

Abstract
Background
Atherosclerosis is a chronic inflammatory disease, with interleukin 6 (IL‐6) as a major player in inflammation cascade. IL‐6 blockade may reduce cardiovascular risk, but current treatments to block IL‐6 also induce dyslipidemia, a finding with an uncertain prognosis.
Methods and Results
We aimed to determine the endothelial function responses to the IL‐6–blocking agent tocilizumab, anti–tumor necrosis factor α, and synthetic disease‐modifying antirheumatic drug therapies in patients with rheumatoid arthritis in a 16‐week prospective study. Sixty consecutive patients with rheumatoid arthritis were enrolled. Tocilizumab and anti–tumor necrosis factor α therapy were started in 18 patients each while 24 patients were treated with synthetic disease‐modifying antirheumatic drugs. Forty patients completed the 16‐week follow‐up period. The main outcome was flow‐mediated dilation percentage variation before and after therapy. In the tocilizumab group, flow‐mediated dilation percentage variation increased statistically significantly from a pre‐treatment mean of (3.43% [95% CI, 1.28–5.58] to 5.96% [95% CI, 3.95–7.97]; P=0.03). Corresponding changes were 4.78% (95% CI, 2.13–7.42) to 6.75% (95% CI, 4.10–9.39) (P=0.09) and 2.87% (95% CI, −2.17 to 7.91) to 4.84% (95% CI, 2.61–7.07) (P=0.21) in the anti–tumor necrosis factor α and the synthetic disease‐modifying antirheumatic drug groups, respectively (both not statistically significant). Total cholesterol increased significantly in the tocilizumab group from 197.5 (95% CI, 177.59–217.36) to 232.3 (201.62–263.09) (P=0.003) and in the synthetic disease‐modifying antirheumatic drug group from 185.8 (95% CI, 169.76–201.81) to 202.8 (95% CI, 176.81–228.76) (P=0.04), but not in the anti–tumor necrosis factor α group. High‐density lipoprotein did not change significantly in any group.
Conclusions
Endothelial function is improved by tocilizumab in a high‐risk population, even as it increases total cholesterol and low‐density lipoprotein levels.
Keywords: dyslipidemia, endothelial function, inflammation, tocilizumab
Subject Categories: Endothelium/Vascular Type/Nitric Oxide, Inflammation, Atherosclerosis, Coronary Artery Disease
Introduction
Atherosclerosis is a chronic inflammatory disease,1 with interleukin (IL) 6, tumor necrosis factor α (TNF‐α), and IL‐1 as major players at the downstream of vascular inflammatory cascade.2, 3 Since JUPITER (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin),4 the relevance of inflammation as a new modifiable cardiovascular risk factor has been increasingly observed. Further, recent genetic polymorphism studies suggest that IL‐6 receptor (IL‐6R) signaling seems to have a causal effect on coronary artery disease (CAD).5
IL‐6 is a cytokine derived from T lymphocytes, macrophages, and adipocytes, and acts via its membrane‐bound or soluble receptor, stimulating C‐reactive protein (CRP), fibrinogen hepatic synthesis, and joint inflammation and accelerating atherosclerosis.6 High IL‐6 concentrations have been associated with increased risk of myocardial infarction (MI) in healthy men.7 Moreover, IL‐6 and its receptor levels have an early peak at the acute phase of MI, likely related to plaque instability.8, 9 Tocilizumab is a monoclonal humanized antibody that blocks soluble and membrane‐bound IL‐6R, significantly reducing CRP concentrations and systemic inflammation parameters.10 However, tocilizumab worsens atherogenic lipid profile, increasing total cholesterol (TC), low‐density lipoprotein (LDL), and triglycerides, a finding with uncertain cardiovascular outcomes.11
Rheumatoid arthritis (RA) is a systemic inflammatory disease that affects ≈1% of the population worldwide.12 IL‐6 and TNF‐α levels are increased in patients with RA, with both playing central roles in RA pathophysiology.2 CAD is a major cause of death in patients with RA,13 with several studies documenting an almost doubling of risk compared with healthy persons even after adjusting for traditional risk factors.14 While tocilizumab is approved for treatment of joint symptoms in patients with moderate to severe RA, it is not known whether the worsening of atherogenic profile observed with tocilizumab will counteract the potential beneficial effect of IL‐6 activity inhibition on cardiac function.
This pilot study aimed to determine the endothelial function response in RA patients after a 16‐week period of tocilizumab therapy, synthetic disease‐modifying antirheumatic drugs (sDMARDs), or anti–TNF‐α treatment, and, secondarily, how these treatments change lipid profile patterns.
Methods
Patients
Sixty patients with RA who attended a university tertiary outpatient clinic between January 2011 and July 2014 were enrolled in a 16‐week prospective consecutive‐patient noninterventional study with blinded outcomes (modified Prospective Randomized Open, Blinded End Point design). All patients were older than 18 years and fulfilled the American College of Rheumatology (1987) criteria for diagnosis and classification of RA.15 Eighteen were selected to start therapy with tocilizumab at a dose of 8 mg/kg IV every 4 weeks. For the control groups, 24 patients were enrolled in treatment with either methotrexate 15 to 25 mg/wk (n=12) or leflunomide 20 mg/d (n=12), while 18 patients were started on etanercept 50 mg/wk (n=14) or adalimumab 40 mg every 2 weeks (n=4). The choice of drug to use was at the discretion of the treating physician, in consultation with the patient. Randomization and patient blinding were not possible in the clinical setting since the drugs needed reimbursement applications to the public health system. No material or financial support was sought from any pharmaceutical company. Patients with a history of acute coronary syndrome in the past 3 months and/or uncontrolled hypertension (systolic blood pressure ≥160 mm Hg or diastolic blood pressure ≥110 mm Hg) were excluded from participation in the study. The study was approved by the Rio de Janeiro State University Hospital's ethics committee, and all participants signed written informed consents according to the Declaration of Helsinki.
