

Abstract
Background
High levels of ketone bodies are associated with improved survival as observed with regular exercise, caloric restriction, and—most recently—treatment with sodium–glucose linked transporter 2 inhibitor antidiabetic drugs. In heart failure, indices of ketone body metabolism are upregulated, which may improve energy efficiency and increase blood flow in skeletal muscle and the kidneys. Nevertheless, it is uncertain how ketone bodies affect myocardial glucose uptake and blood flow in humans. Our study was therefore designed to test whether ketone body administration in humans reduces myocardial glucose uptake (MGU) and increases myocardial blood flow.
Methods and Results
Eight healthy subjects, median aged 60 were randomly studied twice: (1) During 390 minutes infusion of Na‐3‐hydroxybutyrate (KETONE) or (2) during 390 minutes infusion of saline (SALINE), together with a concomitant low‐dose hyperinsulinemic–euglycemic clamp to inhibit endogenous ketogenesis. Myocardial blood flow was measured by 15O‐H2O positron emission tomography/computed tomography, myocardial fatty acid metabolism by 11C‐palmitate positron emission tomography/computed tomography and MGU by 18F‐fluorodeoxyglucose positron emission tomography/computed tomography. Similar euglycemia, hyperinsulinemia, and suppressed free fatty acids levels were recorded on both study days; Na‐3‐hydroxybutyrate infusion increased circulating Na‐3‐hydroxybutyrate levels from zero to 3.8±0.5 mmol/L. MGU was halved by hyperketonemia (MGU [nmol/g per minute]: 304±97 [SALINE] versus 156±62 [KETONE], P<0.01), whereas no effects were observed on palmitate uptake oxidation or esterification. Hyperketonemia increased heart rate by ≈25% and myocardial blood flow by 75%.
Conclusions
Ketone bodies displace MGU and increase myocardial blood flow in healthy humans; these novel observations suggest that ketone bodies are important cardiac fuels and vasodilators, which may have therapeutic potentials.
Keywords: 11C‐palmitate, 18F‐fluorodeoxyglucose, cardiac metabolism, hyperketonemia, myocardial glucose uptake, myocardial perfusion, nuclear medicine, positron emission tomography/computed tomography
Subject Categories: Basic Science Research, Myocardial Biology, Nuclear Cardiology and PET
Introduction
Sparked by the recent report of a 40% reduction in cardiovascular mortality in diabetic patients treated with sodium–glucose linked transporter 2 (SGLT‐2) inhibitors and the findings of upregulated ketone metabolism in heart failure, much scientific interest has lately focused on the potential role of ketone bodies, such as 3‐hydroxybutyrate (3‐OHB), as life‐prolonging cardioprotective fuel substrates.1, 2
During metabolic stress conditions, such as exercise, fasting, and severe acute illness, hepatic ketone body formation (ketogenesis) is stimulated by high levels of glucagon and free fatty acids (FFAs) and low levels of insulin in the portal bed; under such conditions, concentrations of the major ketone body, 3‐OHB, may rise from close to zero to above 5 mmol/L and ketone bodies become an important fuel source. Since most tissues extract ketone bodies in proportion to their delivery,3 hyperketonemia results in a glycolysis‐independent pool of acetyl‐CoA and ultimately energy in the form of ATP. Oxidation of ketone bodies yields more ATP per mole of oxygen consumed than does glucose,4 and ketone body oxidation does not result in uncoupling in the mitochondrial membrane, which is a byproduct of FFA oxidation.5, 6 Ketone bodies may therefore spare hypoxic tissue. As potential results, ketone bodies are neuroprotective,7 improve cognitive performance in patients with cognitive impairment,8 and may ameliorate the effects of acute brain injury and ischemic damage.9
