

Abstract
Background
Valvular interstitial cells (VICs) in the healthy aortic valve leaflet exhibit a quiescent phenotype, with <5% of VICs exhibiting an activated phenotype. Yet, in vitro culture of VICs on tissue culture polystyrene surfaces in standard growth medium results in rapid transformation to an activated phenotype in >90% of cells. The inability to preserve a healthy VIC phenotype during in vitro studies has hampered the elucidation of mechanisms involved in calcific aortic valve disease. This study describes the generation of quiescent populations of porcine VICs in 2‐dimensional in vitro culture and their utility in studying valve pathobiology.
Methods and Results
Within 4 days of isolation from fresh porcine hearts, VICs cultured in standard growth conditions were predominantly myofibroblastic (activated VICs). This myofibroblastic phenotype was partially reversed within 4 days, and fully reversed within 9 days, following application of a combination of a fibroblast media formulation with culture on collagen coatings. Specifically, culture in this combination significantly reduced several markers of VIC activation, including proliferation, apoptosis, α‐smooth muscle actin expression, and matrix production, relative to standard growth conditions. Moreover, VICs raised in a fibroblast media formulation with culture on collagen coatings exhibited dramatically increased sensitivity to treatment with transforming growth factor β1, a known pathological stimulus, compared with VICs raised in either standard culture or medium with a fibroblast media formulation.
Conclusions
The approach using a fibroblast media formulation with culture on collagen coatings generates quiescent VICs that more accurately mimic a healthy VIC population and thus has the potential to transform the study of the mechanisms of VIC activation and dysfunction involved in the early stages of calcific aortic valve disease.
Keywords: aortic valve, calcific aortic valve disease, differentiation, myofibroblasts, quiescence, valvular interstitial cell
Subject Categories: Basic Science Research, Valvular Heart Disease
Introduction
The aortic heart valve is populated primarily by valvular interstitial cells (VICs), a heterogeneous, multipotent1 cell population responsible for maintaining valve homeostasis.2 In healthy leaflets, most VICs exhibit a quiescent (qVIC) phenotype, but injury or stress can trigger their activation into a myofibroblastic (activated [aVIC]) phenotype.3 The aVICs are proliferative and highly contractile and possess an increased capacity to secrete and remodel the extracellular matrix (ECM).4 On completion of the repair process, aVICs are typically eliminated by apoptosis; however, in pathological processes, VICs remain activated and are thought to play an active role in the initiation and progression of valvular disease.5, 6, 7
The ability to replicate VIC activation from a quiescent state in vitro is critically important for studying molecular and cellular features of aortic valve disease initiation and progression. However, although <5% of resident VICs in a healthy aortic valve exhibit an aVIC phenotype,8 >90% of VICs found in standard in vitro culture conditions are aVICs, as these cells appear to undergo a rapid transformation to an aVIC phenotype when cultured on tissue culture polystyrene (TCPS) in growth medium.4, 9 In addition, a microarray comparison between freshly isolated versus cultured porcine VICs revealed >4000 differentially expressed genes as early as 6 days after the beginning of in vitro culture.10 The dominance of the aVIC phenotype in in vitro cultures is likely to significantly diminish their accuracy for studying VIC activation or mimicking healthy valve conditions. To address this significant limitation, recent work by Latif et al identified a fibroblast media formulation (FIB) capable of dedifferentiating aVICs and maintaining a qVIC phenotype in human VICs on TCPS.11 Specifically, the FIB formulation induced a decrease in proliferation and contraction, fibroblastlike morphology, and a reduction in the expression of myofibroblastic proteins compared with culture in standard media.
Porcine hearts, however, remain the most common source of VICs for researchers,12 and porcine VICs are more prone to spontaneous in vitro activation than human VICs; thus, additional treatments beyond the FIB formulation may prove necessary to achieve quiescence in porcine VICs. Moreover, it has not been established how the use of qVIC cultures may be implemented in the setting of a standard experiment to study pathological stimuli. In the current work, we devised an accessible method to reliably generate quiescent populations of porcine VICs in 2‐dimensional (2‐D) in vitro culture. Furthermore, we use these qVIC populations to study phenotype changes following treatment with a known pathological stimulus, transforming growth factor β1 (TGF‐β1), and contrast this against findings obtained using VICs raised in standard culture methods, thereby demonstrating the efficacy of using the qVIC culture method to study VIC pathobiology.
Materials and Methods
VIC Isolation, Culture, and TGF‐β Treatment
Aortic valve leaflets were excised from male porcine hearts, and VICs were immediately isolated via collagenase digestion, as described previously.11, 13 The resulting passage 0 VICs were seeded and expanded on TCPS in low‐glucose DMEM (Sigma‐Aldrich) supplemented with 10% heat‐inactivated FCS (Sigma‐Aldrich), 150 U/mL penicillin/streptomycin (Sigma‐Aldrich), and 2 mmol/L l‐glutamine (Sigma‐Aldrich). At 4 days after isolation, VICs were examined via immunocytochemistry and flow cytometry for expression of myofibroblastic markers. By 4 to 6 days after isolation, VICs had reached ≈90% confluence and were passaged and divided into 3 different culture conditions: (1) Control was VICs on TCPS cultured in standard growth medium, (2) FIB was VICs on TCPS cultured in FIB media (low‐glucose DMEM, 2% heat‐inactivated FCS, 5.25 μg/mL insulin, 10 ng/mL fibroblast growth factor 2 [FGF‐2; PeproTech], 150 U/mL penicillin/streptomycin, 2 mmol/L l‐glutamine), and (3) FIB‐Coll was VICs cultured in FIB media on collagen I–coated TCPS (2 μg/cm2; Advanced BioMatrix).14 This time point is described as day 0 throughout this study, with notations of days 4, 9, and 14 referring to the time elapsed since day 0 seeding into control, FIB, and FIB‐Coll conditions. Day 0, passage 1 VICs were either plated at a 1:10 ratio in TCPS petri dishes or seeded in 24‐well plates at a density of 10 000 cells/cm2 for phenotypic analysis. This passaging and seeding sequence was repeated 2 more times, with full refeeding of media every 48 hours. VIC phenotype was analyzed on days 4, 9, and 14 following the initial switch to the FIB formulation. These time points were selected to align with points at which VICs are usually passaged for experiment seeding.
To assess the response of VICs to a known disease stimulus, VICs cultured for 9 days in control, FIB, or FIB‐Coll conditions were seeded at 10 000 cells/cm2 on TCPS and re‐fed with reduced‐serum media containing 2% FBS supplemented with 0.25 or 1 ng/mL TGF‐β1 (PeproTech) 24 hours after seeding (day 10). VICs were re‐fed and treated again with TGF‐β1 48 hours later. VIC phenotype was analyzed on day 14.
Flow Cytometry and Immunocytochemistry for Myofibroblastic Markers
The protein expression levels of multiple markers associated with a myofibroblastic phenotype were assessed via flow cytometry. For this purpose, VICs (1×105) were resuspended in 100 μL of PBS and incubated for 30 minutes on ice with an unconjugated mouse monoclonal antibody against α‐smooth muscle actin (αSMA; Dako), calponin (Dako), vimentin (Dako), or vinculin (Millipore) or a rabbit polyclonal antibody against smooth muscle protein 22‐α (SM22; Abcam). Excess primary antibody was removed by washing in PBS before addition of fluorescein isothiocyanate–conjugated secondary antibody (Dako) for 30 minutes on ice. Unbound antibody was removed in 2 washes, and cells were fixed in 150 μL 0.05% formaldehyde (BDH) before 10 000 events were acquired on an EPICS XL flow cytometer (Beckman Coulter). Results were expressed as the percentage of cells staining positively above the control level (2%), which was derived using cells with no primary antibody staining or an isotype‐matched antibody control, and the mean fluorescence intensity for each condition was recorded.