Clinical and Laboratory Evaluations
All patient evaluations were carried out by study physicians blinded to the treatment allocation. A baseline evaluation was performed on the first day scheduled to dispense the chosen medication and the second assessment was carried out at the end of 16 weeks. Each patient underwent anthropometric measurements, assessment of disease activity according to Disease Activity Score of 28 joints (DAS28)16 and functional damage evaluated by the Health Assessment Questionnaire—Disability Index (HAQ‐DI). Laboratory parameters included plasma concentrations of TC (mg/dL), high‐density lipoprotein cholesterol (HDL‐C) (mg/dL), LDL cholesterol (LDL‐C) (mg/dL), triglycerides (mg/dL), CRP (mg/dL), erythrocyte sedimentation rate (mm/h), and TC/HDL‐C ratio, all after overnight (12 hours) fasting.
Endothelial function was assessed by flow‐mediated dilation (FMD), conducted prior to drug administration and then again after 16 weeks of treatment. All vascular studies were performed by the same echocardiographist (RB) who was an expert in the method and was blinded to medications and clinical status of patients. The tests were conducted using an ultrasound unit ESAOTE MyLab 60 (Esaote S.p.A, Genoa, Italy) with 11‐MHz linear transducer. According to American College of Cardiology FMD guideline protocol,17 a baseline measurement and another measurement, performed 60 seconds after a 5‐minute cuff occlusion, were used to calculate the FMD percentage variation (FMD%). The cuff was positioned at the forearm, distal to the ultrasound probe placed on the brachial artery and inflated to 200 mm Hg for 5 minutes, inducing ischemia.
Statistical Analysis
Values are presented as means (95% CIs) unless otherwise indicated. Statistical analyses included paired t test for continuous variables and Fisher exact test for categorical variables, as appropriate. Formal statistical comparisons were not planned among the 3 treatment groups because of the nonrandomized nature of the study. STATA statistical software version 11.1 (StataCorp, College Station, TX) was used for all calculations and values of P<0.05 (two‐tailed) were considered statistically significant.
Results
Of the 60 patients enrolled in the study, 18 comprised the tocilizumab group, 18 the anti–TNF‐α group, and 24 the sDMARD group. Twenty patients were lost to follow‐up: 1 (5.56%), 9 (50%), and 10 (41.67%), respectively, in each group.
Baseline characteristics (anthropometric, clinical, and inflammatory parameters) of the patients who completed the 2 assessments are shown in Table 1. The baseline characteristics between patients who dropped out and those who completed the assessments showed very similar characteristics and are described in Table 2.
Table 1.
Baseline Characteristics Among the 3 Treatment Groups
| Variable | Tocilizumab | Anti–TNF‐α | sDMARD |
|---|---|---|---|
| Patients, No. | 17 | 9 | 14 |
| Drug, No. (%) | Tocilizumab=17 (100) |
Etanercept=7 (77.78) Adalimumab=2 (32.32) |
Methotrexate=6 (42.85) Leflunomide=8 (63.15) |
| Age, y | 51.7 [44.52–58.90] | 47.0 [40.02–53.98] | 55.1 [48.60–61.55] |
| Female sex, No. (%) | 15 (88.2) | 7 (77.8) | 11 (78.6) |
| Erosive disease, No. (%) | 17 (100) | 8 (88.89) | 12 (85.71) |
| Rheumatoid factor, No. (%) | 16 (94.12) | 7 (77.78) | 12 (85.71) |
| Disease duration, y | 12.1 [8.39–15.84] | 10.7 [2.89–18.54] | 9.2 [4.63–13.80] |
| Statin users, No. (%) | 1 (5.88) | 4 (44.44) | 1 (7.14) |
| Diabetes mellitus, No. (%) | 1 (5.88) | 1 (11.11) | 0 |
| Family history of CAD, No. (%) | 1 (5.88) | 2 (22.22) | 1 (7.14) |
| Active smokers, No. (%) | 1 (5.88) | 1 (11.11) | 1 (7.14) |
| Ex‐smokers, No. (%) | 7 (41.18) | 3 (33.33) | 3 (21.43) |
| Weight, kg | 69.5 [61.85–77.17] | 64.6 [55.11–74.09] | 72.6 [66.04–79.18] |
| Height, m | 1.61 [1.57–1.65] | 1.60 [1.56–1.64] | 1.62 [1.58–1.66] |
| BMI, kg/m2 | 26.7 [24.00–29.46] | 25.3 [21.09–29.48] | 27.8 [25.15–30.51] |
| Abdominal circumference, cm | 92.4 [85.49–99.22] | 90.1 [81.25–98.97] | 94.4 [88.80–100.06] |
| TC, mg/dL | 197.5 [177.59–217.36] | 185.1 [158.16–212.06] | 185.8 [169.76–201.81] |
| HDL‐C, mg/dL | 62.3 [52.47–72.12] | 58.8 [45.26–72.29] | 52.1 [45.33–58.96] |
| LDL‐C, mg/dL | 116.6 [101.03–132.19] | 108.6 [88.67–128.44] | 111.5 [99.04–123.94] |
| Triglycerides, mg/dL | 92.6 [77.33–107.96] | 88.9 [67.88–109.90] | 110.8 [83.41–138.16] |
| TC/HDL‐C | 3.36 [2.91–3.80] | 3.35 [2.64–4.06] | 3.75 [3.05–4.45] |
| CRP, mg/dL | 3.59 [1.84–5.35] | 1.62 [0.92–2.32] | 2.31 [1.00–3.61] |
| ESR, mm/h | 56.3 [37.33–75.30] | 37.7 [18.64–56.70] | 31.8 [17.78–45.92] |
| DAS28‐CRP | 5.87 [5.29–6.44] | 5.34 [4.22–6.47] | 5.15 [4.43–5.88] |
| HAQ‐DI | 1.88 [1.60–2.15] | 1.69 [1.07–2.32] | 1.23 [0.89–1.56] |
| FMD% | 3.43 [1.28–5.58] | 4.78 [2.13–7.42] | 2.87 [−2.17 to 7.91] |
Values are expressed as mean [95% CI] or percentage as appropriate. Anti–TNF‐α indicates anti–tumor necrosis factor α; BMI, body mass index; CAD, coronary artery disease; DAS28‐CRP, Disease Activity Score of 28 joints, using C‐reactive protein (CRP); ESR, erythrocyte sedimentation rate; FMD%, flow‐mediated dilation percentage variation; HAQ‐DI, Health Assessment Questionnaire—Disability Index; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; sDMARD, synthetic disease‐modifying antirheumatic drug; TC, total cholesterol.
Table 2.
