor quality of RNA. The ratio of the readings at 260 and 280 nm (A260/A280) was also measured in order to check the purity of the isolated RNA. For further and more accurate purity and integrity estimation of the isolated RNA, especially the RNA isolated from mitochondrial fractions, a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA) was used.12
Quantitative Real‐Time Polymerase Chain Reaction
After performing the purity and integrity test, the RNA was reverse transcribed using a miScript Reverse Transcription Kit (Qiagen, Valencia, CA). The polymerase chain reaction (PCR) was performed using a miScript SYBR green PCR kit (Qiagen, Valencia, CA) and detected with a CFX96 detector (Bio‐Rad, Hercules, CA). All reactions were performed in triplicate. Table shows all the primer sequences for the encoded mitochondrial genes.
Table 1.
Primer Sequences for Quantitative Real‐Time Polymerase Chain Reaction
| Primer | Forward (5′‐3′) | Reverse (5′‐3′) |
|---|---|---|
| COX1 | TCCAACTCATCCCTTGACATC | TCCTGCTATGATAGCAAACACT |
| COX2 | CTAATTAGCTCCTTAGTCCTC | TTCGTAGCTTCAGTATCATTG |
| COX3 | ATTCTATTCATCGTCTCGGAA | AAGGCTATGATGAGCTCATGT |
| 16S rRNA | ACCGCAAGGGAAAGATGAAA | GCCACATAGACGAGTTGATTC |
| 12S rRNA | CAAACTGGGATTAGATACCCCACTAT | GAGGGTGACGGGCGGTGTGT |
Myocyte Isolation
For the isolation of adult cardiomyocytes, mouse hearts were quickly removed from the chest after euthanasia, and the aorta was perfused with enzymatic perfusion solution (in mmol/L: 120 NaCl, 5.4 KCl, 1.2 NaH2PO4, 20 NaHCO3, 1.6 MgCl2; glucose [1 mg/mL], 2,3‐butanedione monoxime [BDM, 1 mg/mL], and taurine [0.628 mg/mL]) containing 0.9 mg/mL collagenase type 2 (Worthington Biochemical Co, Lakewood, NJ;, 299 U/mg) and 0.05 mg/mL protease type XIV (Sigma Chemical Co, St. Louis, MO) at 37°C for 9 to 10 minutes.13, 14
Measurement of Sarcomere Shortening and Ca2+ Transients
Dispersed myocytes were filtered and gently centrifuged at 700 rpm for 1 minute. The pellet was resuspended in 1× Tyrode solution (140 NaCl, 5 KCl, 10 HEPES, 1.6 MgCl2; glucose [1 mg/mL]) with increasing Ca2+ up to 1 mmol/L, and cells were then incubated for 20 minutes with 1 mmol/L of the Fura‐2AM (Invitrogen, Molecular Probes, Eugene, OR). After rinsing, cells were placed in a perfusion chamber and stimulated at 0.5 Hz. Changes in sarcomere length and Ca2+ transients were recorded on an inverted fluorescence microscope (Nikon, Tokyo, Japan; TE2000), and the data were analyzed with an IonOptix system, as described previously.14
Langendorff Mouse Heart Preparation
After sufficient anesthesia was achieved with ketamine (19 mg/kg body wt)/xylazine (10 mg/kg body wt) intraperitoneal cocktail, mice were anticoagulated with heparin sodium (500 IU/kg body weight, intravenous injection) (Elkin‐Sinn Inc, Cherry Hill, NJ). Hearts were excised, cannulated, and perfused with Krebs‐Henseleit buffer containing (in mmol/L) NaCl 120, KCl 5.9, MgSO4 1.2, CaCl2 1.25, NaHCO3 25, and glucose 11. The buffer was aerated with 95% O2 and 5% CO2, to give a pH of 7.4 at 37°C as described previously.15 All hearts were perfused to wash out blood and stabilize for 20 minutes, followed by 20 minutes of global ischemia and 120 minutes of reperfusion. Left ventricular diastolic pressure (LVDP) and heart rate were continuously monitored via a water‐filled balloon inserted into the left ventricle. Recovery of contractile function was assessed by measurement of LVDP during reperfusion and was expressed as a percentage of preischemic, pretreatment LVDP. Rate‐pressure product was calculated by multiplying LVDP by heart rate at that particular time point.
At the end of reperfusion, infarct size was measured with 2,3,5‐triphenyltetrazolium chloride (TTC) as described previously.15 Hearts were initially perfused with TTC and then incubated in TTC for an additional 15 minutes at 37°C. The hearts were subsequently fixed in formalin, and 4 to 6 cross‐sectional slices were taken. These slices were imaged on a Leica Stereoscope, and the percentage of infarct (white area) to viable tissue (red area) was analyzed using ImageJ software. Area of infarct was expressed as a percentage of the total ventricles.
Isolated Mitochondria Protocols
Freshly isolated mitochondria were prepared from hearts after perfusion with RNAlater (Sigma‐Aldrich, St. Louis, MO), by differential centrifugation.15 Briefly, at the end of perfusion, the left ventricle was dissected out and placed in Buffer A (in mmol/L: 180 KCl, 2 EGTA, 5 MOPS, 0.2% BSA; pH 7.25). The tissue was then digested with trypsin (0.0001/0.1 g tissue) in 0.7 mL of ice‐cold Buffer B (in mmol/L: 225 Mannitol, 75 sucrose, 5 MOPS, 0.5 EGTA, 2 taurine; pH 7.25) and finally homogenized with Buffer B with a protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN) using a Polytron (Kinematica, Bohemia, NY). To further separate the heart mitochondria from other cellular components and tissue debris, a series of differential centrifugations were performed in a Microfuge 22R centrifuge (Beckman Coulter, Fullerton, CA) at 4°C. The crude pellet was then lysed with QIAzol (Qiagen, Valencia, CA).12
ROS Production Assay
Hydrogen peroxide (H2O2) production from isolated mitochondria was measured fluorimetrically by measurement of oxidation of amplex red to fluorescent resorufin (Life Technologies, Carlsbad, CA). Isolated mouse heart mitochondria or cultured stable H9c2 cells were incubated in buffer containing 120 mmol/L KCl, 1 mmol/L EGTA, 5 mmol/L MOPS, and 5 mmol/L K2HPO4 (pH 7.25). All incubations also contained 50 μmol/L amplex red and 5 U/mL of horseradish peroxidase. The increase in fluorescence at an excitation of 544 nm and an emission of 590 nm was monitored. Standard curves were generated using known amounts of H2O2.12
Mitochondrial Swelling Assay
Mitochondrial permeability transition pore opening was assessed using a mitochondria swelling assay, described previously.15 Briefly, mitochondrial pellets were resuspended in EGTA‐free buffer (in mmol/L: 225 mannitol, 75 sucrose, 5 MOPS, 2 taurine, pH 7.25). About 300 μg of mitochondria were then added to the assay buffer (in mmol/L: 120 KCl, 10 Tris‐HCl, 5 MOPS, 5 KH2PO4, pH 7.25) and energized with glutamate/malate (10/2 mmol/L). The absorbance was monitored at 540 nm. After 2 to 3 minutes, 250 μmol/L calcium was added, and the absorbance was recorded for an additional 8 to 10 minutes.
Electron Microscopy
For transmission electron microscopy the isolated mitochondria were chemically fixed in 3% glutaraldehyde in 0.1 mol/L sodium phosphate buffer (pH 7.3) for 24 hours at 4°C, rinsed in 0.1 mol/L sodium phosphate buffer, and postfixed in 1% osmium tetroxide in the same buffer for 1 hour at room temperature. The samples were dehydrated in a graded series of ethanol, transitioned with toluene, followed by infiltration and embedding in epoxy resin EPON 812 (Polysciences, Warrington, PA). Semithin sections of 500 nm thickness were cut and stained with 1% toluidine blue for visualization by light microscopy. Ultrathin sections of selected areas were cut at a thickness of ~100 nm (gold interference color) with a diamond knife (Diatome, Hatfield, PA), placed on 200 mesh copper grids, and dried at 60°C for 10 minutes. To impart electron contrast, the sections were stained with a saturated solution of uranyl acetate for 10 minutes followed by lead citrate for 2 minutes. The sections were examined with a transmission electron microscope (Philips CM12 TEM) using a tungsten filament operating at an accelerating voltage of 60 keV. Images were acquired using a Morada 11 Megapixel side‐mounted transmission electron microscope CCD camera (Olympus Soft Imaging Solutions, Münster, Germany). Digitally, using iTEM software, we have calculated the long‐side diameter of each mitochondrion.