Cells were also stained via immunocytochemistry for these markers to visualize their expression. VICs seeded on coverslips were washed 2 times with PBS and fixed in 3.7% formaldehyde solution (Sigma‐Aldrich) for 10 minutes. The fixative solution was removed by rinsing 3 times with PBS–1% Tween 20 (PBS‐T; Merck). Cells were permeabilized with Triton X‐100 (0.5% vol/vol in PBS‐T; Sigma‐Aldrich) for 3 minutes, washed 3 times with PBS‐T, and blocked for 30 minutes with bovine serum albumin (BSA, 3% wt/vol; Roche). Directly after blocking, cells were incubated with the primary antibodies (in 1.5% w/v BSA) at room temperature for 90 minutes. Primary antibodies used were αSMA (Dako), vimentin (Dako), SM22 (Abcam), and calponin (Dako). Following washing with PBS‐T, samples were incubated with secondary antibodies labeled with Alexa Fluor 555 or Alexa Fluor 488 in PBS‐T for 60 minutes at room temperature. Coverslips were mounted on glass slides in DAPI (4′,6‐diamidino‐2‐phenylindole) Fluoromount‐G (SouthernBiotech), which stained the cell nuclei, and imaged on an Axiovert200M fluorescent microscope (Zeiss).
VIC phenotype was also assessed in freshly harvested porcine aortic valve leaflets. Within 2 hours of slaughter, leaflets were fixed in formalin and then embedded in paraffin and sectioned into 6‐μm slices. Sections were deparaffinized, and antigen retrieval was performed in citric acid buffer (Vector Laboratories) for 2 hours at 80°C. Samples were incubated overnight in 3% goat serum in PBS followed by 1‐hour incubation with mouse anti‐αSMA (1:250, monoclonal, clone 1A4; Sigma‐Aldrich) diluted in 1% goat serum and fluorescent labeling with goat antimouse Alexa Fluor 488 (1:500; Invitrogen) for 1 hour. Samples were counterstained with DAPI (1:1000; Sigma‐Aldrich) for 5 minutes and imaged using an Olympus IX51 microscope.
Analysis of Cell Morphology
To visualize cell morphology, actin filaments in VIC cultures were stained using a Phalloidin‐Atto 488 fluorescent label (Sigma‐Aldrich). Cells were fixed with 3.7% formaldehyde (Sigma‐Aldrich) for 10 minutes, permeabilized with 0.05% Triton X‐100 (Sigma‐Aldrich) for 3 minutes, and rinsed with PBS‐T prior to incubation with the Phalloidin‐Atto 488 fluorescent label for 60 minutes at room temperature. Coverslips were mounted and stained for cell nuclei with DAPI Fluoromount‐G. Cells were imaged with an Axiovert 200M fluorescent microscope. ImageJ software (National Institutes of Health) was used to calculate the length, width, and area of each cell (30–100 cells per condition, 5 different donor hearts). Aspect ratio was calculated by dividing the width by the length of each cell (40–70 cells per condition, 5 different donor hearts).
Quantification of Proliferation and Apoptosis
Cell proliferation was measured using the Click‐iT EdU Alexa Fluor 488 Imaging Kit (Invitrogen). Briefly, VICs were incubated with EdU for 8 hours, followed by fixation, permeabilization, and labeling with Alexa Fluor 488 via a click reaction, as described by the manufacturer instructions. Samples were counterstained with DAPI, and fluorescent images were captured using an Olympus IX51 microscope. Cells stained positively for EdU were counted using ImageJ software and normalized to the total number of cells indicated by DAPI staining to obtain the percentage of proliferating cells.
Apoptosis was evaluated with the SensoLyte Homogeneous AFC Caspase 3/7 Assay Kit (AnaSpec). This assay relies on the cleavage of a fluorescent caspase 3/7 substrate to yield a quantitative measure of relative apoptotic activity across conditions. Fluorescence readings were obtained using a Tecan Infinite M1000 plate reader and divided by the average total number of cells per well in each condition, as quantified by DAPI staining. The apoptosis levels were then normalized to the average of the readings from the control condition at each time point. In the TGF‐β experiments, apoptosis levels were normalized to the average of the readings from the 0‐ng/mL treatment for each corresponding condition.
Quantification of Myofibroblastic Gene Expression via Quantitative Reverse Transcription–Polymerase Chain Reaction
Quantitative reverse transcription–polymerase chain reaction was used to analyze the gene expression levels of myofibroblastic markers (αSMA [ACTA2], calponin [CNN2], and smooth muscle protein 22‐α [SM22]) and extracellular matrix proteins (fibronectin [FN] and collagen type I [COL1A1]) associated with the aVIC phenotype. Markers associated with an osteoblastic VIC phenotype (alkaline phosphatase [ALP] and osteocalcin [OCN]) were also analyzed. VIC RNA was isolated using an RNEasy Mini Kit (Qiagen), and RNA was reversed transcribed using a High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative reverse transcription–polymerase chain reaction was performed with TaqMan Gene Expression Assays (Applied Biosystems). Analysis of data from days 4, 9, and 14 was performed using the comparative CT (ΔΔCT) method, in which the data for each time point were first normalized to 2 endogenous controls (GAPDH and ACTB) and then to the control condition. To allow comparison across all conditions, analysis of the TGF‐β experiment was performed using the relative standard curve CT method, in which a set of standards was generated for each gene from control samples, with normalization to endogenous expression of GAPDH. Data were expressed as fold change relative to 0‐ng/mL TGF‐β treatment for each condition.
Analysis of Fibronectin Deposition via In Situ ELISA
Fibronectin deposition was assayed by semiquantitative immunocytochemical detection. Cells were fixed in 10% neutral buffered formalin (Sigma‐Aldrich), endogenous peroxidase activity was quenched with 0.3% hydrogen peroxide in methanol for 1 hour, and samples were blocked overnight in 3% goat serum. Samples were then incubated with monoclonal rabbit antifibronectin antibody (polyclonal, 0.4 μg/mL; Santa Cruz Biotechnology) for 2 hours, followed by a series of washes with 1X PBS and labeling with a horseradish peroxidase–linked goat antirabbit secondary antibody (polyclonal, 0.5 μg/mL; Alpha Diagnostic International) for 40 minutes. After washing, samples were incubated in 1‐Step Turbo‐TMB ELISA substrate solution (Thermo Fisher Scientific), and the reaction stopped after 5 minutes through the addition of 2 N sulfuric acid (Thermo Fisher Scientific). Color development was proportional to fibronectin content and measured with a Tecan Infinite M1000 plate reader. Background signal was determined by following the same procedure without incubating in primary antibody solution. This background absorbance was subtracted from the samples for the respective culture condition. Corrected absorbance values were normalized to total cell number determined by DAPI staining and expressed as a fold change over the control condition at each time point.
Quantification of Inflammatory Cytokine Production
The production of inflammatory cytokines by VICs cultured in control and FIB‐Coll conditions was evaluated via ELISAs for interleukin 6 (IL‐6) and IL‐8. VICs were cultured for 9 days in control or FIB‐Coll conditions and then seeded at 10 000 cells/cm2 on TCPS and re‐fed with reduced‐serum media containing 2% FBS. After 5 days of culture, the concentrations of IL‐6 and IL‐8 in VIC culture supernatants were quantified via ELISA, according to the manufacturer's instructions (R&D Systems). A PicoGreen assay (Invitrogen) was used to quantify DNA in the cell culture lysates collected from the aforementioned samples, and inflammatory cytokine concentrations were normalized to DNA amount.
Statistical Analysis
Each experiment was conducted using 6 to 8 individual isolates, for which isolate refers to the VICs isolated from an individual heart. A total of 29 different hearts were used across the entire study. Data are presented as mean±SD, and statistical analyses were performed using GraphPad Prism software. Experimental groups were compared with each other using 2‐way ANOVA, with repeated measures for the time course experiments, in conjunction with the Tukey honestly significant difference post‐test. Data that were not normally distributed were analyzed using a Kruskal–Wallis 1‐way ANOVA followed by a Dunn post hoc test. P values ≤0.01 were considered statistically significant.