Baseline Characteristic Comparison Between Patients Who Completed the 16‐Week Therapy and Patients Who Dropped Out
| Variable | Anti–TNF‐α | sDMARD | ||||
|---|---|---|---|---|---|---|
| Completers | Dropouts | P Value | Completers | Dropouts | P Value | |
| Patients, No. | 9 | 9 | NA | 14 | 10 | |
| Drug, No. (%) |
Etanercept=7 (77.78) Adalimumab=2 (32.32) |
Etanercept=7 (77.78) Adalimumab=2 (32.32) |
NA |
Methotrexate=6 (42.85) Leflunomide=8 (63.15) |
Methotrexate=6 (60) Leflunomide=4 (40) |
0.68 |
| Age, y | 47.0 [40.02–53.98] | 54.1 [49.59–58.63] | 0.07 | 55.1 [48.60–61.55] | 57 [49.96–64.04] | 0.64 |
| Female sex, No. (%) | 7 (77.78) | 9 (100) | 0.47 | 11 (78.57) | 8 (80) | 0.94 |
| Erosive disease, No. (%) | 8 (88.89) | 8 (88.89) | NA | 12 (85.71) | 7 (70) | 0.54 |
| Rheumatoid factor, No. (%) | 7 (77.78) | 9 (100) | 0.47 | 12 (85.71) | 7 (70) | 0.54 |
| Disease duration, y | 10.7 [2.89–18.54] | 16.2 [5.76–26.64] | 0.29 | 9.2 [4.63–13.80] | 6.0 [0.94–11.06] | 0.35 |
| Statins users, No. (%) | 4 (44.44) | 3 (33.33) | 0.50 | 1 (7.14) | 1 (10) | 0.67 |
| Diabetes mellitus, No. (%) | 1 (11.11) | 2 (22.22) | 0.50 | 0 | 0 | NA |
| Family history of CAD, No. (%) | 2 (22.22) | 0 | 0.47 | 1 (7.14) | 1 (10) | 0.67 |
| Active smokers, No. (%) | 1 (11.11) | 1 (11.11) | NA | 1 (7.14) | 2 (20) | 0.58 |
| Ex‐smokers, No. (%) | 3 (33.33) | 3 (33.33) | NA | 3 (21.43) | 3 (30) | 0.58 |
| Weight, kg | 64.6 [55.11–74.09] | 71.9 [59.43–84.37] | 0.30 | 72.6 [66.04–79.18] | 71.0 [64.13–77.78] | 0.68 |
| Height, m | 1.60 [1.56–1.64] | 1.60 [1.55–1.66] | 0.97 | 1.62 [1.58–1.66] | 1.61 [1.55–1.66] | 0.68 |
| BMI, kg/m2 | 25.3 [21.09–29.48] | 27.9 [23.71–32.10] | 0.32 | 27.8 [25.15–30.51] | 27.5 [24.94–29.98] | 0.83 |
| Abdominal circumference, cm | 90.1 [81.25–98.97] | 97.8 [87.94–107.61] | 0.20 | 94.4 [88.80–100.06] | 92.4 [85.61–99.19] | 0.81 |
| TC, mg/dL | 185.1 [158.16–212.06] | 201.9 [179.57–224.21] | 0.29 | 185.8 [169.76–201.81] | 237.5 [199.96–275.04] | 0.004a |
| HDL‐C, mg/dL | 58.8 [45.26–72.29] | 52.7 [42.99–62.34] | 0.41 | 52.1 [45.33–58.96] | 63.3 [49.69–76.91] | 0.08 |
| LDL‐C, mg/dL | 108.6 [88.67–128.44] | 125.7 [108.03–143.31] | 0.16 | 111.5 [99.04–123.94] | 134.7 [99.04–170.40] | 0.19 |
| Triglycerides, mg/dL | 88.9 [67.88–109.90] | 117.8 [93.74–141.82] | 0.05 | 110.8 [83.41–138.16] | 197.4 [7.28–387.52] | 0.50 |
| TC/HDL‐C ratio | 3.35 [2.64–4.06] | 3.98 [3.28–4.68] | 0.16 | 3.75 [3.05–4.45] | 4.22 [2.52–5.93] | 0.77 |
| CRP, mg/dL | 1.62 [0.92–2.32] | 3.10 [1.24–4.95] | 0.11 | 2.31 [1.00–3.61] | 1.68 [0.33–3.03] | 0.41 |
| ESR, mm/h | 37.7 [18.64–56.70] | 47.3 [12.24–82.42] | 0.58 | 31.8 [17.77–45.92] | 41.1 [25.04–57.16] | 0.24 |
| DAS28‐CRP | 5.34 [4.22–6.47] | 6.57 [5.70–7.45] | 0.07 | 5.15 [4.43–5.88] | 5.06 [3.36–6.77] | 0.88 |
| HAQ‐DI | 1.69 [1.07–2.32] | 2.06 [1.56–2.57] | 0.31 | 1.23 [0.89–1.56] | 1.32 [0.25–2.40] | 0.80 |
| FMD% | 4.78 [2.13–7.42] | 1.40 [−6.52 to 9.30] | 0.25 | 2.87 [−2.17 to 7.91] | 3.62 [1.63–5.62] | 0.64 |
Values are expressed as mean [95% CIs] or percentages, as appropriate. Anti–TNF‐α indicates anti–tumor necrosis factor α; BMI, body mass index; CAD, coronary artery disease; DAS28‐CRP, Disease Activity Score of 28 joints, using C‐reactive protein (CRP); ESR, erythrocyte sedimentation rate; FMD%, flow‐mediated dilation percentage variation; HAQ‐DI, Health Assessment Questionnaire—Disability Index; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; NA, not applicable; sDMARD, synthetic disease‐modifying antirheumatic drug; TC, total cholesterol.
Statistically significant.
The assessment of endothelial function by FMD showed a significant improvement only among patients taking tocilizumab therapy (Table 3 and Figure 1). The mean FMD% increased from 3.43% (1.28–5.58) to 5.96% (3.95–7.97) (P=0.03). For the anti–TNF‐α group, there was improvement from 4.78% (2.13–7.42) to 6.75% (4.10–9.39) (P=0.09), and in the sDMARD group, FMD increased from 2.87% (−2.17 to 7.91) to 4.84% (2.61–7.07) (P=0.21).
Table 3.