Western Blot
Stable H9c2 cells, mouse heart, and isolated mitochondrial fractions were lysed with RIPA buffer, and protein content was measured using the Bradford assay. Cell and tissue homogenate protein was separated by 1‐dimensional gel electrophoresis. After transfer to a polyvinylidene difluoride membrane, the membrane was incubated with antibodies that recognize proteins such as mt‐COX1 (Abcam, Cambridge, MA), mt‐COX2 (Life Technologies, Carlsbad, CA), mt‐COX3 (Life Technologies, Carlsbad, CA), COX 5A (Abcam, Cambridge, MA), COX 5B (Abcam, Cambridge, MA), COX VIIa (Abcam, Cambridge, MA), pAkt at Ser 473 (Cell Signaling, Danvers, MA), Akt (Cell Signaling, Danvers, MA), α‐tubulin (Abcam, Cambridge, MA), and VDAC (Abcam, Cambridge, MA) in Tris‐buffered saline (pH 7.4) with 1% TWEEN 20 (TBS‐T) and 5% BSA or nonfat dry milk at 4°C overnight. Membranes were incubated with the appropriate secondary antibody conjugated to horseradish peroxidase IgG in TBS‐T with 5% nonfat dry milk for 1 hour at room temperature. Immunoreactive protein was visualized using an enhanced chemiluminescence analysis kit (EMD Millipore, Temecula, CA).
Statistical Analysis
The results are presented as mean and standard error of the mean (mean±SEM). All other data were analyzed by Wilcoxon signed‐rank test (nonparametric test). For sample size 3 (n=3) we used Student t test to calculate P‐value. P<0.05 was considered statistically significant. For the isolated adult cardiomyocyte measurements, P values were determined using the nonparametric Mann‐Whitney test.
Results
Evaluation of miR‐181‐Sponge Activity
Evaluation of genomic structure indicated that there are 6 mature miRNAs—miR‐181a1, miR‐181a2, miR‐181b1, miR‐181b2, miR‐181c, and miR‐181d—which are encoded by 3 independent sequences located on 3 separate chromosomes16 (Figure 1A). The corresponding premature miRNAs (pre‐miR) lead to expression of 4 sets of mature miRNA, miR‐181—miR‐181a, miR‐181b, miR‐181c, and miR‐181d16—which share the same “seed” sequence “ACAUUCA” (Figure 1A). In human, the 3 paralog precursor transcripts are found on chromosome (Chr)‐1 (miR‐181a1 and miR‐181b1), on Chr‐9 (miR‐181a2 and miR‐181b2), and on Chr‐19 (miR‐181c and miR‐181d).16 However, in mouse, the 3 paralogs are on Chr‐1 (miR‐181a1 and miR‐181b1), on Chr‐2 (miR‐181a2 and miR‐181b2), and on Chr‐8 (miR‐181c and miR‐181d) (Figure 1A). The miR‐181a/b1 cluster is found in an intron of a noncoding RNA host gene (MIR181A1‐HG), the miR‐181a/b2 cluster is found in an intron of the NR6A1 gene, and the miR‐181c/d cluster is found in an uncharacterized sequence on either Chr‐19 (human/rat/monkey) or Chr‐8 (mouse).
Figure 1.
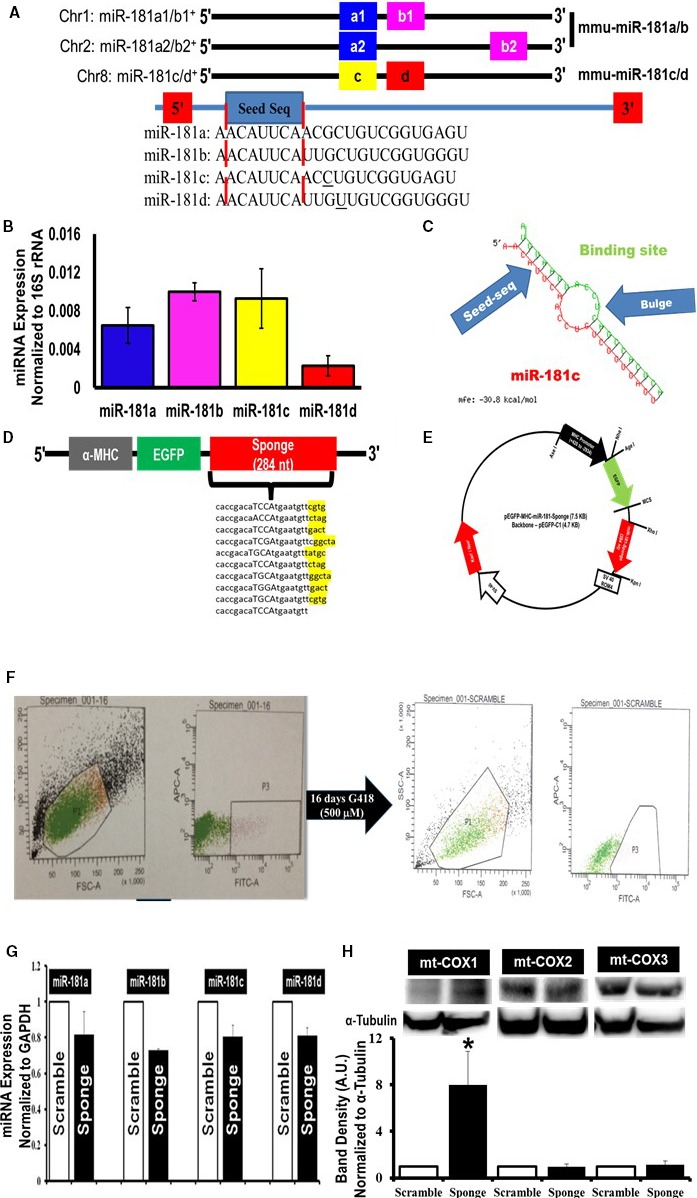
Mitochondrial localization of microRNA‐181 (miR‐181) family and miRNA‐family‐wide knockdown strategy. A, Schematic depiction of mouse miR‐181 family. Two independent paralog transcripts of miR‐181a/b precursors—miR‐181a1/a2 (blue) and miR‐181b1/b2 (pink). The names of the mature miRNAs are denoted in the boxes. The blue box with “seed” labels in the box highlights the conserved sequence among all 4 mature miR‐181, indicated inside the red dotted lines. B, Quantitative polymerase chain reaction (qPCR) (SYBR kit) analysis of miR‐181 family (a, b, c, and d) expression in total RNA from the mitochondrial pellets of C57BL6/J mouse hearts. The miRNA expression was normalized to mitochondrial 16S rRNA. C, Predicted structure of miR‐181 sponge and miR‐181 family. The red side of this structure is miR‐181 family, in this particular case miR‐181c. The green side is 1 of the 10 transcripts of miR‐181 sponge. The binding site shows the antisequence of the seed sequence of miR‐181. The bulge formation gives stability to the structure. The energy required for the stability of this structure is −30.8 kcal/mol. D, Design of an EGFP (enhanced green fluorescent protein) expression construct under the regulation of a heart‐specific promoter, α‐myosin heavy chain (α‐MHC) and containing either the miR‐181 sponge construct (sponge) or scramble sequence (scramble, not shown). The 4 nucleotides indicated with capital letters immediately after the anti‐miR‐181 seed sequence are the spacer, and nucleotides highlighted in yellow after each of the 9 anti‐miR‐181 sequences are the spacer between each anti‐miR‐181 seed sequence. E, Cardiospecific miR‐181 sponge vector map. F, Generation of cardiospecific miR‐181‐sponge‐H9c2 and cardiospecific scramble‐H9c2 from single‐cell clones. Gate P3 in the left part of F represents the green fluorescent protein–positive (GFP +) H9c2 cells, only 8.2% of total number of H9c2 cells, which were sorted and cultured for another 16 days with G418. Gate P3 in the right part of F represents GFP + H9c2 cells, 60% of the total number of cells, which were again sorted and seeded at 5 to 10 cells/well of a 96‐well plate. G, Quantitative PCR data examining miR‐181 family expression in the stable miR‐181‐sponge‐H9c2 (sponge) and the scrambled‐sponge H9c2 (scramble) cells. A slight decrease of the entire miR‐181 family, miR‐181a, b, c, and d, was decreased in the EGFP‐miR‐181‐sponge–expressing group, compared to EGFP‐miR‐181‐scramble‐H9c2 cells. We normalized the data to the GAPDH expression. H, Western blot analysis of mitochondrial cytochrome c oxidase (mt‐COX) expression in miR‐181‐sponge and scramble‐sponge H9c2 cells. The mt‐COX1 (upper band, left side), mt‐COX2 (upper band, in the middle) and mt‐COX3 (upper band, right side) expression was normalized to α‐tubulin (lower bands). Bar graphs show the quantification of protein expression. Protein lysate was prepared from the whole cell pellets. *P<0.05 vs scramble (n=3).