Although less well explored, accumulating evidence indicates that ketone bodies also have beneficial extracerebral effects, notably in the heart. The healthy heart is omnivorous and flexes readily between a mixture of glucose and the less energy‐efficient and reactive oxygen species producing FFAs (≈70%).10 In this setting, it is of interest that ketone bodies have been demonstrated to (1) inhibit myocardial FFA oxidation in pigs,11 (2) inhibit oxidative stress in cell cultures,12 and (3) shift the failing heart in murine models and in humans to ketone body utilization and downregulate enzymes related to fatty acid oxidation.13, 14 Collectively, these observations suggest that increased myocardial oxidation of ketone bodies may be a beneficial adaptive process. Intriguingly, new data from the Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes (EMPA‐REG) trial15 have led several investigators to propose that the 2‐ to 3‐fold increase in ketone body levels observed in type 2 diabetic patients treated with SGLT‐2 inhibitors may explain the surprising and rapid 40% decrease in cardiovascular mortality.2, 16 In this context, it is of interest that insulin resistance and type 2 diabetes are characterized by a 2‐ to 3‐fold increase in cardiovascular mortality together with endothelial dysfunction and coronary microvascular dysfunction with impaired vasodilatory reserve.17, 18 These defects are associated with increased oxidative stress19 and it has been shown that 3‐OHB suppresses oxidative stress in mice12 and increases local cerebral and renal blood flow in humans.20, 21
A negative correlation between uptake of glucose and ketone in the heart has been reported,22 but there is still a lack of studies examining how isolated manipulation of ketone body levels affect myocardial substrate utilization and blood flow in humans. This is most likely because of the relative inaccessibility of the heart and the invasive nature of cardiac catheterization and tissue biopsy techniques. Moreover, the use of labeled ketone body tracers to directly measure rates of disappearance is inherently problematic because of unpredictable loss of label by rapid isotopic exchange in the acetyl‐acetoacetyl CoA pool.23 Given these limitations, we examined alterations of myocardial uptake of glucose and FFA to explore the effects of myocardial ketone body exposure. Ketone bodies suppress lipolysis and ketogenesis, and we used a low‐dose hyperinsulinemic clamp to control these metabolic pathways.
Our study was designed as a randomized cross‐over trial in which either ketone bodies or a saline solution was infused under controlled metabolic conditions to test whether hyperketonemia would result in significantly suppressed myocardial glucose uptake and increased myocardial blood flow.
Research Design and Methods
Study Participants
Eight healthy subjects (3 women) median age 60 (range 50–68) and median body mass index 25.5 (range 21.5–34.6) were recruited and completed the study. All subjects were screened for inclusion in the weeks preceding the studies. Screening included demographic data, clinical history, medical examination, heart rate, and blood pressure. Two additional subjects were originally included (1 woman, 1 man) but withdrew because of claustrophobia during the scans.
The study protocol was performed in accordance with the Declaration of Helsinki and was approved by the local institutional review board (Central Denmark Committee on Health Research [no. 1‐10‐72‐104‐14]). All subjects signed a written informed consent before entering the study.
Study Protocol
The study was a single‐blinded, randomized cross‐over study where subjects were studied under identical euglycemic clamp conditions and with identical positron emission tomography (PET) scan procedures. Study days differed in intervention, which was assigned in a random order. Study day A was a placebo study day where saline (0.9%) was infused (SALINE), whereas study day B included an infusion of 7.5% w/v Na‐3‐β‐hydroxy‐butyrate (3‐OHB) (KETONE) (Figure 1).
Figure 1.
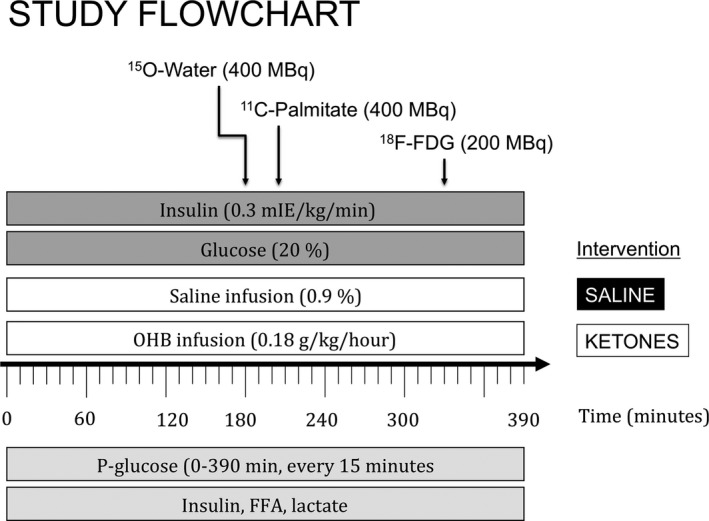
Study protocol. Study days (SALINE and KETONES) were identical except for the intervention, which was either infusion of Na‐betahydroxybutyrate (OHB) or NaCl (0.9%). PET/CT scans were initiated at t=180 minutes at which point quasi‐stable concentrations of metabolites were expected. CT indicates computed tomography; FDG, fluorodeoxyglucose; FFA, free fatty acids.