Results
Porcine VICs Exhibit Myofibroblastic Behavior Shortly After Isolation
The baseline phenotype of porcine VICs cultured in control (ie, normal growth medium) conditions was evaluated via flow cytometry 4 days after isolation, at passage 0. Even at this early time point, a high percentage of VICs expressed the following myofibroblastic markers: vimentin (88%), αSMA (79%), SM22 (41%), and calponin (19%) (Table). These results were confirmed visually through immunocytochemistry (Figure 1). In contrast, most VICs found in healthy porcine aortic valves exhibit a quiescent phenotype (Figure S1). Standard culture methods led to robust and quick transformation of qVICs into an aVIC phenotype.
Table 1.
Mean Percentage of Passage 0 Valvular Interstitial Cells Positive for Myofibroblastic Markers
| Marker | Percentage (SD) |
|---|---|
| α‐Smooth muscle actin | 79.4 (5.1) |
| Calponin | 18.6 (1.9) |
| Smooth muscle protein 22‐α | 41.4 (23.1) |
| Vimentin | 88.2 (4.4) |
Figure 1.
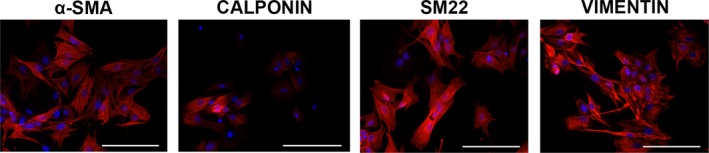
Immunohistochemical staining for activated VIC markers in passage 0 VIC cultures. VICs expressed vimentin and the myofibroblastic markers αSMA, calponin, and SM22 after culture in control conditions. Detected proteins are stained red, and nuclei are stained blue. Scale bar represents 100 μm. αSMA indicates α‐smooth muscle actin; SM22, smooth muscle protein 22‐α; VIC, valvular interstitial cell.
VICs Cultured in FIB or FIB‐Coll Conditions Do Not Exhibit Myofibroblastic Morphology
The morphology of VICs cultured in control, FIB, and FIB‐Coll conditions was evaluated, as myofibroblastic differentiation of cells is typically accompanied by a large increase in cell area and an evenly spread morphology. Staining with phalloidin resulted in the visualization of clear differences in cell shape at all time points between VICs cultured in FIB or FIB‐Coll conditions compared with control VICs (Figure 2A). On day 0, which corresponds to 4 days after the initial harvest of VICs from intact leaflets, VICs already demonstrated the spread, flattened appearance that is characteristic of myofibroblasts. At subsequent time points, control VICs continued to exhibit this myofibroblastic morphology, whereas VICs cultured in FIB or FIB‐Coll exhibited a thinner, spindled shape with long extensions. Quantification of cell shape and aspect ratio confirmed these observations and revealed that, as early as 4 days after initiation of FIB or FIB‐Coll treatment, VICs exhibited a 2‐fold decrease in area and aspect ratio (Figure 2B and 2C). No significant differences were noted between FIB and FIB‐Coll conditions. These trends were sustained throughout the course of the study.
Figure 2.
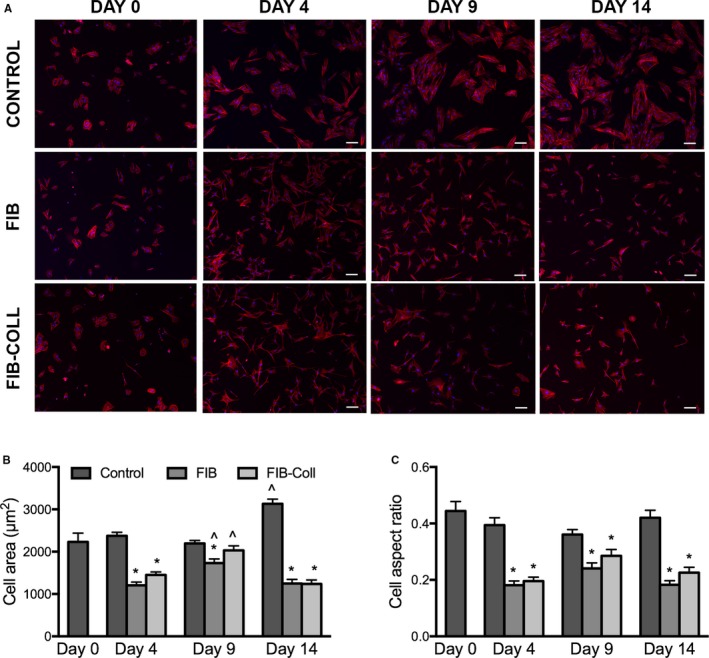
VICs cultured in FIB or FIB‐Coll conditions exhibit a more elongated and spindled morphology compared with control VICs. A, Staining for phalloidin (red), nuclei are counterstained blue. Scale bar represents 100 μm. Quantification of (B) cell shape and (C) aspect ratio for VICs cultured in control, FIB, or FIB‐Coll conditions. *P<0.01 compared with control VICs at the same time point. ^ P<0.01 compared with day 4 VICs cultured in the same condition. n=6. FIB indicates fibroblast media formulation; FIB‐Coll, fibroblast media formulation with culture on collagen coatings; VIC, valvular interstitial cell.
FIB‐Coll Decreases VIC Proliferation and Apoptosis
Cell quiescence is generally associated with low rates of proliferation and apoptosis.15, 16 These behaviors were assessed in control, FIB, and FIB‐Coll conditions at time points of 4, 9, and 14 days following the transition to FIB or FIB‐Coll conditions. At all time points, VICs cultured in FIB medium exhibited significantly lower proliferation than those cultured in control medium (Figure 3A). At day 4, no additional change in proliferation was conferred by the FIB‐Coll condition relative to FIB alone. However, significant differences between FIB and FIB‐Coll emerged by day 9, at which point VIC proliferation in FIB‐Coll was significantly lower than that in FIB (18% versus 28%). At day 14, this difference was even more pronounced (11% for Fib‐Coll versus 32% for FIB).
Figure 3.
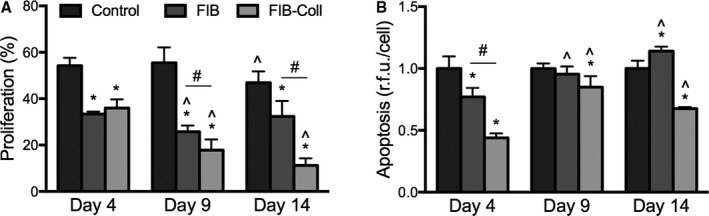
VIC proliferation and apoptosis decrease after culture in FIB‐Coll. A, Quantification of the percentage of proliferating cells after 8 hours of incubation with EdU. B, Evaluation of apoptosis levels through a caspase 3/7 activity assay. *P<0.01 compared with control VICs at the same time point. ^ P<0.01 compared with day 4 VICs cultured in the same condition. # P<0.01 for comparisons indicated on graph. n=6 to 8. FIB indicates fibroblast media formulation; FIB‐Coll, fibroblast media formulation with culture on collagen coatings; VIC, valvular interstitial cell.
VICs exhibited lower caspase 3/7 activity, indicative of lower levels of apoptosis, in FIB medium compared with control medium at day 4 (Figure 3B). At both days 9 and 14, however, apoptosis levels in FIB medium were equivalent to those observed in the control condition. In contrast, VICs cultured in FIB‐Coll exhibited significantly lower apoptosis compared with control media at all time points.
Myofibroblastic Marker Expression Is Reduced by FIB and FIB‐Coll
The gene expression of proteins associated with VIC activation and myofibroblastic differentiation was assessed via quantitative reverse transcription–polymerase chain reaction. At all time points, culture in FIB or FIB‐Coll conditions led to significantly lower expression of ACTA2, SM22, and CNN2 (Figure 4A–4C). At day 4, a 3‐fold decrease in ACTA2 and SM22 mRNA levels was observed in FIB and FIB‐Coll relative to control medium. Meanwhile, VIC culture in FIB‐Coll was significantly more effective than FIB in decreasing the expression of CNN2. At days 9 and 14, both FIB and FIB‐Coll exhibited similar abilities to decrease expression of myofibroblastic markers, yielding a 4‐fold decrease in ACTA2, SM22, and CNN2 relative to the control.