Previously, we discovered miR‐181c in heart mitochondria based on microarray data from rat heart‐derived mitochondria.12 Here, we measured miR‐181a, b, c, and d using quantitative PCR in the mitochondrial fraction of C57BL6/J (WT) mouse heart. Expression of mature miRNAs was normalized to the mitochondrial 16S rRNA (Figure 1B). Because the entire miR‐181 family of miRNAs appears to be present in the mitochondria, we hypothesized that knocking down the entire miR‐181 family may provide a better understanding of the underlying mechanism by which miR‐181 modulates cardiovascular health. Therefore, we constructed a heart‐targeted miR‐181‐sponge to sequester and thereby lower the activity of the entire miR‐181 family. Previously, a similar miR‐sponge method has been validated in in vitro and in vivo studies in other tissues.17, 18, 19, 20
We designed the miR‐181 sponge using the plasmid Enhanced Green Fluorescent Protein‐C1 vector backbone based on design principles previously published.17, 18 Specifically, our sponge vector contained 10 repeated antisense miR‐181 seed sequences in the 3′‐UTR at the end of the EGFP ORF (Figure 1C). In addition, we cloned in a rat α‐myosin heavy chain promoter to drive expression of the EGFP‐UTR (Figure 1D). As a control, we also created an α‐MHC promoter‐EGFP vector that contained a scrambled sequence of equivalent length and composition in place of the miR‐181 antisense binding region (Figure 1E). We generated 2 groups of stable H9c2 cells expressing either the EGFP‐scramble UTR or the EGFP‐miR‐181‐sponge (Figure 1F). In stably transfected EGFP‐miR‐181‐sponge–expressing cells, levels of the entire miR‐181 family were moderately decreased relative to EGFP‐scrambled–expressing cells (Figure 1G), consistent with other miRNA sponge studies.21 Because the major function of sponge mRNA is to serve as a competitive inhibitor of miR‐181 for endogenous targets, we expect relief of the inhibitory effect of miR‐181c mitochondrial target gene mt‐COX1. By Western blot, only mt‐COX1 demonstrated increased expression in the EGFP‐miR‐181‐sponge‐H9c2–expressing cells compared to scramble‐H9c2 cells (Figure 1H). Because mt‐COX2 and mt‐COX3 are found directly after mt‐COX1 on the polycistronic primary transcript,12, 22 we analyzed whether the EGFP‐sponge‐H9c2–expressing cells altered the expression of these genes. Western blot showed that expression of mt‐COX2 and mt‐COX3 was not altered (Figure 1H). We used α‐tubulin as normalization control for the Western blots. The effect was only on mt‐COX1.
Decreased miR‐181 Expression Alters Mitochondrial Function
Because miR‐181c targets mt‐COX1 and overexpression results in complex IV dysfunction and increased ROS production,12 we measured ROS production in the EGFP‐miR‐181‐sponge and EGFP‐scrambled stable H9c2 cells. EGFP‐miR‐181‐sponge‐H9c2 cells significantly reduced the rate of ROS generation compared to scramble‐H9c2 cells (Figure 2A). We measured ROS under state 4 conditions (without adding excess ADP to the medium).11 Because decreased miR‐181 expression can affect the electron transport chain, we hypothesized that manipulation of miR‐181 might alter metabolism. To examine this we measured OCR and anaerobic acid production in intact cells. OCR was measured at baseline and after addition of oligomycin, a complex V inhibitor, followed by uncoupler, carbonilcyanide p‐triflouromethoxyphenylhydrazone, iodoacetate (inhibitor of glycolysis), and rotenone (complex I inhibitor) plus antimycin (complex III inhibitor) (Figure 2B). We did not observe any differences in mitochondrial respiration between scrambled‐ and miR‐sponge–expressing cells (Figure 2C). The ECAR is a measure of glycolysis as pH change is due to lactic acid production under anaerobic conditions (Figure 2D). However, miR‐181‐sponge‐H9c2 cells displayed a significant reduction in ECAR (Figure 2E), indicating that increasing the expression of miR‐181 target(s) by sequestering miR‐181 may decrease glycolysis. Consistent with this finding, a previous study found that miR‐181a/b targets cytosolic PTEN and thereby regulates glycolysis in thymocytes.23
Figure 2.
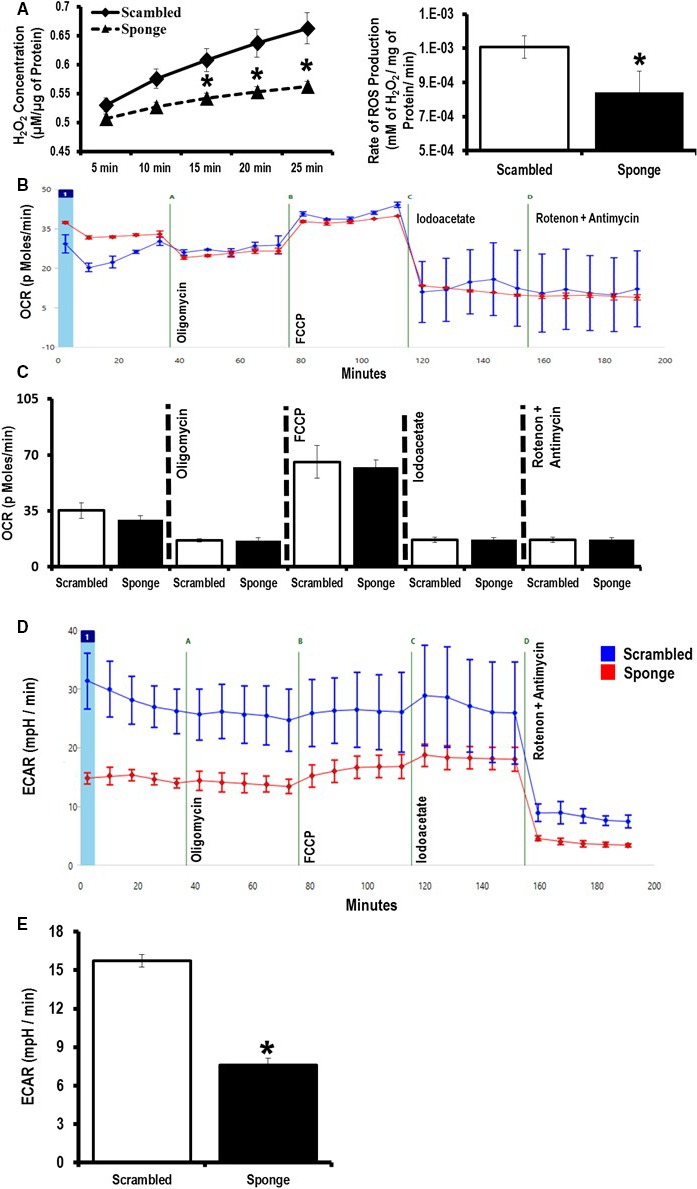
Effect of microRNA (miR)‐181‐sponge on mitochondrial function and cellular metabolism. A, Measurement of H2O2 production as an indicator of reactive oxygen species (ROS) generated from the miR‐181‐sponge and scrambled sponge H9c2 cell lines. B, Representative trace for the mitochondrial O2 consumption rate (OCR). C, Analysis of OCR under basal and after addition of oligomycin, carbonilcyanide p‐triflouromethoxyphenylhydrazone (FCCP), iodoacetate, and rotenone/antimycin in miR‐181‐sponge compared to scramble‐expressed H9c2 cells. D, Representative trace for extracellular acidification rates (ECAR). E, Analysis of glycolysis using ECAR, in miR‐181‐sponge compared to scramble‐expressed H9c2 cells. *P<0.05 vs scramble (n=8).
Decreased miR‐181‐Expression Regulates Cellular Metabolism by Targeting Cytosolic Genes
Our ECAR data suggest that miR‐181a/b may have a conventional cytosolic target(s) in the heart. Because miR‐181a/b has been shown to target PTEN,23 we measured PTEN expression by Western blot and found increased PTEN expression in the EGFR‐miR‐181‐sponge cells compared to EGFP‐scrambled–expressing cells (Figure 3A and 3B). Because PTEN is a negative regulator of the PI3K‐Akt signaling cascade,24 we looked at Akt phosphorylation at the Ser 473 site in EGFP‐miR‐181‐sponge–expressing cells. We observed a significant decrease in pAkt (Ser 473) in miR‐181‐sponge‐H9c2 cells compared to EGFP‐scramble–expressing cells (Figure 3A and 3C).
Figure 3.