All subjects fasted overnight for 10 hours before the study days. At t=−30 minutes, 2 catheters were inserted: the first in an antecubital vein used for infusions and the second in a dorsal heated hand vein used for arterialized blood sampling. At t=0 minute, a continuous infusion of insulin (0.3 mIE/kg per minute [Humulin, Eli Lily, US]) and glucose (200 g/L [Fresenius Kabi]) was initiated with the glucose infusion rate continually adjusted to maintain euglycemia (5 mmol/L). On study day A, a continuous infusion of saline (0.9%) was initiated at t=0 minutes, whereas a continuous infusion of OHB (0.18 g/kg per hour) was initiated at t=0 on study day B. All infusions were maintained throughout the study days and were discontinued at t=390 minutes when all PET scans had been completed. PET scans were performed at t=180 minutes (15O‐H2O), t=210 minutes (11C‐palmitate), and at t=330 minutes (18F‐fluorodeoxyglucose [FDG]). Blood samples were drawn at t=0, 60, 120, 180, 240, 300, 360, and 390 minutes for analysis of FFAs and insulin and every 15 minutes for analysis of p‐glucose levels. Lactate levels were measured at t=120, 240, and 390 minutes. Blood pressure and heart rate were measured every hour through the study day.
Blood Samples
All blood samples excluding glucose were stored at −20°C and analyzed in the same assay after all subjects had completed both study days. FFAs were quantified using the in vitro enzymatic colorimetric method assay NEFA‐HR(2) (Wako Chemicals GmbH, Germany), glucose and lactate concentrations were measured using a YSI 2300 model STAT plus (YSI Incorporated, Netherlands), and insulin was measured by ELISA. Serum concentrations of 3‐OHB were measured using hydrophilic interaction liquid chromatography tandem mass spectrometry (HILIC‐MS/MS).24
PET Scan Protocols
PET/CT scans were conducted using a Siemens Biograph 64 PET/CT (Siemens, Erlangen, Germany). Injections of radiotracers were done in the antecubital catheter used for infusions. For arterial activity samplings (11C‐palmitate), a catheter was inserted in the contralateral radial artery at t=120 minutes. Patients were placed with the heart in the field of view and a low‐dose CT scan was obtained for attenuation and anatomic localization purposes. At t=180 minutes, 400 MBq 15O‐H2O was administered and simultaneously, a 6‐minute list mode PET scan was begun with the following time‐frame sequence: 1×10, 8×5, 4×20, 2×15, 3×20, 2×30, and 2×60 s. Images were reconstructed using a 3‐dimensional (D) iterative algorithm (3 iterations, 21 subsets, 5‐mm Gaussian postfilter), applying all appropriate corrections for normalization, dead time, random coincidences, scatter, and attenuation.
The 11C‐palmitate scan was initiated at t=210 minutes allowing for sufficient decay of residual 15O‐H2O activity. Thus, 11C‐palmitate was injected as a bolus (283±65 MBq [SALINE] or 274±62 MBq [KETONE]) and a 50‐minute list mode scan (frame structure 6×5, 6×10, 3×20, 5×30, 5×60, 8×150, 4×300 s) was performed with 37 manual blood samples taken during the scan. Plasma as well as whole‐blood activity were measured in a well counter (Cobra II; Packard Instruments Co.) cross‐calibrated to the tomograph. Data were reconstructed using a 3D iterative algorithm (3 iterations, 21 subsets, 5‐mm Gaussian postfilter). Blood and dynamic PET data were decay corrected to scan start. After completion of the 11C‐palmitate scan, subjects were moved to a bed in the adjoining room to await the final scan.