Endothelial Function, Inflammatory Parameters, and Disease Activity Scores After 16 Weeks of Therapy
| Variable | Tocilizumab | Anti–TNF‐α | sDMARD | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | Post‐Treatment | P Value | Baseline | Post‐Treatment | P Value | Baseline | Post‐Treatment | P Value | |
| FMD% | 3.43% [1.28–5.58] | 5.96% [3.95–7.97] | 0.03a | 4.78% [2.13–7.42] | 6.75% [4.10–9.39] | 0.09 | 2.87% [−2.17 to 7.91] | 4.84% [2.61–7.07] | 0.21 |
| CRP, mg/dL | 3.59 [1.84–5.35] | 0.16 [0.02–0.31] | <0.001a | 1.62 [0.92–2.32] | 1.07 [0.16–1.98] | 0.05 | 2.31 [1.00–3.61] | 1.30 [0.26–2.33] | 0.07 |
| ESR, mm/h | 56.3 [37.33–75.30] | 7.9 [0.81–15.07] | <0.001a | 37.7 [18.64–56.70] | 28.2 [9.22–47.22] | 0.15 | 31.8 [17.77–45.92] | 30.5 [16.00–44.92] | 0.82 |
| DAS28‐ESR | 6.73 [6.15–7.32] | 4.00 [3.39–4.61] | <0.001a | 5.90 [4.69–7.12] | 4.74 [3.71–5.76] | 0.03a | 5.62 [4.88–6.36] | 4.04 [3.25–4.83] | 0.001a |
| DAS28‐CRP | 5.87 [5.29–6.44] | 4.02 [3.54–4.51] | <0.001a | 5.34 [4.22–6.47] | 4.35 [3.40–5.29] | 0.049a | 5.15 [4.43–5.88] | 3.53 [2.78–4.29] | 0.001a |
| HAQ‐DI | 1.88 [1.60–2.15] | 1.40 [1.04–1.77] | 0.004a | 1.69 [1.07–2.32] | 1.37 [0.73–2.02] | 0.10 | 1.22 [0.89–1.56] | 0.80 [0.38–1.23] | 0.04a |
Values are expressed as mean [95% CI]. Anti–TNF‐α indicates anti–tumor necrosis factor α; DAS28‐CRP, Disease Activity Score of 28 joints, using C‐reactive protein (CRP); ESR, erythrocyte sedimentation rate; FMD%, flow‐mediated dilation percentage variation; HAQ‐DI, Health Assessment Questionnaire—Disability Index; sDMARD, synthetic disease‐modifying antirheumatic drug.
Statistically significant.
Figure 1.
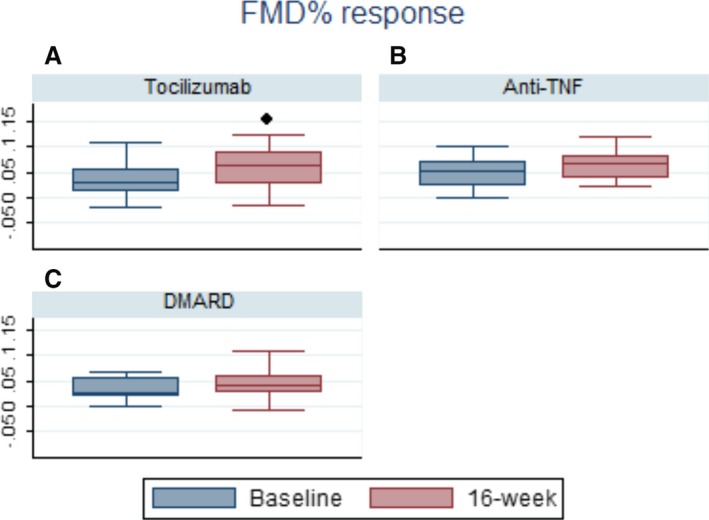
Flow‐mediated dilation percentage variation (FMD%) after 16 weeks of therapy. ● P<0.05; 16‐week represents second assessment. A, Box plot graphic: Tocilizumab FMD% variation. B, Box plot graphic. Anti–tumor necrosis factor (Anti‐TNF) FMD%. C, Box plot graphic. Synthetic disease‐modifying antirheumatic drug (sDMARD) FMD%.
The lipid profile behavior after 16 weeks of treatment with tocilizumab demonstrated a significant increase in TC, rising from 197.5 (177.59–217.36) to 232.3 (201.62–263.09) (P=0.003). The same pattern was seen with LDL‐C (from 116.6 [95% CI, 101.03–132.19] to 137.0 [95% CI, 113.57–160.41], P=0.03), triglycerides (92.6 [95% CI, 77.33–107.96] to 157.7 [95% CI, 116.55–198.86], P<0.001), and TC/HDL‐C ratio (3.36 [95% CI, 2.91–3.80] to 3.84 [95% CI, 3.25–4.43], P=0.02). HDL did not change significantly, from (62.3 [95% CI, 52.47–72.12] to 63.8 [95% CI, 52.96–74.69], P=0.63). In both control groups, there were no statistically significant changes, except for TC, which rose from (185.8 [95% CI, 169.76–201.81] to 202.8 [95% CI, 47.62–63.38], P=0.04) in the sDMARD cohort. The major results are depicted in Figure 2 and the complete data on lipid changes for the 3 groups are available in Table 4.
Figure 2.
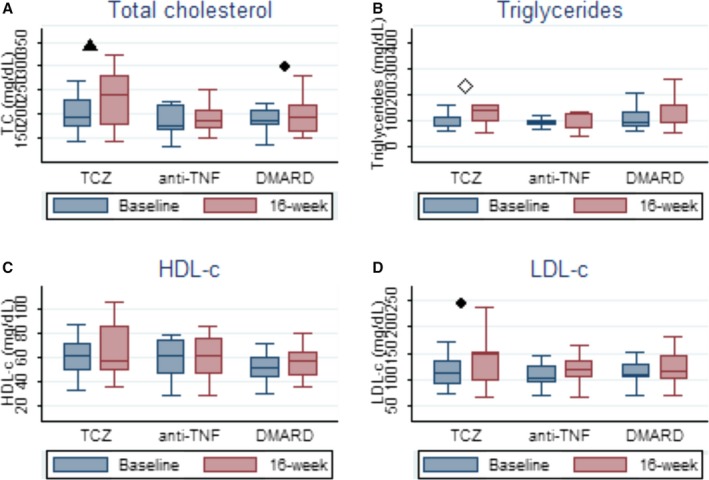
Changes in plasma lipoproteins and triglycerides behavior after 16 weeks of therapy. A, Box plot graphic. Total cholesterol levels. B, Box plot graphic. Triglycerides levels. C, Box plot graphic. High‐density lipoprotein cholesterol (HDL‐C) levels. D, Box plot graphic. Low‐density lipoprotein (LDL‐C) levels. ● P<0.05; ▲ P<0.01and ≥0.001; ◊ P<0.001; 16‐week represents second assessment. anti‐TNF indicates anti–tumor necrosis factor; DMARD, synthetic disease‐modifying antirheumatic drug; TC, total cholesterol; TCZ, tocilizumab.