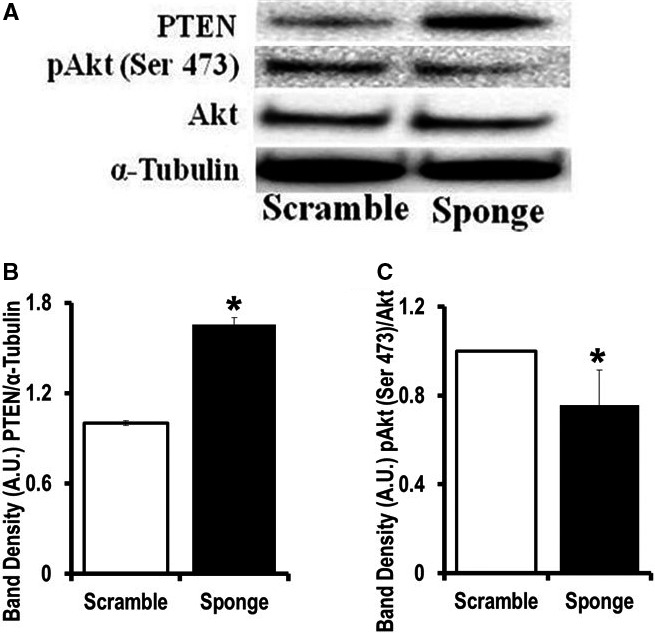
MicroRNA (miR)‐181‐sponge‐expressing cells have upregulated PTEN expression. A, Western blot analysis of phosphatase and tensin homolog (PTEN), phospho‐Akt Ser 473, and total Akt expression in the miR‐181‐sponge‐H9c2 and scramble‐sponge–expressing cells. Lysates were probed with the indicated antibodies. α‐Tubulin is a loading control. B and C, Band‐densitometry is indicated. B, *P<0.05 vs EGFP‐scramble‐H9c2. C, *P<0.05 vs scramble (n=4).
miR‐181‐Sponge Protects Against Oxidative Stress
Our work suggests a reciprocal relationship between miR‐181c activity and ROS production, so we postulated that the miR‐181 sponge would protect cells against ROS‐induced injury. To test this hypothesis, we treated EGFP‐miR‐181‐sponge‐H9c2 cells and EGFP‐scramble‐H9c2 cells with DOX, which is known to cause cell injury through mitochondrial ROS production.25 We used lactic acid dehydrogenase release as an index of cell injury, and we observed no lactic acid dehydrogenase release in EGFP‐miR‐181‐sponge‐H9c2 cells (Sp) and in EGFP‐scramble‐H9c2 cells (Scr) after 48 hours of culture (Figure 4A). DOX treatment resulted in greater cell death in the scramble (Scr+DOX) than in the sponge‐containing cells (Sp+DOX) (Figure 4A). This could be the result of lowering miR‐181c in the mitochondria, resulting in less ROS production, but there could also be an effect related to lowering miR‐181a/b in the cytosol, resulting in increased PTEN expression and less activity of the PI3K pathway. To eliminate the effect of miR‐181a/b sequestration in the cytosol, we used the PI3K‐inhibitor LY294002 (LY): 20 μmol/L LY treatment for 48 hours did not induce lactic acid dehydrogenase release in the 2 control groups, (Scr+LY) and (Sp+LY) (Figure 4A). We did not observe any significant effect of LY treatment in either the scramble or sponge‐treated groups, suggesting that differences in PTEN activity and PI3K pathway activity were not responsible for the difference in cell injury between the EGFP‐miR‐181‐sponge‐H9c2 cells (Sp+DOX) compared to EGFP‐scramble‐H9c2 cells (Scr+DOX). These data are consistent with previous reports that suggested that DOX treatment does not alter the PI3K signaling pathway.26 To assess the potential role of PTEN upregulation, we used insulin (200 nmol/L) to stimulate PI3K signaling. We hypothesized that the upregulation of PTEN in the EGFP‐miR‐181‐sponge‐H9c2 cells would decrease Akt phosphorylation during insulin‐induced PI kinase activation. Using Western blot, we measured the phosphorylation of Akt at Thr 308 as an index of PI3 kinase activity. We observed an almost 3‐fold less increase in p‐Akt (Thr 308)/Akt in EGFP‐miR‐181‐sponge‐H9c2 cells after 5 minutes of insulin treatment compared to EGFP‐scramble‐H9c2 cells with insulin treatment (Figure 4B, left panel). The total Akt level was unaltered (Figure 4B, right panel). We used untreated EGFP‐miR‐181‐sponge‐H9c2 cells as a negative control to demonstrate the effect of insulin treatment in PI3K pathway activation. This indicates a functional role for the miR‐181 family regulating important signaling pathways in the heart, notably with detrimental results by targeting mt‐COX1 in the mitochondria (miR‐181c)11, 12 or with protection by targeting PTEN in the cytoplasm (miR‐181a/b).
Figure 4.
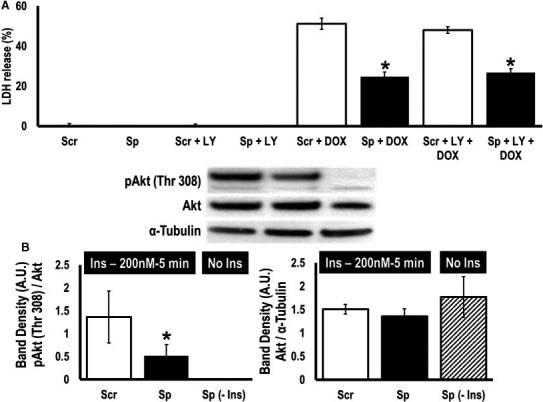
MicroRNA (miR)‐181‐sponge protects cells against oxidative stress induced by doxyrubicin. A, Analysis of doxorubicin (DOX) treatment on cellular viability. Lactic acid dehydrogenase release, in scramble‐sequence (Scr)‐ and miR‐181‐sponge (Sp)‐expressing cells treated with 10 μmol/L with/without LY294002 (LY) for 48 hours. *P<0.05 vs corresponding scramble (n=8). B, Western blot analysis of pAkt Thr 308, and total Akt expression in the scramble‐sponge (Scr)‐ and miR‐181‐sponge (Sp)‐expressing cells pretreated with insulin (200 nmol/L) for 5 minutes after 16 hours of serum starvation. We have used miR‐181‐sponge untreated group (Sp‐Ins) as a negative control for insulin treatment. Lysates were probed with the indicated antibodies. Total Akt was used as a normalization control for pAkt (Thr 308). We also used α‐tubulin to normalize total Akt expression. *P<0.05 vs scramble (n=4).
Loss of miR‐181 Family miRNAs Modulates Cardiac Function
We also wanted to study the effects of miR‐181 family miRNAs in vivo. To better understand the dual roles of the miR‐181 family in cardiac myocytes in vivo, we used 2 sets of knockout mice, miR‐181a/b−/− and miR‐181c/d−/−. These knockout mice have been characterized previously.23 These 2 sets of knockout mice are on a C57BL6/J background,23 so we used C57BL6/J WT mice for our control group. We measured miR‐181a, b, and c expression in the heart tissue of these 2 sets of knockout mice (Figure 5A). Based on our discussion with Qiagen R&D (primers provider), the miR‐181a‐5p primer can detect miR‐181c‐5p. This is because the sequences are so similar with just 1 bp deletion at bp position 12 from the 3′‐end of the primer. The homology between miR‐181a and miR‐181c is almost 99%. Similarly, the miR‐181b‐5p primer can also detect miR‐181d‐5p (first mismatch is at bp position 9 from the 3′‐end of the primer sequence). Thus, it is possible that the miR‐181a primer will detect miR‐181c, and the miR‐181b primer will detect miR‐181d in our mouse model. Furthermore, our miR‐181a/b−/− mice have deletion of MIR181A1‐HG segment, and the miR‐181a2/b2 cluster is still intact in the heart tissue of this miR‐181a/b−/− animals. This could explain the ~40% deletion of miR‐181a and miR‐181b in the heart tissue of our miR‐181a/b−/− mouse model (Figure 5A). On the other hand, ~50% expression for the miR‐181c in the miR‐181c/d−/− mouse hearts is due to the cross‐reactivity of the miR‐181c primer to the miR‐181a (off‐target effect). Nevertheless, miR‐181a/b−/− showed a significant decrease in miR‐181a and miR‐181b, and miR‐181c/d−/− showed a significantly lower miR‐181c expression in the heart (Figure 5A). Using echocardiography to assess cardiac function in vivo (Figure 5B), we have observed a significant decrease in both fractional shortening and ejection fraction in 12‐week‐old miR‐181a/b−/− animals compared to WT mice (Figure 5C and 5D). In contrast, ejection fraction and fractional shortening were not different between the miR‐181c/d−/− and WT mice (Figure 5B and 5C). Whereas miR‐181c/d−/− and WT were comparable in heart weight to tibial length ratio, miR‐181a/b−/− mice showed evidence of hypertrophy at 12 weeks of age (Figure 5E).