Figure 4.
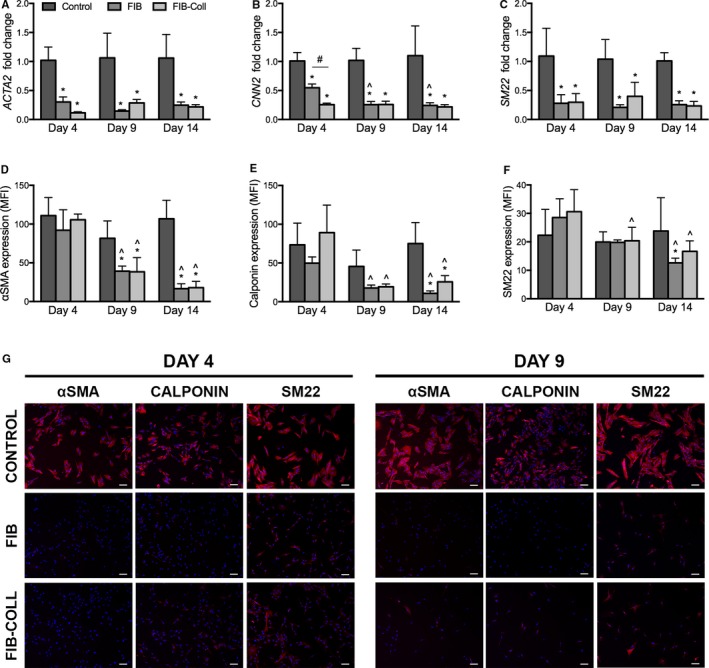
Expression of myofibroblastic markers decreases dramatically after culture in FIB and FIB‐Coll. A through C, Gene expression analysis of (A) ACTA2, (B) CNN2, and (C) SM22 after 4, 9, and 14 days of culture in control, FIB, or FIB‐Coll conditions. D through F, Evaluation of protein expression for the same markers: (D) αSMA, (E) calponin, and (F) SM22. MFI levels were obtained through flow cytometry. G, Immunostaining for myofibroblastic markers. Scale bar represents 100 μm. *P<0.01 compared with control VICs at the same time point. ^ P<0.01 compared with day 4 VICs cultured in the same condition. # P<0.01 for comparison indicated on graph. n=6. αSMA indicates α‐smooth muscle actin; FIB, fibroblast media formulation; FIB‐Coll, fibroblast media formulation with culture on collagen coatings; MFI, mean fluorescence intensity; SM22, smooth muscle protein 22‐α; VIC, valvular interstitial cell.
To confirm these findings at the protein level, myofibroblastic marker expression was analyzed via flow cytometry (Figure 4D–4F, Figure S1). At day 4, no statistically significant differences in αSMA, calponin, or SM22 mean fluorescence intensity were found across conditions. At day 9, VICs exhibited a 2‐fold decrease in αSMA expression in both FIB and FIB‐Coll conditions compared with the control, ultimately yielding a 5‐fold decrease by day 14 (Figure 4D). Similarly, both FIB and FIB‐Coll led to a decrease in calponin expression levels by day 14 (Figure 4E). SM22 levels were significantly reduced in FIB compared with the control at day 14, whereas FIB‐Coll appeared to have no effect on the fluorescence intensity of this marker at any time point. The percentage of VICs positive for αSMA, calponin, and SM22 also decreased after day 9 and beyond in FIB and FIB‐Coll (Figure S2). Because the reported mean fluorescence intensity refers to the expression levels only in the cells that stained positively, immunocytochemistry was also performed to better characterize VIC phenotype. This analysis revealed stark differences in myofibroblastic protein expression between the control and FIB or FIB‐Coll conditions (Figure 4G, Figure S3) that confirmed the polymerase chain reaction and flow cytometry findings. As early as day 4, a decrease in staining for αSMA, calponin, and SM22 in VICs cultured in FIB and FIB‐Coll was observed. This decrease was sustained at days 9 and 14.
ECM Production Is Reduced by FIB and FIB‐Coll
A myofibroblastic phenotype is typically associated with increased ECM production. Results shown in Figure 5 demonstrate that collagen gene expression by VICs cultured in FIB or FIB‐Coll was lower at all time points compared with the control condition (Figure 5A). On day 4, however, culture in FIB‐Coll was more effective than FIB alone in suppressing COL1A1 expression, yielding a 0.2 fold change in expression relative to control, compared with 0.7 for FIB. By day 9, there was no longer a significant difference in the expression levels between the FIB and FIB‐Coll conditions. This trend was maintained at day 14, at which point both FIB and FIB‐Coll had decreased COL1A1 expression by 5‐fold relative to the control. Protein levels of collagen were not assessed because of interference created by the collagen coating in the FIB‐Coll condition.
Figure 5.
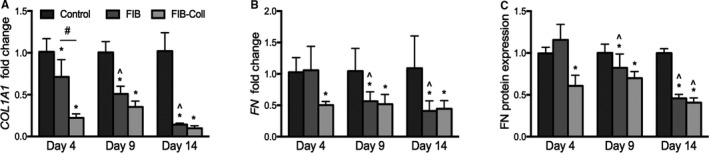
Extracellular matrix production decreases after culture in FIB and FIB‐Coll. A, COL1A1 gene expression was evaluated after 4, 9, and 14 days of culture in control, FIB, or FIB‐Coll conditions. Fibronectin (B) gene and (C) protein expression were also assessed. *P<0.01 compared with control VICs at the same time point. ^ P<0.01 compared with day 4 VICs cultured in the same condition. # P<0.01 for comparison indicated on graph. n=6. FIB indicates fibroblast media formulation; FIB‐Coll, fibroblast media formulation with culture on collagen coatings; VIC, valvular interstitial cell.
There was no difference in FN gene expression (Figure 5B) or protein production (Figure 5C) between control and FIB medium after 4 days in culture. In contrast, VICs cultured in FIB‐Coll exhibited a significant decrease in FN gene expression and fibronectin deposition at that same time point. By day 9, both FIB and FIB‐Coll conditions yielded a statistically significant reduction in FN gene expression and protein deposition compared with the control. At day 14, the difference between either of the FIB treatments and the control was >2‐fold for both gene expression and protein deposition.
qVICs Generated via Culture in FIB‐Coll Exhibit Increased Sensitivity to TGF‐β1 Treatment
We next examined whether the qVICs generated by FIB or FIB‐Coll treatment exhibited differential responsiveness to a known stimulus of myofibroblastic differentiation, TGF‐β1, compared with the aVICs generated via standard culture conditions. VICs were cultured in control, FIB, or FIB‐Coll conditions until the day 9 time point described in previous sections. At this point, instead of continuing in FIB or FIB‐Coll conditions, all VICs were seeded on TCPS and treated with 0, 0.25, or 1 ng/mL TGF‐β1 in reduced‐serum media for 5 days. Treatment with TGF‐β1 had no effect on cell morphology in VICs that had been raised in control or FIB conditions (Figure 6A–6C). In contrast, qVICs generated via FIB‐Coll culture exhibited an increase in aspect ratio following treatment with 1 ng/mL TGF‐β1. Similarly, the proliferation of VICs generated by culture in control or FIB conditions was unaffected by TGF‐β1 treatment (Figure 6D), whereas both TGF‐β1 doses significantly increased the proliferation of qVICs raised in FIB‐Coll culture. Combined with the morphology data, these findings suggested that qVICs generated via FIB‐Coll culture underwent myofiboblastic differentiation on TGF‐β1 treatment. Although TGF‐β1 induced apoptosis in VICs raised in all 3 conditions, the most dramatic increase in VIC apoptosis relative to no treatment was observed in qVICs generated via FIB‐Coll culture.
Figure 6.