At t=315 minutes, a second low‐dose CT scan was acquired using the same protocol as previously described. At t=330 minutes, ≈200 MBq 18F‐FDG was administered and a 50‐minute list mode scan (frame structure 1×10, 8×5, 4×10, 3×20, 5×30, 5×60, 4×150, 4×300, and 1×600 s) was performed using 3D iterative reconstruction (3 subsets, 21 iterations, 4‐mm Gaussian postfilter). Voxel size for all 3 scans was 4×4×4 mm.
Tracer Kinetics
Perfusion: Myocardial blood flow (MBF), expressed in mL/g per minute, was calculated using Cardiac VUer.25 In brief, the arterial and venous blood pool were segmented automatically and used to extract the arterial input function, which was then used to generate parametric images of MBF. Then, these parametric images were used for reorientation to short‐axis images on which the left ventricle was then defined automatically and segmented according to the recommendations of the American Heart Association. MBF is reported for the left ventricle as a whole, but also for the 3 vascular territories (left anterior descending, left circumflex, and right coronary).
Myocardial fatty acid metabolism
Using in‐house developed software, myocardial fatty acid metabolism was analyzed using a 3‐tissue compartment model in which 3 rate constants need to be fitted. In this model, the efflux 11CO2 rate is fixed to the oxidation rate and a slow esterification compartment is included. Macroparameters myocardial fatty acid oxidation (MFAO), myocardial fatty acid esterification (MFAE), and myocardial fatty acid uptake (MFAU) were defined according to the suggestions by Bergmann et al26:
Segmentation of the blood pool, reorientation of the images, and definition of the left ventricle was performed in a way similar to the analysis of the perfusion scans. For the all calculations and parametric images, the input function was corrected for 11C‐metabolites using arterial blood samples as validated by Guiducci et al.27
Myocardial glucose uptake
Myocardial glucose uptake (MGU) rate from the dynamic 18F‐FDG scan was estimated by Patlak analysis28 using Cardiac VUer. Irreversible uptake rate, Ki, was first estimated from the relation
where Ci is the myocardial 18F‐FDG radioactivity and Cp is the plasma 18F‐FDG radioactivity at time t. Plots of Ci(t)/Cp(t) versus ∫Cp(t)dt/Cp(t) were fitted to straight lines by conventional least‐squares methods and the slopes of the best fits were taken as estimates of Ki. Absolute myocardial glucose uptake rate (nmol/g per minute) was calculated using the equation
where pGlc is plasma glucose concentration and LC is the lumped constant. The lumped constant was assumed not to differ between visits (as demonstrated by previous work performed during similar study conditions)29 and was therefore fixed at 1.
Statistics
Normal distribution was assessed by inspection of QQ‐plots and data were ln‐transformed when appropriate. Curves are presented as means±SE, whereas all other data involving group comparisons are presented as means±SD. Statistical comparisons between study days were performed by a paired t test or a mixed‐model repeated‐measures ANOVA when appropriate (time series). The mixed model was a 2‐way ANOVA with time and treatment as factor variables and the time versus treatment interaction as the term of interest. The level of significance was set at P<0.05. All statistical analyses were performed using SPSS version 21.
Results
Hemodynamics, MBF, and pH
Blood pressure and heart rate during the study days are presented in Figure 2. On the SALINE study day, blood pressure and heart rate remained stable, whereas heart rate increased ≈25% from 62±13 to 78±8 mm Hg during the KETONE study day (repeated measurements treatment×time, P<0.001).We observed a uniform 75% increase in resting MBF in all vascular territories (at t=180 minutes) as shown in Table 1. Arterial pH was measured in a subset of patients (n=4) and increased from 7.42 (range 7.41–7.43) to 7.50 (range 7.46–7.52), P<0.01.
Figure 2.