Figure 5.
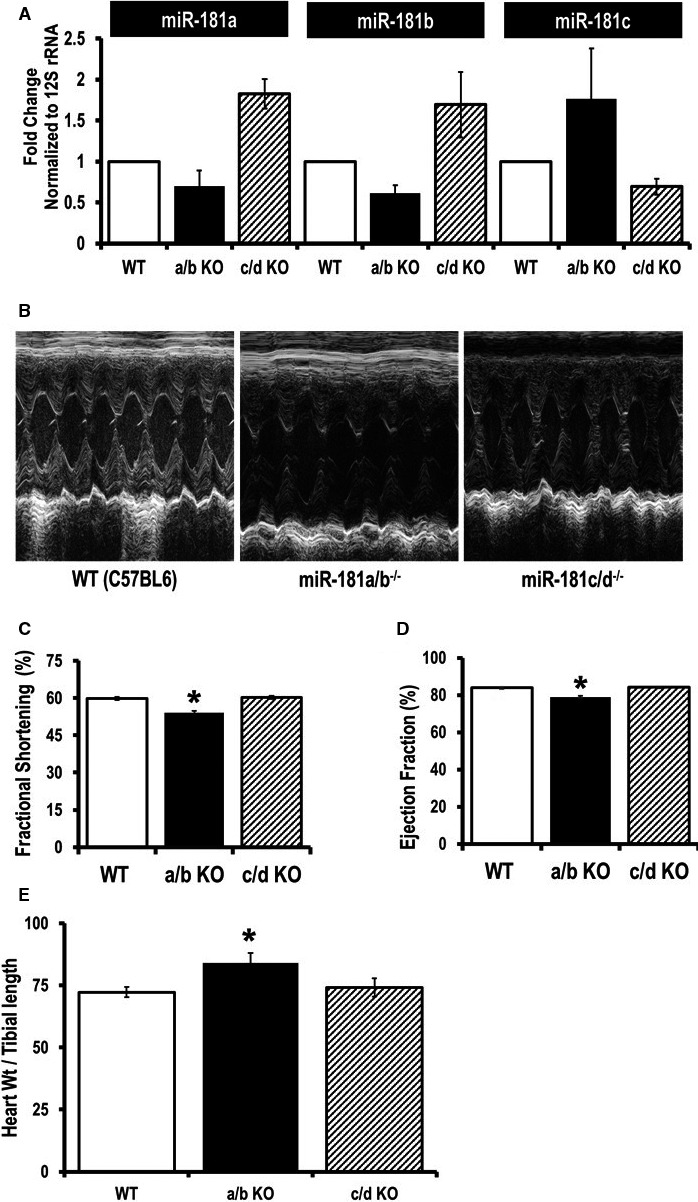
Examination of cardiac function in miR‐181a/b−/− and miR‐181c/d−/− mice. A, Quantitative polymerase chain reaction (SYBR kit) analysis of miR‐181a, b, and c expression in total RNA from the heart tissues of miR‐181a/b−/− (a/b KO), and miR‐181c/d−/− (c/d KO) mice. The miRNA expression was normalized to mitochondrial 16S rRNA. B, Two‐dimensional M‐mode and Doppler echocardiography were performed on nonanesthetized 12 week old mice: wild type (WT C57BL6) mice (left), miR‐181a/b−/− mice (middle panel), and miR‐181c/d−/− mice (right). C, Percentage fractional shortening and (D) ejection fraction were calculated using the software of the echocardiography instrument (n=8). E, Signs of hypertrophy (heart weight/tibial length) were measured (n=5). *P<0.05 vs WT.
miR‐181a/b Deficiency Inhibits PI3K Signaling Through Upregulation of PTEN in the Heart
RNA‐Seq data have identified significantly higher PTEN mRNA levels in miR‐181a/b−/− mouse thymocytes compared to WT mice.23 Because upregulation of PTEN was detected in EGFR‐miR‐181‐sponge‐H9c2 cells, we hypothesized that the lower fractional shortening and ejection fraction in miR‐181a/b−/− mice was due to chronic inhibition of the PI3K pathway. To investigate this, we analyzed PTEN expression and Akt phosphorylation by Western blot in miR‐181 knockout mice (Figure 6A and 6B). We observed a significant increase in PTEN expression in the miR‐181a/b−/− heart (Figure 6A). We also identified a significant decrease in pAkt (Ser 473) in miR‐181a/b−/− hearts compared to the wild type (WT) heart, suggesting that the loss of miR‐181a/b inhibits the PI3K pathway (Figure 6B). We then determined the expression of the entire miR‐181 family in the total heart homogenate of a WT (C57Bl6/J) mice. Quantitative PCR data showed a significantly higher expression of miR‐181a and miR‐181b in the heart compared to miR‐181c expression (Figure 6C). Figure 6C can clarify 2 very important facts about the miR‐181 family in the heart: (1) if we compare Figure 6C with Figure 1B, we can say almost the entire expression profile of miR‐181a and miR‐181b is in the cytoplasmic fraction, and (2) almost 50% expression of miR‐181c in our miR‐181c/d−/− mouse hearts is due to the cross‐reactivity of the miR‐181c primer with the highly abundant miR‐181a in the heart tissue (Figure 5A). Taken together, our data demonstrate that miR‐181a/b targets PTEN in the heart. Thus, in the miR‐181a/b−/− hearts, significant upregulation of PTEN results in the inhibition of the PI3K pathway, which may explain the cardiac dysfunction in these mice (Figure 5B through 5D). In contrast, no difference in PTEN expression was observed between the WT and miR‐181c/d−/− groups. However, pAkt was significantly higher in the miR‐181c/d−/− mouse hearts compared to the WT group, suggesting that lower ROS in the miR‐181c/d−/− mouse heart might play an important role in activating the PI3K/Akt pathway.
Figure 6.
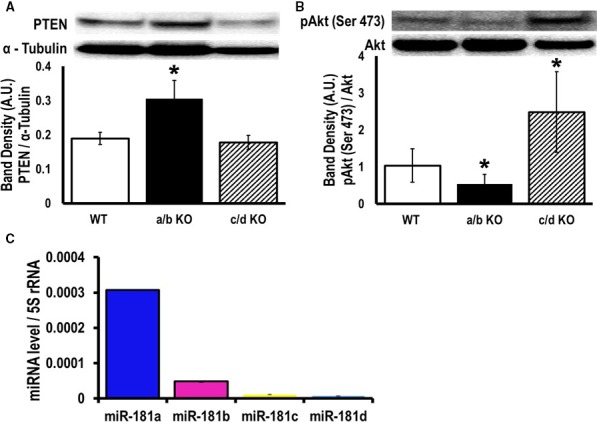
MicroRNA (miR)‐181a/b inhibits PI3K pathway activity in the miR‐181a/b−/− mouse heart through modulation of phosphatase and tensin homolog (PTEN). Western blot analysis of (A) PTEN, (B) phosphorylation of Akt (pAkt) Ser 473, and total Akt expression in the heart from wild type (WT), miR‐181a/b−/−, and miR‐181c/d−/− knockout mice whole‐heart homogenates were probed with the indicated antibodies. α‐Tubulin is a loading control. Band densitometry is shown in the bar graph. C, Quantitative polymerase chain reaction (SYBR kit) analysis of miR‐181 family (a, b, c, and d) expression in total RNA from the heart homogenate of WT mouse hearts. miRNA expression was normalized to 5S r RNA. *P<0.05 vs WT, n=4.
miR‐181a/b Affects Cardiomyocyte Contraction by Targeting PTEN
Global miR‐181a/b−/− mice have an immune phenotype involving absence of mature NKT and severe defects in lymphoid development and T‐cell homeostasis,23 which could confound the cardiac phenotype. To determine if the changes in fractional shortening (Figure 5C) and ejection fraction (Figure 5D) were due to intrinsic defects in cardiomyocytes, we isolated adult cardiomyocytes from miR‐181a/b−/− hearts and measured sarcomere shortening and Ca2+ transients. The miR‐181a/b−/− myocytes showed a significantly shorter peak amplitude of Ca2+ transients compared to WT (C57BL/6J) myocytes and displayed slower velocities in the times to 50% and 90% relaxation. However, their time to peak amplitude was not different (Figure 7A through 7C).
Figure 7.