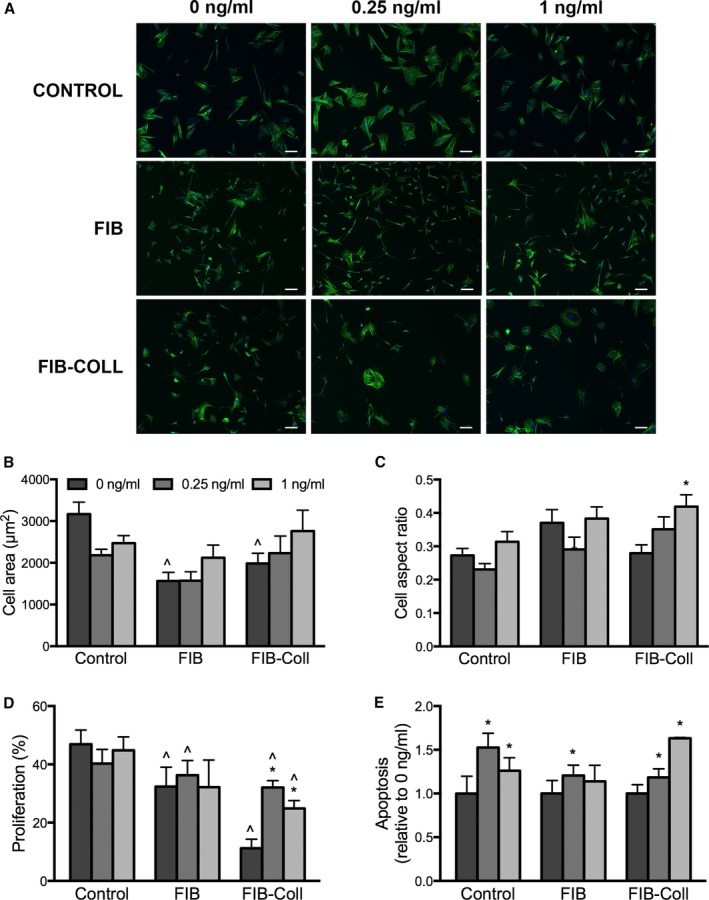
TGF‐β1 induces changes in cell morphology, proliferation, and apoptosis in VICs generated via FIB‐Coll culture. VICs raised in control, FIB, or FIB‐Coll culture were treated with 0, 0.25, or 1 ng/mL TGF‐β1 for 5 days on tissue culture polystyrene. A, Staining for phalloidin (green), nuclei are counterstained blue. Scale bar represents 100 μm. Quantification of (B) cell shape, (C) aspect ratio, (D) proliferation, and (E) apoptosis. *P<0.01 compared with 0 ng/mL in the same condition (control, FIB, or FIB‐Coll). ^ P<0.01 compared with control VICs treated with the same amount of TGF‐β1. n=6 to 8. FIB indicates fibroblast media formulation; FIB‐Coll, fibroblast media formulation with culture on collagen coatings; TGF‐β1, transforming growth factor β1; VIC, valvular interstitial cell.
Quantification of gene and protein expression levels of myofibroblastic markers (Figure 7A–7C) yielded findings that were consistent with our data on cell shape and proliferation. Neither dose of TGF‐β1 was able to induce myofibroblastic differentiation of control VICs, as indicated by unchanged ACTA2 and SM22 expression compared with untreated cultures. Meanwhile, qVICs generated via FIB culture exhibited some myofibroblastic characteristics in response to TGF‐β1 treatment; although ACTA2 expression remained unchanged at both TGF‐β1 doses, the higher dose (1 ng/mL) of TGF‐β1 increased the expression of SM22. Consistent with the results for morphology, proliferation, and apoptosis, qVICs raised in FIB‐Coll culture exhibited the greatest sensitivity to TGF‐β1‐induced myofibroblastic differentiation. Treatment of these qVICs with only 0.25 ng/mL TGF‐β1 increased expression of ACTA2 and SM22 by >5‐ and >4‐fold, respectively, compared with untreated qVICs. These observations were also reflected by immunofluorescent staining for αSMA and SM22 (Figure 7C), with which qVICs generated in FIB‐Coll culture displayed a noticeable increase in SM22 on TGF‐β1 treatment, whereas VICs from control or FIB conditions appeared relatively unchanged. The increase in αSMA protein in TGF‐β1‐treated qVICs from FIB‐Coll culture was less prominent, perhaps because αSMA is typically produced at a slightly later time than SM22 during myofibroblastic differentiation.17
Figure 7.
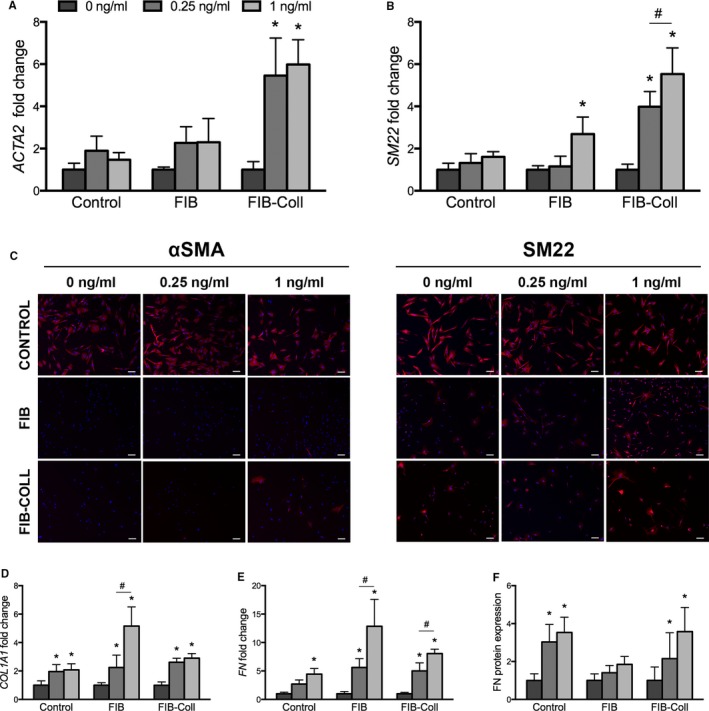
VICs raised in FIB‐Coll culture respond more strongly to TGF‐β1 treatment than VICs raised in FIB and control culture. Myofibroblastic marker and ECM expression were also analyzed in response to TGF‐β1 treatment. The extent of VIC activation was evaluated through gene expression analysis of (A) ACTA2 and (B) SM22 and (C) immunofluorescent staining for αSMA and SM22. Red indicates positive staining for each marker; nuclei are counterstained blue. Scale bar represents 100 μm. ECM production was assessed through quantitative reverse transcriptase–polymerase chain reaction for (D) COL1A1 and (E) FN, as well as an in situ ELISA for (F) fibronectin protein deposition. *P<0.01 compared with 0 ng/mL in the same condition (control, FIB, or FIB‐Coll). # P<0.01 for comparison indicated on graph. n=6. αSMA indicates α‐smooth muscle actin; ECM, extracellular matrix; FIB, fibroblast media formulation; FIB‐Coll, fibroblast media formulation with culture on collagen coatings; SM22, smooth muscle protein 22‐α; TGF‐β1, transforming growth factor β1; VIC, valvular interstitial cell.
TGF‐β1 is known as a potent stimulus of ECM production in many cell types; this was also the case for all conditions in the current study (Figure 7D–7F). Control VICs exhibited an increase in COL1A1 and FN gene expression after treatment with the higher dose (1 ng/mL) of TGF‐β1. Meanwhile, VICs conditioned through FIB or FIB‐Coll were responsive to both the low and high doses of TGF‐β1, resulting in significantly increased COL1A1 and FN mRNA levels compared with untreated cells. Analysis of fibronectin protein production showed significant increases in fibronectin production by VICs raised in control or FIB‐Coll conditions.
Finally, it is important to note that VICs generated via FIB or FIB‐Coll culture and not treated with TGF‐β1 maintained a qVIC phenotype throughout the course of the 5‐day experiment, even in the absence of insulin, FGF‐2, and a collagen coating. These VICs continued to exhibit smaller cell area (Figure 6B), proliferated at lower rates (Figure 6D), expressed lower levels of myofibroblastic markers (Figure 7C, Figure S4A and S4B), and produced less ECM (Figure S4C and S4D) compared with control VICs raised in standard growth medium.