Table 4.
Changes in Lipid Profile After 16 Weeks of Therapy
| Variable | Tocilizumab | Anti–TNF‐α | sDMARD | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | Post‐Treatment | P Value | Baseline | Post‐Treatment | P Value | Baseline | Post‐Treatment | P Value | |
| TC, mg/dL | 197.5 [177.59–217.36] | 232.3 [201.62–263.09] | 0.003a | 185.1 [158.16–212.06] | 192.9 [164.97–220.81] | 0.36 | 185.8 [169.76–201.81] | 202.8 [176.81–228.76] | 0.04a |
| HDL‐C, mg/dL | 62.3 [52.47–72.12] | 63.8 [52.96–74.69] | 0.63 | 58.8 [45.26–72.29] | 60.4 [44.75–76.14] | 0.58 | 52.1 [45.33–58.96] | 55.5 [47.62–63.38] | 0.15 |
| LDL‐C, mg/dL | 116.6 [101.03–132.19] | 137.0 [113.57–160.41] | 0.03a | 108.6 [88.67–128.44] | 115.1 [90.87–139.35] | 0.50 | 111.5 [99.04–123.94] | 121.4 [101.66–141.11] | 0.09 |
| Triglycerides, mg/dL | 92.6 [77.33–107.96] | 157.7 [116.55–198.86] | <0.001a | 88.9 [67.88–109.90] | 86.7 [59.39–113.943] | 0.85 | 110.8 [83.41–138.16] | 129.5 [85.45–173.55] | 0.19 |
| TC/HDL‐C ratio | 3.36 [2.91–3.80] | 3.84 [3.25–4.43] | 0.02a | 3.35 [2.64–4.06] | 3.51 [2.64–4.38] | 0.51 | 3.75 [3.05–4.45] | 3.86 [3.08–4.64] | 0.44 |
Values are expressed as mean [95% CIs]. Anti–TNF‐α indicates anti–tumor necrosis factor α; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; sDMARD, synthetic disease‐modifying antirheumatic drug; TC, total cholesterol.
Statistically significant.
Inflammatory parameters showed significant differences in the tocilizumab‐treated patients, as expected. Mean CRP levels were reduced after 16 weeks of therapy, dropping from 3.59 to 0.16 (P<0.001). For the anti–TNF‐α and sDMARD groups, the CRP results showed a smaller reduction, from 1.69 to 1.07 (P=0.05) and from 2.31 to 1.30 (P=0.07), respectively. As shown in Table 3, the tocilizumab‐treated patients had higher baseline CRP levels and erythrocyte sedimentation rates compared with other groups.
Disease activity, as measured by DAS28‐CRP, decreased after therapy as expected, with statistically significant differences in all 3 groups (Figure 3). Complete results on endothelial function changes and inflammatory and disease activity parameters are described in Table 3.
Figure 3.
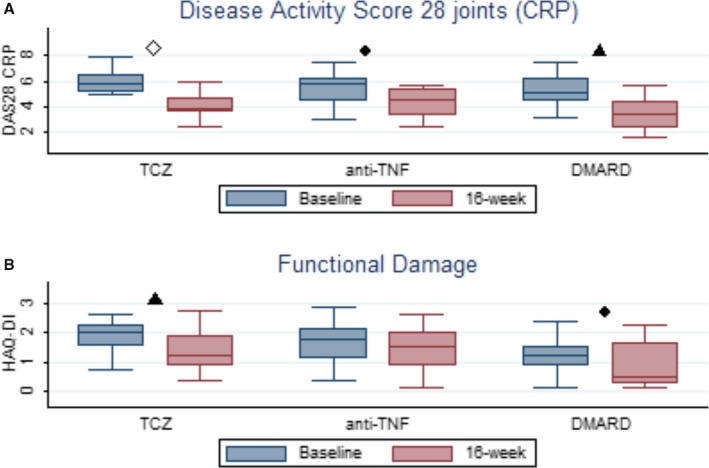
Changes in disease activity scores and functional damage after 16 weeks of therapy. A, box plot graphic for disease activity score changes. B, Box plot graphic for functional damage behavior. ● P<0.05; ▲ P<0.01and ≥0.001; ◊ P<0.001; 16‐week represents second assessment. anti‐TNF indicates anti–tumor necrosis factor; CRP, C‐reactive protein; DMARD, synthetic disease‐modifying antirheumatic drugs; TCZ, tocilizumab.
Discussion
Inflammation and atherosclerosis share a similar pathophysiologic pathway, and treatments that lower systemic inflammatory markers show a beneficial effect on atherosclerotic complications.18 However, this observation is confounded by the fact that reductions in inflammatory markers are also often accompanied by improvement in “traditional” risk factors such as atherogenic cholesterol levels. For example, the JUPITER trial4 showed a remarkable reduction in cardiovascular mortality in patients with “normal” LDL levels, presumably related to rosuvastatin's effect on inflammation, but the LDL levels in rosuvastatin‐treated patients also dropped dramatically, providing an alternate explanation for the reduced cardiac mortality. In this study, we attempted to isolate the anti‐inflammatory effect from the anti‐cholesterol effect by evaluating an anti‐inflammatory treatment that, in fact, worsens the atherogenic lipid profile, and tested its effect on endothelial function in a population with high cardiovascular risk. We show that a treatment that significantly reduces IL‐6 activity even while worsening the atherogenic lipid profile still provides dramatic improvement in endothelial function in a high‐risk RA population.