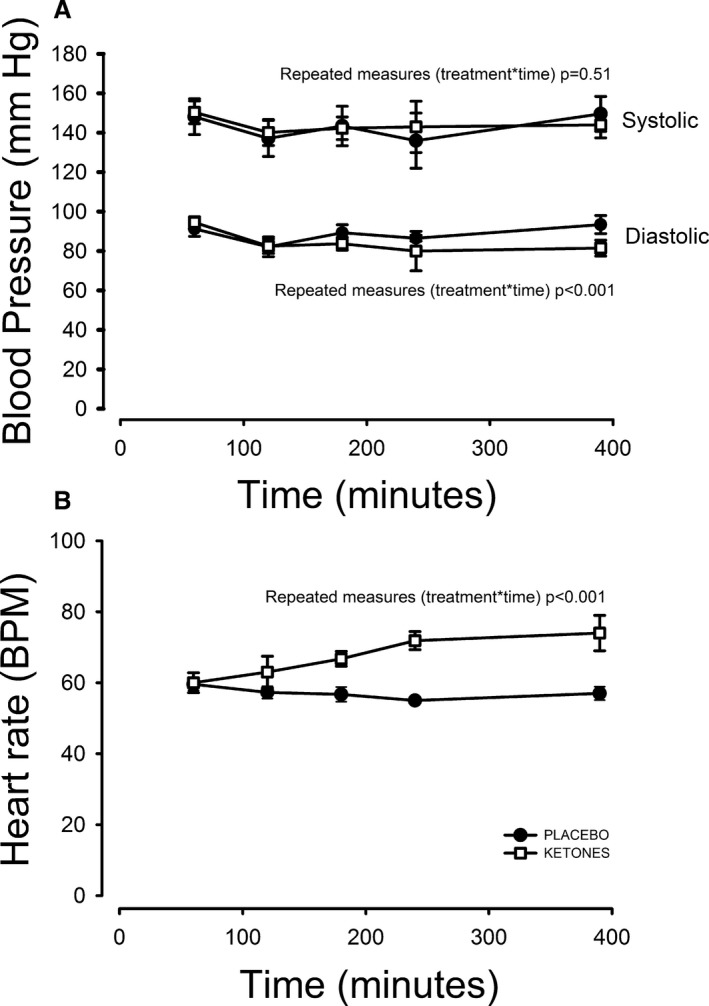
Hemodynamics. Systolic (treatment×time, P=0.54) blood pressure was comparable on the 2 study days (A), whereas heart diastolic blood pressure (treatment×time, P<0.001) decreased and heart rate (treatment×time, P<0.001) increased significantly during the KETONES study day (B). BPM indicates beats per minute.
Table 1.
Mean MBF (SD)
| Vascular Territory | MBF (mL/g per minute) | P Value | |
|---|---|---|---|
| SALINE | KETONE | ||
| LAD | 0.81 (0.20) | 1.47 (0.29) | <0.0001 |
| Cx | 0.87 (0.21) | 1.61 (0.34) | <0.0001 |
| RCA | 0.79 (0.16) | 1.36 (0.29) | <0.0001 |
| Global | 0.83 (0.19) | 1.46 (0.27) | <0.0001 |
Cx indicates left circumflex; LAD, left anterior descending; MBF, myocardial blood flow; RCA, right coronary artery.
Hormones and Metabolites
As seen in Figure 3A, circulating OHB was below the detection threshold during the SALINE study day. 3‐OHB increased rapidly during the KETONE study day, reaching mean values ≈3 mmol/L at t=180 minutes with a further increase to a ≈4 mmol/L plateau phase at t=300 minutes. FFA levels decreased rapidly during both study days, reaching an almost complete suppression at t=120 minutes. A tendency towards lower levels of FFAs was observed during the KETONE day even though this did not reach statistical significance (Figure 3B). Average circulating lactate levels were increased ≈35% by the ketone infusion (Table 2).
Figure 3.