Discussion
The spontaneous activation of VICs in standard in vitro culture on TCPS is a major limiting factor in the study of healthy valve biology and calcific aortic valve disease (CAVD). Previously, a media formulation (FIB) capable of deactivating VICs had been reported; however, this formulation had been tested only on cells isolated from human valves,11 which experience less robust myofibroblastic activation in culture. Moreover, the utility of these cells in studying VIC response to pathological stimuli has not been demonstrated. In the current study, culture of porcine VICs on TCPS in FIB media significantly reduced several markers of VIC activation, including proliferation, apoptosis, αSMA expression, and matrix production, relative to standard growth medium. A combination of FIB with culture on collagen coatings (FIB‐Coll) achieved similar results and an even greater reduction of proliferation, apoptosis, and matrix production compared with FIB media alone. To our knowledge, this is the first time that such robust VIC deactivation has been achieved in 2‐dimensional culture. Moreover, VICs generated via FIB‐Coll culture exhibited increased responsiveness to TGF‐β1, a known inducer of myofibroblastic activation, compared with either standard VIC cultures or those treated with FIB media alone. This method of maintaining a porcine qVIC phenotype provides a valuable platform for mimicking healthy valve behavior and for studying disease stimuli in aortic valve disease.
We demonstrated that a large percentage of porcine VICs cultured under standard conditions express myofibroblastic markers shortly after isolation, prior to passaging. This VIC activation could be reversed by culture in FIB or FIB‐Coll conditions, where characteristics of a quiescent phenotype emerged after only 4 days in culture, including changes in cell shape and a decrease in proliferation, apoptosis, myofibroblastic gene expression, and ECM production. By day 9, the reduction in all of those features had become more pronounced, and the effect on myofibroblastic marker expression could be detected at the protein level. At day 14, no further effect of the FIB or FIB‐Coll treatment on VIC phenotype was observed for most metrics analyzed. Perhaps even more strikingly, the quiescent features induced by FIB and FIB‐Coll were sustained even after 5 days in culture without the full FIB formulation or a collagen coating (Figure S4). Because the myofibroblast phenotype may be transient in a diseased valve and segue into inflammatory activity and/or osteogenic differentiation, markers for these further downstream phenotypes were also evaluated; both the production of inflammatory cytokines and the osteogenic marker alkaline phosphatase were found to be significantly decreased in FIB and FIB‐Coll cultures relative to the control (Figure S5). Consequently, we developed a robust method to efficiently generate qVICs in <2 weeks that can then be used for further experiments without significant phenotypic drift.
This dramatic change in VIC phenotype warrants closer examination of the culture conditions selected for this study. Latif et al published a full rationale for the selection of the cocktail present in FIB media.11 Briefly, supplementation with 2% FBS was selected over the traditional 10% concentration to reduce the amount of growth factors present in the medium. In addition, insulin and FGF‐2 were selected for their reported ability to regulate growth18 and decrease VIC αSMA expression,19 respectively. Meanwhile, collagen has been previously associated with decreased VIC pathology in both 2D and 3D studies.14, 20, 21 Our experiments found that collagen coating alone can decrease the expression of contractile proteins but significantly increases VIC proliferation and thus is unable to yield a qVIC phenotype (Figure S6); others have similarly found that collagen is not sufficient to yield qVIC cultures.11, 14 We hypothesized, however, that the combination of a collagen coating with soluble factors (ie, FIB‐Coll condition) might provide additional deactivation cues. This hypothesis was confirmed by the data: Although the FIB formulation alone was sufficient to generate significant reductions in myofibroblast markers, culture in FIB‐Coll achieved the same results, sometimes sooner (4 versus 9 days for ECM production) and at other times more dramatically (eg, proliferation and apoptosis), compared with FIB alone. Moreover, qVICs generated via FIB‐Coll culture exhibited markedly higher sensitivity to TGF‐β1 treatment. Consistent with previous work,14, 20, 21, 22 these findings point to an important role for collagen in sustaining healthy VIC function and valve homeostasis. Further study is needed to elucidate the mechanisms by which these soluble and insoluble cues are able to modulate VIC phenotype; a preliminary investigation of vinculin expression suggests that focal adhesion‐related pathways may be involved (Figure S6C).
Although this is not the first report of successful deactivation of porcine VICs in in vitro culture, it is the only method described for 2D culture and thus is easily implemented for large‐scale VIC expansion and subsequent seeding for experiments. Other approaches for reducing αSMA expression have involved 3D VIC culture in soft hydrogels.10, 23, 24, 25 Interestingly, VIC encapsulation in synthetic hydrogels decorated with peptides derived from collagen I led to the lowest percentage of αSMA‐positive VICs compared with fibronectin‐ and elastin‐derived peptides.23 In many of these cases, the ability to induce the quiescent phenotype has been hypothesized to depend on the soft mechanical properties of these materials.26, 27 Within our system, the presence of both soluble (insulin and FGF‐2) and insoluble (collagen) biochemical cues appeared to be sufficient to overcome the effect of culture on stiff TCPS substrates. Although the aforementioned 3D systems provide elegant platforms for investigating VIC behavior in response to microenvironmental stimuli, they are not intended as a means of regular generation of qVIC populations for subsequent seeding and experimentation. Most laboratories do not possess the expertise to synthesize the biomaterials necessary for these systems, and the costs associated with these methods are too high for the continuous generation of qVICs. Furthermore, the difficulties associated with isolating cells from 3D matrices without incurring cellular damage limit the utility of VICs cultured in this manner for follow‐up experiments. In contrast, the FIB‐Coll method presented in this paper is easy to use and inexpensive; only small quantities of FGF‐2 and collagen are necessary to achieve the desired effect. In addition, it can easily be scaled up or down according to the desired application. Any laboratory already performing valve‐related research could readily adopt this approach. The ability of the geographically diverse author team to generate reproducible, consistent results in experiments performed in 3 different countries and using independent sources of pig hearts also serves as a testament to the reliability and robustness of the FIB‐Coll approach.
As discussed previously, although aVICs are rare in healthy valves (Figure S1), existing in vitro studies have been performed predominantly on aVICs and, therefore, may not accurately represent the VIC response to pathological stimuli in the native valve environment. Our hypothesis that qVICs would be more sensitive to a myofibroblastic stimulus was borne out by administration of TGF‐β1 to VICs raised in FIB, FIB‐Coll, or control conditions. Increased levels of TGF‐β1 are found in CAVD,28 and this molecule is widely studied as an inducer of VIC pathology to better understand its role in disease progression.28, 29, 30 Our control condition represents the standard approach that is used to grow VICs, including those used in studies involving TGF‐β treatment29, 30, 31; however, these cells are already predominantly aVICs and remain unaltered with respect to morphology, proliferation, and myofibroblastic gene expression when treated with TGF‐β, thereby illustrating the serious problem in using VICs raised in standard growth medium to study VIC pathology. Meanwhile, VICs raised in FIB‐Coll conditions were highly sensitive to TGF‐β1, exhibiting changes in cell aspect ratio and large increases in cell proliferation, apoptosis, myofibroblastic gene and protein expression, and ECM production. These responses were elicited by 0.25‐ and 1‐ng/mL dosages of TGF‐β1; there do not appear to be any previous reports of a TGF‐β1 dose as low as 0.25 ng/mL being capable of inducing VIC activation.14, 29, 31, 32, 33, 34, 35 In contrast to VICs grown in standard culture conditions, which exhibited a diminished response to this disease stimulus, qVICs generated via FIB‐Coll culture required only a small perturbation with TGF‐β1 to elicit dysfunctional behavior. It is likely that the control VICs would have responded to much higher doses of TGF‐β1 (eg, 5 ng/mL),32, 36, 37 but the physiological relevance of a majority aVIC population receiving high doses of TGF‐β1 is questionable. Consequently, qVICs generated via FIB‐Coll culture may provide a more accurate model to elucidate the mechanisms of VIC activation and dysfunction, which have been elusive because of the lack of a starting healthy population. The ability to generate qVICs can be applied to the study of CAVD etiology by enabling the creation of culture environments that are appropriate for studying disease onset (ie, predominantly qVICs) or different stages of disease progression (ie, varying ratios of qVIC:aVIC, depending on extent of disease). The differential responsiveness of qVICs and aVICs to TGF‐β1 in our study emphasizes the importance of using the VIC phenotype that is most appropriately matched to the biological question or disease stage being investigated.