Our results provide further support for findings from Mendelian randomization studies suggesting that IL‐6R signaling seems to have a causal role in the development of CAD. Mendelian randomization is an innovative approach that evaluates the interactions of genotype polymorphisms, phenotype, and risk of coronary heart disease. It relies on the paradigm that people with a genetic susceptibility that exposes them to abnormal levels of a risk factor causally related to atherosclerosis will eventually manifest an increased risk of coronary heart disease.19 Two groups of researchers studied the population distribution and effect of Asp358Ala variant in the IL‐6R gene, IL6R, a polymorphism that decreases IL‐6 signaling and results in a significant systemic anti‐inflammatory effect. One study evaluated the frequency of Asp358Ala in 51 441 patients with coronary heart disease and in 136 226 controls and found that for every copy of 358Ala inherited, the risk of CAD was reduced by 3.4% (95% CI, 1.8–5.0).20 The second study in 25 458 CAD cases and 100 740 controls found a risk reduction of 5% (95% CI, 3–7).5 While the results of these studies open an intriguing possibility for the use of IL‐6R blockade as a novel therapeutic approach to prevent CAD in the future, both sets of investigators also cautioned that the currently available IL‐6R–blocking treatment, tocilizumab, while showing a pattern of inflammatory biomarkers similar to that associated with the 358Ala allele, also demonstrated a strikingly different proatherogenic lipid profile, perhaps as an off‐target effect of the drug. Whether the anti‐inflammatory effect “overcomes” the proatherogenic effects remained an open question, one that we have addressed with our study.
The causal association of CRP and other inflammatory markers with CAD has been a source of intense controversy. While it is known that high CRP levels are often associated with increased risk of CAD,21 the observation is confounded by the fact that people with elevated CRP levels often also have increased levels of “traditional” risk factors such as central obesity, hypertension, hypertriglyceridemia, and low HDL‐C concentrations.22 The JUPITER trial4 showed that aggressive statin use lowers CAD risk as well as CRP levels, but Mendelian randomization studies indicate that this is unlikely to be a causal relationship.23 Indeed, treatments such as darapladib24 and varespladib25 which strongly reduce phospholipase A2 (another inflammatory marker), do not have any significant effect on reducing CAD, a finding predicted by Mendelian randomization analyses.26 Similarly, Mendelian randomization studies have predicted that direct targeting of CRP is unlikely to have any beneficial effect on CAD.23 Consistent with these observations, we show that while CRP levels were reduced in all 3 treatment groups, only IL‐6 inhibition showed a significant benefit in FMD improvement and a strong correlation between reduction in CRP and increase in FMD.
Other recent studies on the effect of IL‐6 inhibition and FMD have come to somewhat varying conclusions from our study. A pilot study with 11 patients with RA compared with healthy controls observed an improvement in endothelial function by FMD after 6 months of tocilizumab.27 A more robust randomized clinical trial, MEASURE,3 designed primarily to assess the effects of tocilizumab on aortic stiffness by pulse wave velocity and the drug impact on small and dense LDL, could not demonstrate a significant difference between tocilizumab‐ and methotrexate‐treated patients. However, the authors attributed this to “technical problems” in the measurement of endothelial function by different observers at multiple centers. On the other hand, the lipid behavior end points showed significantly increased levels of TC, triglycerides, and LDL, and TC/HDL‐C ratio, similar to what we observed. The authors argued that this higher risk lipid profile pattern could be balanced by beneficial changes in “new” cardiovascular biomarkers, such as serum amyloid A, paraoxonase‐1, d‐dimer, and fibrinogen, but provided no evidence.
It is interesting to note that while Mendelian randomization studies seem to suggest that the proatherogenic effect of tocilizumab is an off‐target drug effect, other studies have argued that the worsening lipid profile observed with tocilizumab is actually a “return to baseline” of TC, lipoproteins subunits, and triglycerides when inflammation is controlled, allowing “normal” liver synthesis of these biomarkers.5 Our study appears to support the former hypothesis. While both anti–TNF‐α and tocilizumab therapies reduced RA disease activity equally, only the tocilizumab‐treated group showed an increase in atherogenic lipidemic profile. All 3 treatment groups showed strong reduction in RA disease activity, coupled with striking reductions in inflammatory markers, but only the tocilizumab‐treated patients developed significant dyslipidemia. Nevertheless, we demonstrate that the strong anti‐inflammatory effect due to anti–IL‐6 suppression results in a net beneficial effect on endothelial function, in spite of increases in LDL and triglycerides. However, it should be noted that our study was limited to 16 weeks' duration, and it is difficult to predict whether the anti‐inflammatory effect would continue to dominate the proatherogenic effects over the long term.
Study Strengths and Limitations
Our study has several limitations and strengths. While we enrolled consecutively‐seen patients in a busy tertiary care setting, it was not possible for us to randomize treatment allocation since the expensive medications required formal application for reimbursement to the state public health system. While most baseline characteristics of cardiovascular risk were similar among the 3 treatment groups, the tocilizumab‐treated patients had somewhat higher CRP levels and erythrocyte sedimentation rates at baseline. We used a surrogate outcome of changes in FMD rather than directly measuring cardiovascular outcomes. However, changes in FMD measurement have a strong correlation with future cardiovascular outcomes—in fact, Inaba et al28 showed a decrease of 13% (95% CI, 9–17) in the risk of future cardiovascular events for every 1% increase in FMD. In comparison to observational studies based on convenience sampling and unblinded assessments, our use of a modified Prospective Randomized Open, Blinded End Point design—with consecutive‐patient entry, protocol‐driven prospective follow‐ups, and blinded end point assessments—lend particular strengths to our findings. We did not measure HDL functionality and particle size, which have been shown to be more predictive of cardiovascular outcomes compared with plasma levels of HDL cholesterol.29 Within the limitations of a nonrandomized design and a small sample size (particularly in the anti‐TNF subgroup), we provide strong hypothesis‐generating evidence that a reduction of inflammation can have significant beneficial effect on CAD outcome, even if the traditional risk factors (eg, lipid profile) continue to worsen.
Two major trials designed to elucidate the real impact of inflammation control over CAD are underway. CIRT (the Cardiovascular Inflammation Reduction Trial)30 enrolled patients with prior MI and diabetes mellitus or metabolic syndrome to undergo methotrexate therapy or placebo to prevent secondary cardiovascular events. CANTOS (the Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study)31 uses canakinumab, a monoclonal antibody against IL‐1, in post‐MI patients who have high CRP levels despite traditional secondary prevention therapy for CAD. Their results may start a new anti‐inflammatory approach in CAD secondary prevention.