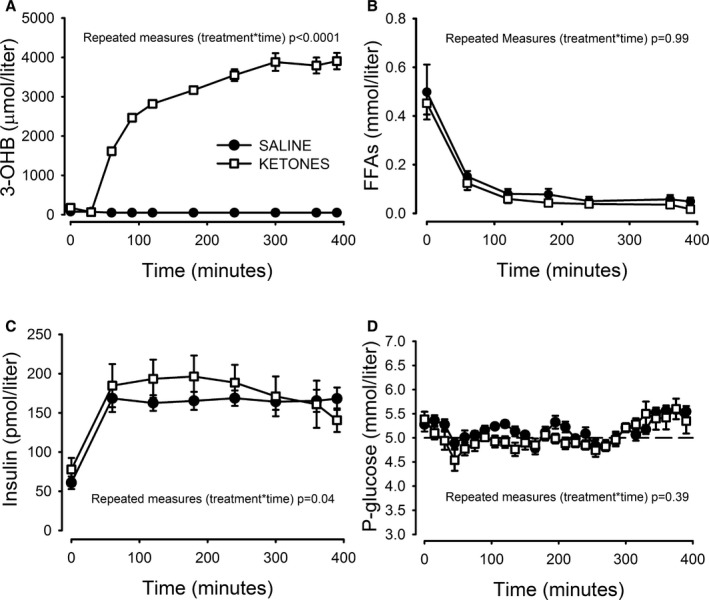
Hormones and metabolites. A, Circulating 3‐OHB concentrations were below detection threshold (50 μmol/L) during the SALINE study day and increased to between 3 and 4 mmol/L between t=120 and 390 during the KETONE study day. By design, FFA concentrations (B) were suppressed to ≈50 μmol/L during both study days by the HE‐clamp, which increased insulin levels (C) to between 160 and 180 pmol/L. Glucose levels (D) were clamped ≈5 mmol/L through the study days, although a tendency to increasing glucose concentrations was observed from t=300 minutes and onwards. P‐values refer to the mixed model treatment vs time interaction. N=8, error bars are ±SE. FFA indicates free fatty acids; HE, hyperinsulinemic–euglycemic; 3‐OHB, 3‐hydroxybutyrate.
Table 2.
Average Hormone and Metabolite Concentrations (t=180–390 Minutes)
| Hormones and Metabolites | P Value | ||
|---|---|---|---|
| SALINE | KETONE | ||
| 3‐OHB, mmol/L | 0.05 (0.00) | 3.78 (0.47) | <0.0001 |
| FFA, mmol/L | 0.06 (0.05) | 0.04 (0.03) | 0.14 |
| Insulin, pmol/L | 176 (45) | 178 (63) | 0.89 |
| Lactate, pmol/L | 1.05 (0.14) | 1.44 (0.35) | 0.02 |
| Glucose, mmol/L | 5.2 (0.1) | 5.1 (0.2) | 0.07 |
N=8, mean values (SD). FFA indicates free fatty acids; OHB, hydroxybutyrate.
The hyperinsulinemic–euglycemic clamp increased insulin levels by a factor of 3 with no difference in mean levels from t=180 to 390 (Table 2). A dynamic difference was evident on the KETONE study day, as insulin levels increased more in the early stages while a late‐onset decrease was observed from t=240 minutes onwards (Figure 3C). Analyzed by repeated measurements, the treatment versus time interaction was significant. Finally, p‐glucose levels were clamped ≈5 mmol/L during both study days, with a tendency to increase during the last hour of the investigations, coinciding with the 18F‐FDG PET/CT scan during which tight control of p‐glucose is difficult (Figure 3D).
Insulin Sensitivity
Insulin sensitivity as assessed by a low‐dose HE‐clamp was not significantly different between study days, although a trend towards greater glucose infusion rates was observed during the ketone body infusion (Figure 4). No correlation was observed between peripheral glucose uptake and MGU (data not shown).
Figure 4.