Conclusions
We developed a facile method to reliably and robustly generate porcine qVIC cultures, which are intended to mimic the healthy VIC population in the aortic valve and thus improve our ability to study pathological stimuli involved in CAVD. The culture conditions described in this work complement existing methods to generate osteoblastic VICs,38, 39 a phenotype thought to be responsible for leaflet mineralization in the late stages of CAVD.4 This work has significant implications for the valve research community, as the ability to generate the VIC phenotypes involved in CAVD progression (qVICs, aVICs, and osteoblastic VICs) is a critical advancement necessary for deciphering the mechanisms of this disease. As the field continues to develop more complex platforms for cell culture involving combinations of environmental cues, researchers will need to carefully assess the correct VIC phenotypes to be used depending on the pathological event being studied, for example, lipid oxidation (early in CAVD)40 versus angiogenesis (later in CAVD). The importance of using the correct VIC phenotype will become increasingly important as these models transition from tools for basic research and discovery to drug testing applications; for example, multiple studies have reported that statin drugs elicit differential responses in aVICs versus osteoblastic VICs.41, 42 Careful evaluation not only of the biochemical and physical cues present in each model but also of the starting phenotype of the VICs under study will be necessary to mimic healthy and diseased valve behavior and to reliably predict biological responses to pharmacological treatments.
Sources of Funding
This work was supported by the National Heart, Lung, and Blood Institute and the National Institute of Biomedical Imaging and Bioengineering at the National Institutes of Health (HL093281 and EB019508; to Masters), a predoctoral fellowship from the American Heart Association (15PRE 22170006; to Porras) as well as the Magdi Yacoub Institute.
Disclosures
None.
Supporting information
Figure S1. VICs are quiescent in freshly explanted porcine aortic valve leaflets. Immunofluorescent staining for αSMA demonstrated the absence of activated VICs in healthy aortic valves: αSMA (green), DAPI (blue). Scale bar represents 200 μm. αSMA indicates α‐smooth muscle actin; DAPI, 4′,6‐diamidino‐2‐phenylindole; VIC, valvular interstitial cell.
Figure S2. The percentage of VICs positive for myofibroblastic markers decreases after culture in FIB or FIB‐Coll. Quantification of (A) α‐smooth muscle actin, (B) calponin, and (C) smooth muscle protein 22‐α expression via flow cytometry. *P<0.05 compared with control VICs at the same time point. ^ P<0.05 compared with day 4 VICs cultured in the same condition. VIC, valvular interstitial cell.
Figure S3. Myofibroblastic marker expression after 14 days of culture in control, FIB, or FIB with culture on collagen coatings. Myofibroblastic markers are stained red; nuclei are stained blue with DAPI. Scale bar represents 100 μm. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; FIB, fibroblast media formulation.
Figure S4. Valvular interstitial cells generated via FIB or FIB‐Coll remained quiescent for 5 days following removal of FIB or FIB‐Coll conditions. Quantification of myofibroblastic (α‐smooth muscle actin and smooth muscle protein 22‐α) and extracellular matrix (Col1A1 and FN) gene expression. *P<0.05 compared with control, ^ P<0.05 for indicated comparison. FIB, fibroblast media formulation; FIB‐Coll, fibroblast media formulation with culture on collagen coatings.
Figure S5. VICs cultured in fibroblast media formulation with culture on collagen coatings exhibited decreased inflammatory cytokine production and osteogenic activity relative to control. The inflammatory activity of VICs was assessed using ELISAs for (A) IL‐6 and (B) IL‐8. The expression of (C) alkaline phosphatase (ALP) and (D) osteocalcin (OCN) genes by VICs in different culture environments was evaluated as a measure of osteogenic activity. The “standard” is a positive control generated by culture of VICs on uncoated tissue culture polystyrene and treatment with osteogenic medium (DMEM with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mmol/L l‐glutamine, 10 mmol/L β‐glycerophosphate, and 50 μg/mL ascorbic acid). IL indicates interleukin; VIC, valvular interstitial cell.
Figure S6. Culture on collagen alone was insufficient to yield a quiescent phenotype. A, VIC proliferation on collagen was significantly increased relative to the control and conditions containing FIB media, although (B) VICs on collagen exhibited decreased expression of contractile proteins relative to the control. C, An initial increase in vinculin expression was noted in FIB with culture on collagen coatings at Day 4, but then significantly decreased at later time points. FIB, fibroblast media formulation; VIC, valvular interstitial cell.
(J Am Heart Assoc. 2017;6:e005041. DOI: 10.1161/JAHA.116.005041.)
Contributor Information
Najma Latif, Email: n.latif@imperial.ac.uk.
Kristyn S. Masters, Email: kmasters@wisc.edu.
References
- 1. Chen JH, Yip CY, Sone ED, Simmons CA. Identification and characterization of aortic valve mesenchymal progenitor cells with robust osteogenic calcification potential. Am J Pathol. 2009;174:1109–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen JH, Simmons CA. Cell‐matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108:1510–1524. [DOI] [PubMed] [Google Scholar]
- 3. Rajamannan NM, Evans FJ, Aikawa E, Grande‐Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O'Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: calcific aortic valve disease‐2011 update. Circulation. 2011;124:1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li C, Xu S, Gotlieb AI. The progression of calcific aortic valve disease through injury, cell dysfunction, and disruptive biologic and physical force feedback loops. Cardiovasc Pathol. 2013;22:1–8. [DOI] [PubMed] [Google Scholar]
- 6. Spadaccio C, Mozetic P, Nappi F, Nenna A, Sutherland F, Trombetta M, Chello M, Rainer A. Cells and extracellular matrix interplay in cardiac valve disease: because age matters. Basic Res Cardiol. 2016;111:16. [DOI] [PubMed] [Google Scholar]
- 7. Yip CY, Simmons CA. The aortic valve microenvironment and its role in calcific aortic valve disease. Cardiovasc Pathol. 2011;20:177–182. [DOI] [PubMed] [Google Scholar]
- 8. Latif N, Sarathchandra P, Chester AH, Yacoub MH. Expression of smooth muscle cell markers and co‐activators in calcified aortic valves. Eur Heart J. 2015;36:1335–1345. [DOI] [PubMed] [Google Scholar]
- 9. Taylor PM, Allen SP, Yacoub MH. Phenotypic and functional characterization of interstitial cells from human heart valves, pericardium and skin. J Heart Valve Dis. 2000;9:150–158. [PubMed] [Google Scholar]
- 10. Wang H, Tibbitt MW, Langer SJ, Leinwand LA, Anseth KS. Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity‐regulated PI3K/AKT pathway. Proc Natl Acad Sci USA. 2013;110:19336–19341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Latif N, Quillon A, Sarathchandra P, McCormack A, Lozanoski A, Yacoub MH, Chester AH. Modulation of human valve interstitial cell phenotype and function using a fibroblast growth factor 2 formulation. PLoS One. 2015;10:e0127844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bowler MA, Merryman WD. In vitro models of aortic valve calcification: solidifying a system. Cardiovasc Pathol. 2015;24:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson CM, Hanson MN, Helgeson SC. Porcine cardiac valvular subendothelial cells in culture: cell isolation and growth characteristics. J Mol Cell Cardiol. 1987;19:1185–1193. [DOI] [PubMed] [Google Scholar]
- 14. Rodriguez KJ, Masters KS. Regulation of valvular interstitial cell calcification by components of the extracellular matrix. J Biomed Mater Res A. 2009;90:1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yao G. Modelling mammalian cellular quiescence. Interface Focus. 2014;4:20130074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013;14:329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2010;43:146–155. [DOI] [PubMed] [Google Scholar]