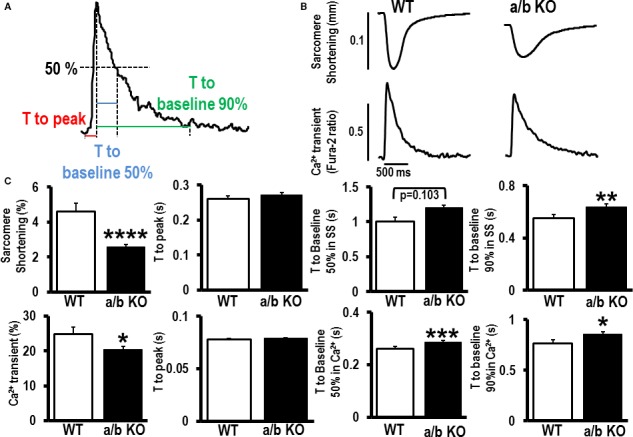
Role of microRNA (miR)‐181a/b in the fundamental changes in cardiomyocyte contractility. A, Definitions for calcium (Ca2+) transient analysis. B, Representative sarcomere shortening and Ca2+ transients for miR‐181a/b−/− (a/b KO) mouse myocytes compared to wild type (WT) C57BL/6J adult myocytes. C, Quantification of peak amplitude of sarcomere shortening and Ca2+ transients, time to peak, time to 50% and 90% return to baseline measured with WT (n=20) and a/b KO (n=73‐75) mouse myocytes. Data are mean±SEM; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
We further examined the contractile properties of miR‐181a/b−/− by measuring sarcomere shortening with optical video microscopy. The miR‐181a/b−/− myocytes showed significantly shorter amplitude of sarcomere shortening compared to WT myocytes and displayed slower velocity in time to 90% relaxation. The miR‐181a/b−/− and WT myocytes showed similar characteristics in the time to peak of sarcomere shortening and time to 50% relaxation (Figure 7B and 7C).
The miR‐181 Family Plays a Role in Myocyte Death After Ischemia
The miR‐181a/b knockout mice demonstrated a detrimental cardiac phenotype, suggesting a beneficial role for miR‐181a/b in cardiac function associated with suppression of cytosolic PTEN expression. Because PI3K activation can be associated with cardioprotection, we examined whether ischemia/reperfusion injury would be enhanced in the miR‐181a/b−/− mice with suppressed PI3K activity. We also examined whether lower ROS production in miR‐181c/d−/− hearts would be associated with protection against ischemia/reperfusion injury. We examined cardiac cell death (necrosis) following 20 minutes of global ischemia followed by 2 hours of reperfusion in WT and miR‐181a/b−/−, miR‐181c/d−/− knockout mice. The amount of necrosis was assessed by TTC staining, in the 3 groups of mice. The miR‐181a/b−/− mice showed a significant increase in cell death with 20 minutes of ischemia followed by 2 hours of reperfusion; on the other hand, the miR‐181c/d−/− showed significantly less necrosis compared to WT (Figure 8A).
Figure 8.
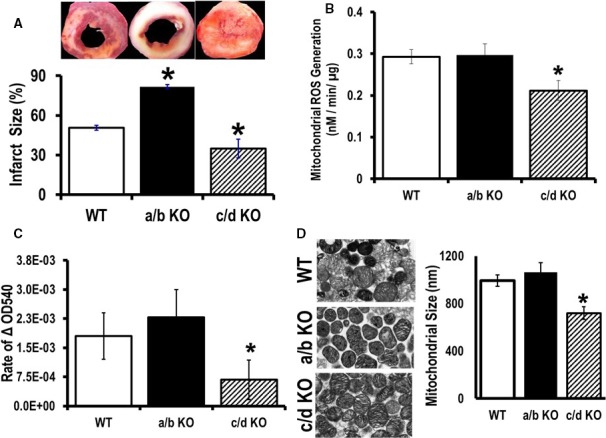
Cardiopretective effect of microRNA (miR)‐181c/d−/− from ischemia/reperfusion injury. A, Infarct size was calculated after 20 minutes of global ischemia, followed by 2 hours of reperfusion from 3 groups of animals: wild type (WT), miR‐181a/b−/−, and miR‐181c/d−/−. B, Rate of reactive oxygen species (ROS) generation and (C) mitochondrial swelling was measured from isolated heart mitochondria from the 3 groups of mice: WT, miR‐181a/b−/− (a/b KO), and miR‐181c/d−/− (c/d KO), using glutamate/malate as a substrate. D, Electron microscopy of mitochondria isolated from mouse hearts. Representative pictures of the 3 groups of animals, WT, miR‐181a/b−/− (a/b KO), and miR‐181c/d−/− (c/d KO), is shown on the left side. Transmission electron microscope measurement of average longitudinal mitochondrion size is represented by a bar graph on the right side. *P<0.05 vs WT (n=6).
We previously reported that miR‐181c overexpression can activate ROS production in heart mitochondria.11, 12 It has been well documented that reperfusion of ischemic zones in the heart initiates excessive mitochondrial ROS production and oxidative damage.24 Whereas isolated mitochondria from miR‐181a/b−/− and WT were comparable in ROS generation, heart mitochondria from miR‐181c/d−/− mice showed significantly lower ROS generation at baseline (without any stress) compared to WT mice (Figure 8B). Previously we have shown that overexpression of miR‐181c leads to dysfunction of complex IV, which activates ROS generation and O2 consumption, involving increased matrix [Ca2+].11 ROS and mitochondrial matrix [Ca2+] can influence mitochondrial function by regulating mitochondrial permeability transition pore opening.24 Isolated heart mitochondria from miR‐181c/d−/− mice showed a markedly reduced rate of mitochondrial swelling compared to WT mitochondria (Figure 8C). This could be due to less ROS accumulation in miR‐181c/d−/− heart mitochondria. There were no statistical differences in the rate of mitochondrial swelling between WT and miR‐181a/b−/− groups (Figure 8C). Therefore, loss of miR‐181c can protect the heart from ischemia‐reperfusion injury by regulating mitochondrial permeability transition pore opening. Furthermore, we employed transmission electron microscopy to check the morphology of the isolated mitochondria from the 3—WT, miR‐181a/b−/−, and miR‐181c/d−/−— groups of animals. Isolated heart mitochondria from miR‐181c/d−/− mice showed a significantly smaller size compared with either WT or miR‐181a/b−/− groups (Figure 8D). There were no differences in mitochondrial size between WT and miR‐181a/b−/− groups (Figure 8D).
Critical Insights in the Heart Mitochondria in miR‐181c/d−/− Mice
The most critical components of mitochondrial respiratory complex IV are the 3 mitochondrially encoded subunits of cytochrome c oxidase (mt‐COX1, mt‐COX2, and mt‐COX3); the remaining subunits (IV, Va, Vb, VIa, VIb, VIc, VIIa, VIIb, VIIc, and VIII) are encoded by nuclear genes, synthesized in the cytosol, and imported into the mitochondria.1, 22 Thus, we measured both the mitochondrial COX IV mRNA expressions and the protein expression of both mitochondrial and nuclear components of complex IV in miR‐181a/b−/− and miR‐181c/d−/− mice. Western blot data demonstrate that all the mitochondrial genes in complex IV are significantly higher in miR‐181c/d−/− heart mitochondria compared to WT heart mitochondria (Figure 9A and 9B). We have also identified some of the most important nuclear gene components of complex IV as also being elevated in miR‐181c/d−/− mouse hearts, with the exception of COX 5B (Figure 9A and 9C). Figure 9 suggests that miR‐181c/d−/− mouse heart mitochondria contain higher complex IV expression compared to WT animals. The miR‐181c directly influences mt‐COX1 expression. However, the higher expression of other components of complex IV suggests that chronic loss of miR‐181c/d results in an increase in complex IV relative to VDAC. We did not observe any significant differences in complex IV expression in the mitochondria isolated from miR‐181a/b−/− animals.
Figure 9.