Sources of Funding
Pedro Ernesto Rheumatology Research Center paid for the laboratory tests. The funding source has no involvement in the design or conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication. No financial or material support was sought or obtained from any pharmaceutical company.
Disclosures
Dr Pinheiro has received honoraria for consultancies from AbbVie, AstraZeneca, BMS Hospira, Janssen, Pfizer, Roche, RuiYi, and Sanofi‐Aventis. Dr Usnayo was a GlaxoSmithKline Brazil employee from September 2015 until March 2016. The other authors do not have any conflicts of interests.
(J Am Heart Assoc. 2017;6:e005038. DOI: 10.1161/JAHA.116.005038.)
References
- 1. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 2. Sattar N, McCarey DW, Capell H, McInnes IB. Explaining how “high‐grade” systemic inflammation accelerates vascular risk in rheumatoid arthritis. Circulation. 2003;108:2957–2963. [DOI] [PubMed] [Google Scholar]
- 3. McInnes IB, Thompson L, Giles JT, Bathon JM, Salmon JE, Beaulieu AD, Codding CE, Carlson TH, Delles C, Lee JS, Sattar N. Effect of interleukin‐6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo‐controlled study. Ann Rheum Dis. 2013;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AM, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 5. The interleukin‐6 receptor as a target for prevention of coronary heart disease: a Mendelian randomisation analysis. Lancet. 2012;379:1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davies R, Choy E. Clinical experience of IL‐6 blockade in rheumatic diseases—implications on IL‐6 biology and disease pathogenesis. Semin Immunol. 2014;26:97–104. [DOI] [PubMed] [Google Scholar]
- 7. Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin‐6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–1772. [DOI] [PubMed] [Google Scholar]
- 8. Anderson DR, Poterucha JT, Mikuls TR, Duryee MJ, Garvin RP, Klassen LW, Shurmur SW, Thiele GM. IL‐6 and its receptors in coronary artery disease and acute myocardial infarction. Cytokine. 2013;62:395–400. [DOI] [PubMed] [Google Scholar]
- 9. Gabriel AS, Martinsson A, Wretlind B, Ahnve S. IL‐6 levels in acute and post myocardial infarction: their relation to CRP levels, infarction size, left ventricular systolic function, and heart failure. Eur J Intern Med. 2004;15:523–528. [DOI] [PubMed] [Google Scholar]
- 10. Jones G, Sebba A, Gu J, Lowenstein MB, Calvo A, Gomez‐Reino JJ, Siri DA, Tomšič M, Alecock E, Woodworth T, Genovese MC. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawashiri S‐Y, Kawakami A, Yamasaki S, Imazato T, Iwamoto N, Fujikawa K, Aramaki T, Tamai M, Nakamura H, Ida H, Origuchi T, Ueki Y, Eguchi K. Effects of the anti‐interleukin‐6 receptor antibody, tocilizumab, on serum lipid levels in patients with rheumatoid arthritis. Rheumatol Int. 2011;31:451–456. [DOI] [PubMed] [Google Scholar]
- 12. Alamanos Y, Voulgari PV, Drosos AA. Incidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Rheumatology criteria: a systematic review. Semin Arthritis Rheum. 2006;36:182–188. [DOI] [PubMed] [Google Scholar]
- 13. Prior P, Symmons DP, Scott DL, Brown R, Hawkins CF. Cause of death in rheumatoid arthritis. Rheumatology. 1984;23:92–99. [DOI] [PubMed] [Google Scholar]
- 14. Maradit‐Kremers H, Crowson CS, Nicola PJ, Ballman KV, Roger VL, Jacobsen SJ, Gabriel SE. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population‐based cohort study. Arthritis Rheum. 2005;52:402–411. [DOI] [PubMed] [Google Scholar]
- 15. van Gestel AM, Haagsma CJ, van Riel PL. Validation of rheumatoid arthritis improvement criteria that include simplified joint counts. Arthritis Rheum. 1998;41:1845–1850. [DOI] [PubMed] [Google Scholar]
- 16. Prevoo ML, van't Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight‐joint counts development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. 1995;38:44–48. [DOI] [PubMed] [Google Scholar]
- 17. Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard‐Herman M, Herrington D, Vallance P, Vita J, Vogel R. Guidelines for the ultrasound assessment of endothelial‐dependent flow‐mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–265. [DOI] [PubMed] [Google Scholar]
- 18. Dixon WG, Watson KD, Lunt M, Hyrich KL, Silman AJ, Symmons DP. Reduction in the incidence of myocardial infarction in patients with rheumatoid arthritis who respond to anti‐tumor necrosis factor α therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2007;56:2905–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boekholdt SM, Stroes ES. The interleukin‐6 pathway and atherosclerosis. Lancet. 2012;379:1176–1178. [DOI] [PubMed] [Google Scholar]