- 18. Strassburger K, Tiebe M, Pinna F, Breuhahn K, Teleman AA. Insulin/IGF signaling drives cell proliferation in part via Yorkie/YAP. Dev Biol. 2012;367:187–196. [DOI] [PubMed] [Google Scholar]
- 19. Cushing MC, Mariner PD, Liao JT, Sims EA, Anseth KS. Fibroblast growth factor represses Smad‐mediated myofibroblast activation in aortic valvular interstitial cells. FASEB J. 2008;22:1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu X, Masters KS. Regulation of valvular interstitial cell calcification by adhesive peptide sequences. J Biomed Mater Res A. 2010;93:1620–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez KJ, Piechura LM, Porras AM, Masters KS. Manipulation of valve composition to elucidate the role of collagen in aortic valve calcification. BMC Cardiovasc Disord. 2014;14:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hutson HN, Marohl T, Anderson M, Eliceiri K, Campagnola P, Masters KS. Calcific aortic valve disease is associated with layer‐specific alterations in collagen architecture. PLoS One. 2016;11:e0163858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gould ST, Anseth KS. Role of cell‐matrix interactions on VIC phenotype and tissue deposition in 3D PEG hydrogels. J Tissue Eng Regen Med. 2016;10:E443–E453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hjortnaes J, Camci‐Unal G, Hutcheson JD, Jung SM, Schoen FJ, Kluin J, Aikawa E, Khademhosseini A. Directing valvular interstitial cell myofibroblast‐like differentiation in a hybrid hydrogel platform. Adv Healthc Mater. 2015;4:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mabry KM, Payne SZ, Anseth KS. Microarray analyses to quantify advantages of 2D and 3D hydrogel culture systems in maintaining the native valvular interstitial cell phenotype. Biomaterials. 2016;74:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang H, Haeger SM, Kloxin AM, Leinwand LA, Anseth KS. Redirecting valvular myofibroblasts into dormant fibroblasts through light‐mediated reduction in substrate modulus. PLoS One. 2012;7:e39969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang H, Leinwand LA, Anseth KS. Cardiac valve cells and their microenvironment–insights from in vitro studies. Nat Rev Cardiol. 2014;11:715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jian B, Narula N, Li QY, Mohler ER III, Levy RJ. Progression of aortic valve stenosis: TGF‐beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465; discussion 465‐6. [DOI] [PubMed] [Google Scholar]
- 29. Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor‐beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95:253–260. [DOI] [PubMed] [Google Scholar]
- 30. Liu AC, Gotlieb AI. Transforming growth factor‐beta regulates in vitro heart valve repair by activated valve interstitial cells. Am J Pathol. 2008;173:1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benton JA, Kern HB, Anseth KS. Substrate properties influence calcification in valvular interstitial cell culture. J Heart Valve Dis. 2008;17:689–699. [PMC free article] [PubMed] [Google Scholar]
- 32. Benton JA, Fairbanks BD, Anseth KS. Characterization of valvular interstitial cell function in three dimensional matrix metalloproteinase degradable PEG hydrogels. Biomaterials. 2009;30:6593–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duan B, Yin Z, Hockaday Kang L, Magin RL, Butcher JT. Active tissue stiffness modulation controls valve interstitial cell phenotype and osteogenic potential in 3D culture. Acta Biomater. 2016;36:42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hutcheson JD, Ryzhova LM, Setola V, Merryman WD. 5‐HT(2B) antagonism arrests non‐canonical TGF‐beta1‐induced valvular myofibroblast differentiation. J Mol Cell Cardiol. 2012;53:707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Osman L, Yacoub MH, Latif N, Amrani M, Chester AH. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation. 2006;114:I547–I552. [DOI] [PubMed] [Google Scholar]
- 36. Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–942. [DOI] [PubMed] [Google Scholar]
- 37. Quinlan AM, Billiar KL. Investigating the role of substrate stiffness in the persistence of valvular interstitial cell activation. J Biomed Mater Res A. 2012;100:2474–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monzack EL, Masters KS. Can valvular interstitial cells become true osteoblasts? A side‐by‐side comparison. J Heart Valve Dis. 2011;20:449–463. [PMC free article] [PubMed] [Google Scholar]
- 39. Yang X, Fullerton DA, Su X, Ao L, Cleveland JC Jr, Meng X. Pro‐osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll‐like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. 2009;53:491–500. [DOI] [PubMed] [Google Scholar]
- 40. Porras AM, Shanmuganayagam D, Meudt JJ, Krueger CG, Hacker TA, Rahko PS, Reed JD, Masters KS. Development of aortic valve disease in familial hypercholesterolemic swine: implications for elucidating disease etiology. J Am Heart Assoc. 2015;4:e002254 DOI: 10.1161/JAHA.115.002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monzack EL, Masters KS. A time course investigation of the statin paradox among valvular interstitial cell phenotypes. Am J Physiol Heart Circ Physiol. 2012;303:H903–H909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu B, Elmariah S, Kaplan FS, Cheng G, Mohler ER III. Paradoxical effects of statins on aortic valve myofibroblasts and osteoblasts: implications for end‐stage valvular heart disease. Arterioscler Thromb Vasc Biol. 2005;25:592–597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. VICs are quiescent in freshly explanted porcine aortic valve leaflets. Immunofluorescent staining for αSMA demonstrated the absence of activated VICs in healthy aortic valves: αSMA (green), DAPI (blue). Scale bar represents 200 μm. αSMA indicates α‐smooth muscle actin; DAPI, 4′,6‐diamidino‐2‐phenylindole; VIC, valvular interstitial cell.
Figure S2. The percentage of VICs positive for myofibroblastic markers decreases after culture in FIB or FIB‐Coll. Quantification of (A) α‐smooth muscle actin, (B) calponin, and (C) smooth muscle protein 22‐α expression via flow cytometry. *P<0.05 compared with control VICs at the same time point. ^ P<0.05 compared with day 4 VICs cultured in the same condition. VIC, valvular interstitial cell.
Figure S3. Myofibroblastic marker expression after 14 days of culture in control, FIB, or FIB with culture on collagen coatings. Myofibroblastic markers are stained red; nuclei are stained blue with DAPI. Scale bar represents 100 μm. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; FIB, fibroblast media formulation.
Figure S4. Valvular interstitial cells generated via FIB or FIB‐Coll remained quiescent for 5 days following removal of FIB or FIB‐Coll conditions. Quantification of myofibroblastic (α‐smooth muscle actin and smooth muscle protein 22‐α) and extracellular matrix (Col1A1 and FN) gene expression. *P<0.05 compared with control, ^ P<0.05 for indicated comparison. FIB, fibroblast media formulation; FIB‐Coll, fibroblast media formulation with culture on collagen coatings.
Figure S5. VICs cultured in fibroblast media formulation with culture on collagen coatings exhibited decreased inflammatory cytokine production and osteogenic activity relative to control. The inflammatory activity of VICs was assessed using ELISAs for (A) IL‐6 and (B) IL‐8. The expression of (C) alkaline phosphatase (ALP) and (D) osteocalcin (OCN) genes by VICs in different culture environments was evaluated as a measure of osteogenic activity. The “standard” is a positive control generated by culture of VICs on uncoated tissue culture polystyrene and treatment with osteogenic medium (DMEM with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mmol/L l‐glutamine, 10 mmol/L β‐glycerophosphate, and 50 μg/mL ascorbic acid). IL indicates interleukin; VIC, valvular interstitial cell.
Figure S6. Culture on collagen alone was insufficient to yield a quiescent phenotype. A, VIC proliferation on collagen was significantly increased relative to the control and conditions containing FIB media, although (B) VICs on collagen exhibited decreased expression of contractile proteins relative to the control. C, An initial increase in vinculin expression was noted in FIB with culture on collagen coatings at Day 4, but then significantly decreased at later time points. FIB, fibroblast media formulation; VIC, valvular interstitial cell.