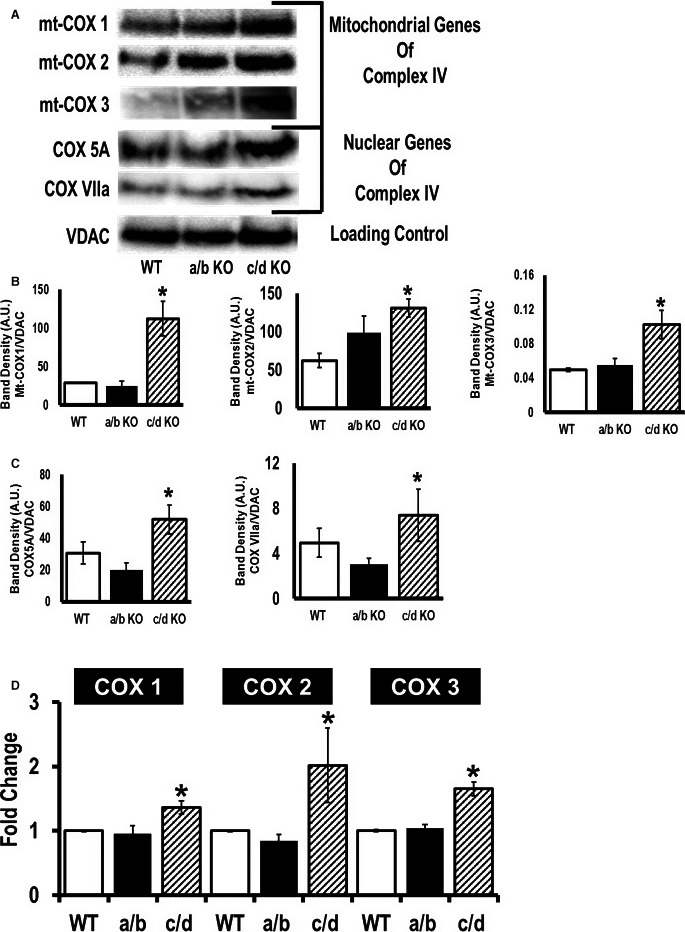
Higher mitochondrial complex IV expression leads to cardioprotection from ischemia/reperfusion injury in microRNA (miR)‐181c/d−/− mice. Western blot analysis of mitochondrial cytochrome c oxidase subunit 1 (mt‐COX1), mitochondrial cytochrome c oxidase subunit 2 (mt‐COX2), mitochondrial cytochrome c oxidase subunit 3 (mt‐COX3), cytochrome c oxidase subunit 5A (COX 5A), cytochrome c oxidase subunit 5B (COX 5B), and cytochrome c oxidase subunit VIIa (COX VIIa) expression in the mitochondrial fraction of the heart from 3 different groups of mice, wild type (WT), miR‐181a/b−/−, and miR‐181c/d−/−. Mitochondrial extracts were probed with the indicated antibodies. VDAC (lower band) is a loading control. A, Representative bands are shown in different groups based on the genomic locations. B, Band‐densitometry of mitochondrial genes, mt‐COX1, mt‐COX2, and mt‐COX3, were tabulated in the bar graphs. C, Band densitometry of nuclear genes COX 5A, COX 5B, and COX VIIa, were tabulated in the bar graphs. D, Quantitative polymerase chain reaction (SYBR kit) analysis of the complex IV genes, encoded by the mitochondrial genome, using total RNA from the hearts of C57BL6/J (WT), miR‐181a/b−/− (a/b), and miR‐181c/d−/− (c/d) mice. The mt‐COX1, mt‐COX2, and mt‐COX3 expressions were normalized to mitochondrial 16S rRNA. *P<0.05 vs WT, n=4.
Most of the subunits that form the mitochondrial respiratory chain complexes are encoded by nuclear genes except for some of the subunits of complexes I, III, and IV, which are encoded by mitochondrial DNA. All 13 mitochondrial genes are sequentially transcribed from the H2‐strand of mitochondrial DNA. Because this is a polycistronic primary transcript, regulation of 1 mRNA can alter the other transcripts. Our previous study showed that overexpressing miR‐181c can affect Several complex IV mitochondrial subunits, mt‐COX1, mt‐COX2, and mt‐COX3,11 even though miR‐181c targets only mt‐COX1.12 However, the Western blot data showed a significant increase in all 3 mitochondrial genomic complex IV subunits, mt‐COX1, mt‐COX2, and mt‐COX3, in miR‐181c/d−/− mice compared to the WT group (Figure 9A and 9B). Therefore, using quantitative PCR, we analyzed expression of other mitochondrial complex IV genes in the hearts from the 3 groups of animals, WT, miR‐181a/b−/−, and miR‐181c/d−/−. The miR‐181c/d−/− group showed a significant increase in all 3—mt‐COX1, mt‐COX2, and mt‐COX3—all derived from the same polycistronic primary transcript from the mitochondrial genome, compared to the WT group (Figure 9D). Figure 9D illustrates the higher translation of all 3 mitochondrial complex IV subunits in miR‐181c/d−/− hearts. There were no difference in the expression of these mitochondrial genes between WT and miR‐181a/b−/− groups (Figure 9D).
Discussion
In the present study we have determined that the nuclear‐encoded miR‐181 family members play a role in cardiac function by regulating target genes both in the cytoplasm and within the mitochondria. Specifically, we find that miR‐181a/b regulates PTEN expression in the cytoplasm, whereas miR‐181c regulates mt‐COX1 in the mitochondria.
Over the past few years, there have been numerous studies that have pointed to the significant role of miRNAs in heart diseases by regulating nuclear genes.27, 28 However, almost all of the studies demonstrated that miRNAs target mRNA in the cytoplasm of cardiomyocytes. Previously, both in vitro12 and in vivo,11 we have demonstrated a pivotal role for mitochondrial miR‐181c in cardiac dysfunction. We11, 12 and others,29 have identified a significant role for miR‐181 in the heart during end‐stage heart failure. We identified miR‐181c in the mitochondria based on microarray data.12 All the miR‐181 family share the same “seed” sequence and can potentially target the same mt‐COX1 mRNA in the mitochondrial compartment. Even though a small portion of miR‐181a and miR‐181b are present in the mitochondrial compartment, in the present study we have detected the entire miR‐181 family in heart mitochondria. As we explored the underlying mechanism by which miR‐181c might be involved in the detrimental effects in the heart, we observed an unexpected finding that some family members of miR‐181 (miR‐181a/b) had a protective effect by targeting PTEN in the cytosol, whereas other family members (miR‐181c) enhanced damage by overproduction of ROS via targeting mt‐COX1 in the mitochondrial compartment of myocytes.
Even though we have detected the multiple members of the miR‐181 family in mouse heart mitochondria, our current data suggest that only miR‐181c is functional in the mitochondrial compartment. That miR‐181a, b, and d can be detected in the mitochondrial pellets is mainly due to the primer specificity issue (miR‐181a primer can detect miR‐181c, and miR‐181b primer can detect miR‐181d, and vice versa). We have validated PTEN, a cytosolic protein, as a target of miR‐181a and b in the heart. The current study also highlighted ~1000‐fold higher expression of miR‐181a and b, compared to miR‐181c, in the heart tissue. The present data showed that the major target of miR‐181a and b expression in the heart is PTEN in the cytoplasm. This phenomenon can be observed better in the miR‐181a/b−/− mice, as these mice showed significant cardiac dysfunction at baseline. Additionally, in agreement with other findings,23 we found that by suppressing miR‐181a/b activity, the EGFP‐miR‐181‐sponge lowered the rate of glycolysis. This metabolic regulation is a cytosolic phenomenon. Indeed, here we show that sequestering the miR‐181 family, using the miR‐181‐sponge approach, results in a significant increase in PTEN levels. PTEN is an important regulator of the PI3K/Akt pathway. Numerous studies have demonstrated the cardioprotective role of the PI3K signaling pathway.24, 30 Furthermore, cross talk between mitochondrial function and PI3K signaling has been previously shown.4, 24, 30 We have previously shown that miR‐181c is mainly localized in the mitochondrial fraction of the heart.12 The current study has also shown the mitochondrial functional changes in the miR‐181c/d−/− mouse heart and not in the miR‐181a/b−/− heart.
Recently, it has been demonstrated that during muscle differentiation, muscle‐specific miR‐1 (skeletal and cardiac muscle) targets mt‐COX1 and/or ND‐1 in the mitochondrial fraction. The miR‐1 is able to stimulate mitochondrial translation of multiple mtDNA‐encoded transcripts, while targeting Mef2 and/or HDAC4 in the cytoplasm, repressing its nuclear DNA.31 The authors proposed a mechanism by which miR‐1 shuttles between the cytoplasm and mitochondria. When the RISC complex involves Ago2 and its binding partner GW182 together with miR‐1, miR‐1 is active in the cytoplasm, but when the GW182—detached Ago2 protein complex along with miR‐1—translocates into the mitochondria, it targets mitochondrial genes (mt‐COX1 and/or ND‐1). The mechanism by which GW182 gets detached from Ago2 is largely unknown; however, it is clear that Ago2 is required for miRNA‐mediated gene regulation in the mitochondrial compartment.31, 32
Decreasing miR‐181 family expression in the heart demonstrated that different family members are active in different cellular compartments and have different phenotypic effects: loss of miR‐181c in the mitochondrial compartment shows protective effects while miR‐181a/b in the cytosolic compartment shows detrimental effects during ischemia/reperfusion injury. The miR‐181a/b can affect glycolysis. Thymocytes isolated from miR‐181a/b−/− mice have impaired metabolism, which results in an impaired capacity for cell growth and proliferation.23 The PI3K/Akt signaling pathway is one of the major cellular metabolism pathways that not only control cellular growth and proliferation but also are involved in ischemic preconditioning and protect cells from apoptotic cell death.24 Thus, 20 minutes of ischemia followed by 2 hours of reperfusion caused markedly more necrosis in the miR‐181a/b−/− mouse heart. Although the reperfusion phase is the initiator of oxidative damage, early reperfusion is essential to minimize ischemic injury. Mitochondrial ETC activity plays an important role in cell survival during reperfusion.1 This may explain the cardioprotective effect observed in miR‐181c/d−/− mouse hearts in response to ischemia/reperfusion injury.