- 20. IL6R Genetics Consortium Emerging Risk Factors Collaboration; Sarwar N, Butterworth AS, Freitag DF Gregson J, Willeit P, Gorman DN, Gao P, Saleheen D, Rendon A, Nelson CP, Braund PS, Hall AS, Chasman DI, Tybjærg‐Hansen A, Chambers JC, Benjamin EJ, Franks PW, Clarke R, Wilde AA, Trip MD, Steri M, Witteman JC, Qi L, van der Schoot CE, de Faire U, Erdmann J, Stringham HM, Koenig W, Rader DJ, Melzer D, Reich D, Psaty BM, Kleber ME, Panagiotakos DB, Willeit J, Wennberg P, Woodward M, Adamovic S, Rimm EB, Meade TW, Gillum RF, Shaffer JA, Hofman A, Onat A, Sundström J, Wassertheil‐Smoller S, Mellström D, Gallacher J, Cushman M, Tracy RP, Kauhanen J, Karlsson M, Salonen JT, Wilhelmsen L, Amouyel P, Cantin B, Best LG, Ben‐Shlomo Y, Manson JE, Davey‐Smith G, de Bakker PI, O'Donnell CJ, Wilson JF, Wilson AG, Assimes TL, Jansson JO, Ohlsson C, Tivesten Å, Ljunggren Ö, Reilly MP, Hamsten A, Ingelsson E, Cambien F, Hung J, Thomas GN, Boehnke M, Schunkert H, Asselbergs FW, Kastelein JJ, Gudnason V, Salomaa V, Harris TB, Kooner JS, Allin KH, Nordestgaard BG, Hopewell JC, Goodall AH, Ridker PM, Hólm H, Watkins H, Ouwehand WH, Samani NJ, Kaptoge S, Di Angelantonio E, Harari O, Danesh J. Interleukin‐6 receptor pathways in coronary heart disease: a collaborative meta‐analysis of 82 studies. Lancet. 2012;379:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emerging Risk Factors Collaboration; Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J . C‐reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta‐analysis. Lancet. 2010;375:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ridker PM, Buring JE, Cook NR, Rifai N. C‐reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8‐year follow‐up of 14 719 initially healthy American women. Circulation. 2003;107:391–397. [DOI] [PubMed] [Google Scholar]
- 23. C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC); Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, Shah T, Engert JC, Clarke R, Davey‐Smith G, Nordestgaard BG, Saleheen D, Samani NJ, Sandhu M, Anand S, Pepys MB, Smeeth L, Whittaker J, Casas JP, Thompson SG, Hingorani AD, Danesh J. Association between C reactive protein and coronary heart disease: Mendelian randomisation analysis based on individual participant data. BMJ. 2011;342:d548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. STABILITY Investigators; White HD, Held C, Stewart R, Tarka E, Brown R, Davies RY, Budaj A, Harrington RA, Steg PG, Ardissino D, Armstrong PW, Avezum A, Aylward PE, Bryce A, Chen H, Chen MF, Corbalan R, Dalby AJ, Danchin N, De Winter RJ, Denchev S, Diaz R, Elisaf M, Flather MD, Goudev AR, Granger CB, Grinfeld L, Hochman JS, Husted S, Kim HS, Koenig W, Linhart A, Lonn E, López‐Sendón J, Manolis AJ, Mohler ER 3rd, Nicolau JC, Pais P, Parkhomenko A, Pedersen TR, Pella D, Ramos‐Corrales MA, Ruda M, Sereg M, Siddique S, Sinnaeve P, Smith P, Sritara P, Swart HP, Sy RG, Teramoto T, Tse HF, Watson D, Weaver WD, Weiss R, Viigimaa M, Vinereanu D, Zhu J, Cannon CP, Wallentin L . Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med. 2014;370:1702–1711. [DOI] [PubMed] [Google Scholar]
- 25. Nicholls SJ, Kastelein JP, Schwartz GG, Bash D, Rosenson RS, Cavender MA, Brennan DM, Koenig W, Jukema JW, Nambi V, Wright RS, Menon V, Lincoff AM, Nissen SE. Varespladib and cardiovascular events in patients with an acute coronary syndrome: the VISTA‐16 randomized clinical trial. JAMA. 2014;311:252–262. [DOI] [PubMed] [Google Scholar]
- 26. Holmes MV, Simon T, Exeter HJ, Folkersen L, Asselbergs FW, Guardiola M, Cooper JA, Palmen J, Hubacek JA, Carruthers KF, Horne BD, Brunisholz KD, Mega JL, van Iperen EP, Li M, Leusink M, Trompet S, Verschuren JJ, Hovingh GK, Dehghan A, Nelson CP, Kotti S, Danchin N, Scholz M, Haase CL, Rothenbacher D, Swerdlow DI, Kuchenbaecker KB, Staines‐Urias E, Goel A, van't Hooft F, Gertow K, de Faire U, Panayiotou AG, Tremoli E, Baldassarre D, Veglia F, Holdt LM, Beutner F, Gansevoort RT, Navis GJ, Mateo Leach I, Breitling LP, Brenner H, Thiery J, Dallmeier D, Franco‐Cereceda A, Boer JM, Stephens JW, Hofker MH, Tedgui A, Hofman A, Uitterlinden AG, Adamkova V, Pitha J, Onland‐Moret NC, Cramer MJ, Nathoe HM, Spiering W, Klungel OH, Kumari M, Whincup PH, Morrow DA, Braund PS, Hall AS, Olsson AG, Doevendans PA, Trip MD, Tobin MD, Hamsten A, Watkins H, Koenig W, Nicolaides AN, Teupser D, Day IN, Carlquist JF, Gaunt TR, Ford I, Sattar N, Tsimikas S, Schwartz GG, Lawlor DA, Morris RW, Sandhu MS, Poledne R, Maitland‐van der Zee AH, Khaw KT, Keating BJ, van der Harst P, Price JF, Mehta SR, Yusuf S, Witteman JC, Franco OH, Jukema JW, de Knijff P, Tybjaerg‐Hansen A, Rader DJ, Farrall M, Samani NJ, Kivimaki M, Fox KA, Humphries SE, Anderson JL, Boekholdt SM, Palmer TM, Eriksson P, Paré G, Hingorani AD, Sabatine MS, Mallat Z, Casas JP, Talmud PJ. Secretory phospholipase A2‐IIA and cardiovascular disease: a Mendelian randomization study. J Am Coll Cardiol. 62:1966–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Protogerou AD, Zampeli E, Fragiadaki K, Stamatelopoulos K, Papamichael C, Sfikakis PP. A pilot study of endothelial dysfunction and aortic stiffness after interleukin‐6 receptor inhibition in rheumatoid arthritis. Atherosclerosis. 2011;219:734–736. [DOI] [PubMed] [Google Scholar]
- 28. Inaba Y, Chen J, Bergmann S. Prediction of future cardiovascular outcomes by flow‐mediated vasodilatation of brachial artery: a meta‐analysis. Int J Cardiovasc Imaging. 2010;26:631–640. [DOI] [PubMed] [Google Scholar]
- 29. Santos‐Gallego CG, Badimon JJ, Rosenson RS. Beginning to understand high‐density lipoproteins. Endocrinol Metab Clin North Am. 2014;43:913–947. [DOI] [PubMed] [Google Scholar]
- 30. Everett BM, Pradhan AD, Solomon DH, Paynter N, MacFadyen J, Zaharris E, Gupta M, Clearfield M, Libby P, Hasan AAK, Glynn RJ, Ridker PM. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166:199–207.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin‐1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti‐inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J. 2011;162:597–605. [DOI] [PubMed] [Google Scholar]