In conclusion, we have demonstrated that miR‐181 family members that share the same seed sequence can have dramatically different phenotypic effects on cardiac function. Furthermore, our work highlights the finding that compartmental localization of miRNA expression is critical for the phenotypic effects. There is growing interest in miRNA‐therapeutics using tiny locked nucleic acids to target the seed sequence and thereby sequester miRNA families.33, 34 However, our study highlights that we should be cautious as different members of the same miRNA family may function differently. Our findings strengthen our understanding of the role of miRNAs in the pathogenesis of heart failure and may provide insight into future therapies and diagnostics.
Sources of Funding
This work was supported by grants from the NIH, HL39752 (Steenbergen), HL114721 (Kohr), and by a Scientist Development Grant from the American Heart Association 14SDG18890049 (Das).
Disclosures
None.
Acknowledgments
The rat cardiospecific promoter was generously provided by Dr Jeffery D. Molkentin at Cincinnati Children's Hospital.
(J Am Heart Assoc. 2017;6:e004694. DOI: 10.1161/JAHA.116.004694.)
References
- 1. Chen JQ, Cammarata PR, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta. 2009;1793:1540–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lam MP, Scruggs SB, Kim TY, Zong C, Lau E, Wang D, Ryan CM, Faull KF, Ping P. An MRM‐based workflow for quantifying cardiac mitochondrial protein phosphorylation in murine and human tissue. J Proteomics. 2012;75:4602–4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Das S, Steenbergen C. Mitochondrial adenine nucleotide transport and cardioprotection. J Mol Cell Cardiol. 2012;52:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steenbergen C, Das S, Su J, Wong R, Murphy E. Cardioprotection and altered mitochondrial adenine nucleotide transport. Basic Res Cardiol. 2009;104:149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. [DOI] [PubMed] [Google Scholar]
- 6. Bandiera S, Ruberg S, Girard M, Cagnard N, Hanein S, Chretien D, Munnich A, Lyonnet S, Henrion‐Caude A. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One. 2011;6:e20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barrey E, Saint‐Auret G, Bonnamy B, Damas D, Boyer O, Gidrol X. Pre‐microRNA and mature microRNA in human mitochondria. PLoS One. 2011;6:e20220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bian Z, Li LM, Tang R, Hou DX, Chen X, Zhang CY, Zen K. Identification of mouse liver mitochondria‐associated miRNAs and their potential biological functions. Cell Res. 2010;20:1076–1078. [DOI] [PubMed] [Google Scholar]
- 9. Kren BT, Wong PY, Sarver A, Zhang X, Zeng Y, Steer CJ. MicroRNAs identified in highly purified liver‐derived mitochondria may play a role in apoptosis. RNA Biol. 2009;6:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sripada L, Tomar D, Prajapati P, Singh R, Singh AK, Singh R. Systematic analysis of small RNAs associated with human mitochondria by deep sequencing: detailed analysis of mitochondrial associated miRNA. PLoS One. 2012;7:e44873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Das S, Bedja D, Campbell N, Dunkerly B, Chenna V, Maitra A, Steenbergen C. miR‐181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One. 2014;9:e96820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Das S, Ferlito M, Kent OA, Fox‐Talbot K, Wang R, Liu D, Raghavachari N, Yang Y, Wheelan SJ, Murphy E, Steenbergen C. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res. 2012;110:1596–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bassani RA, Bers DM. Na‐Ca exchange is required for rest‐decay but not for rest‐potentiation of twitches in rabbit and rat ventricular myocytes. J Mol Cell Cardiol. 1994;26:1335–1347. [DOI] [PubMed] [Google Scholar]
- 14. Lee DI, Vahebi S, Tocchetti CG, Barouch LA, Solaro RJ, Takimoto E, Kass DA. PDE5A suppression of acute beta‐adrenergic activation requires modulation of myocyte beta‐3 signaling coupled to PKG‐mediated troponin I phosphorylation. Basic Res Cardiol. 2010;105:337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage‐dependent anion channel phosphorylation. Circ Res. 2008;103:983–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ji J, Yamashita T, Budhu A, Forgues M, Jia HL, Li C, Deng C, Wauthier E, Reid LM, Ye QH, Qin LX, Yang W, Wang HY, Tang ZY, Croce CM, Wang XW. Identification of microRNA‐181 by genome‐wide screening as a critical player in EpCAM‐positive hepatic cancer stem cells. Hepatology. 2009;50:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ebert MS, Sharp PA. MicroRNA sponges: progress and possibilities. RNA. 2010;16:2043–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Loya CM, Lu CS, Van Vactor D, Fulga TA. Transgenic microRNA inhibition with spatiotemporal specificity in intact organisms. Nat Methods. 2009;6:897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie J, Ameres SL, Friedline R, Hung JH, Zhang Y, Xie Q, Zhong L, Su Q, He R, Li M, Li H, Mu X, Zhang H, Broderick JA, Kim JK, Weng Z, Flotte TR, Zamore PD, Gao G. Long‐term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods. 2012;9:403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bak RO, Hollensen AK, Mikkelsen JG. Managing microRNAs with vector‐encoded decoy‐type inhibitors. Mol Ther. 2013;21:1478–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Montoya J, Lopez‐Perez MJ, Ruiz‐Pesini E. Mitochondrial DNA transcription and diseases: past, present and future. Biochim Biophys Acta. 2006;1757:1179–1189. [DOI] [PubMed] [Google Scholar]
- 23. Henao‐Mejia J, Williams A, Goff LA, Staron M, Licona‐Limon P, Kaech SM, Nakayama M, Rinn JL, Flavell RA. The microRNA miR‐181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity. 2013;38:984–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia‐reperfusion injury. Physiol Rev. 2008;88:581–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Belmonte F, Das S, Sysa‐Shah P, Sivakumaran V, Stanley B, Guo X, Paolocci N, Aon MA, Nagane M, Kuppusamy P, Steenbergen C, Gabrielson K. ErbB2 overexpression upregulates antioxidant enzymes, reduces basal levels of reactive oxygen species, and protects against doxorubicin cardiotoxicity. Am J Physiol Heart Circ Physiol. 2015;309:H1271–H1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abdul‐Ghani R, Serra V, Gyorffy B, Jurchott K, Solf A, Dietel M, Schafer R. The PI3K inhibitor LY294002 blocks drug export from resistant colon carcinoma cells overexpressing MRP1. Oncogene. 2006;25:1743–1752. [DOI] [PubMed] [Google Scholar]
- 27. van Rooij E, Marshall WS, Olson EN. Toward microRNA‐based therapeutics for heart disease: the sense in antisense. Circ Res. 2008;103:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dorn GW II, Matkovich SJ, Eschenbacher WH, Zhang Y. A human 3′ miR‐499 mutation alters cardiac mRNA targeting and function. Circ Res. 2012;110:958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Naga Prasad SV, Duan ZH, Gupta MK, Surampudi VS, Volinia S, Calin GA, Liu CG, Kotwal A, Moravec CS, Starling RC, Perez DM, Sen S, Wu Q, Plow EF, Croce CM, Karnik S. Unique microRNA profile in end‐stage heart failure indicates alterations in specific cardiovascular signaling networks. J Biol Chem. 2009;284:27487–27499. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30. Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol. 2007;69:51–67. [DOI] [PubMed] [Google Scholar]
- 31. Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y, Huang J, Zhao X, Zhou J, Yan Y, Zhang H, Guo P, Sun H, Guo L, Zhang Y, Fu XD. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. 2014;158:607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Latronico MV, Condorelli G. The might of microRNA in mitochondria. Circ Res. 2012;110:1540–1542. [DOI] [PubMed] [Google Scholar]
- 33. Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, Hansen HF, Koch T, Pappin D, Hannon GJ, Kauppinen S. Silencing of microRNA families by seed‐targeting tiny LNAs. Nat Genet. 2011;43:371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rossi JJ. Stopping RNA interference at the seed. Nat Genet. 2011;43:288–289. [DOI] [PubMed] [Google Scholar]