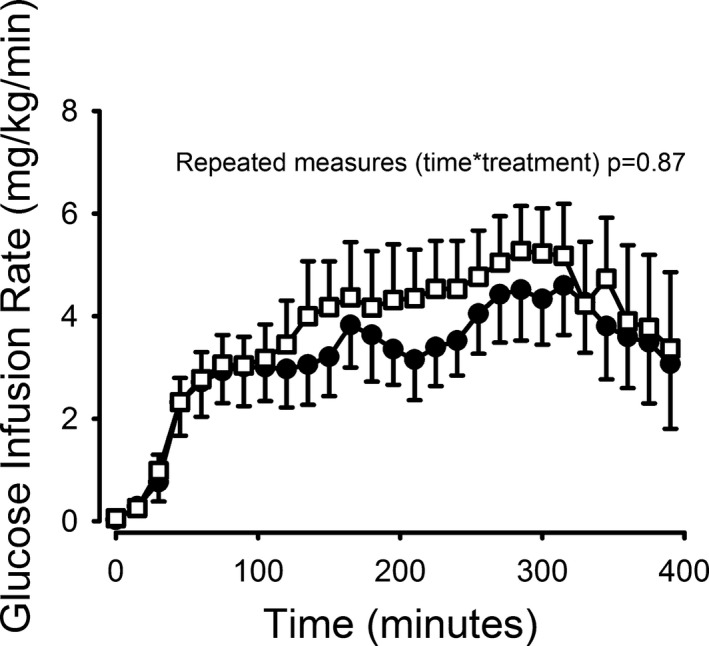
Glucose infusion rates reached plateau levels at t=180 minutes and started decreasing from t=300 onwards. No difference in insulin sensitivity was observed. N=8, error bars are ±SE for the GIR curves. Open boxes = KETONES, closed circles = SALINE. GIR indicates glucose infusion rates.
Myocardial 18F‐FDG Uptake
Experimental hyperketonemia roughly halved myocardial glucose uptake (304±97–155±61 nmol/g per minute [P=0.03]) exclusively because of a reduction in relative uptake rate (Ki) (Figure 5). No correlation was observed between plateau 3‐OHB levels obtained and the reduction in MGU.
Figure 5.
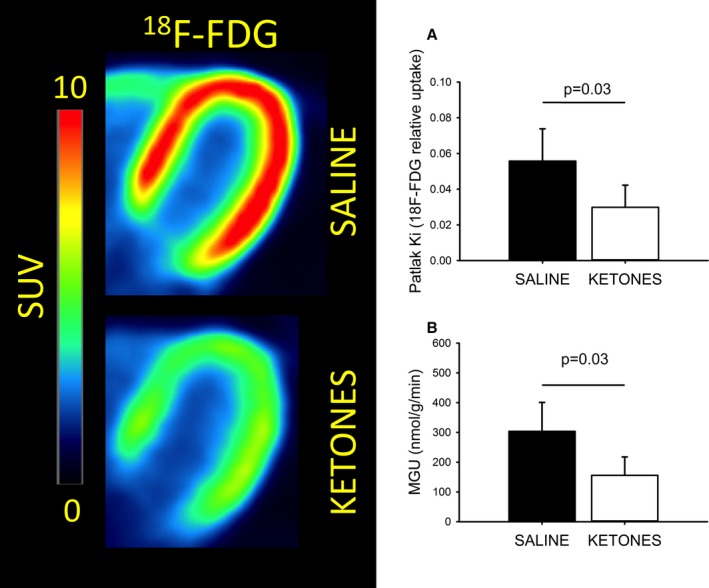
Myocardial 18F‐FDG uptake. Summed images (t=10–50 minutes) from a representative subject are presented in the left panel. Infusion of OHB significantly decreased 18F‐FDG uptake in all subjects, resulting in significantly lower relative uptake rates (Ki) when compared with the SALINE study day (A). Since blood glucose levels were clamped at ≈5 mmol/L, the resulting absolute MGU was roughly halved (B). N=8, error bars are SD. FDG indicates fluorodeoxyglucose; MGU, myocardial glucose uptake; OHB, hydroxybutyrate; SUV, standardized uptake value.
Myocardial 11C‐Palmitate Metabolism
Oxidation and uptake rates were unaltered by the ketone body infusion, whereas a small but significant increase was seen in the re‐esterification transfer rate (Table 3).
Table 3.
Mean FMAO, MFAE, MFAU (SD) (SD)
| Transfer Rate | 11C‐Palmitate Relative Transfer Rates | P Value | |
|---|---|---|---|
| SALINE | KETONE | ||
| MFAO | 0.10 (0.02) | 0.10 (0.05) | 0.84 |
| MFAE | 0.06 (0.02) | 0.08 (0.03) | <0.01 |
| MFAU | 0.16 (0.03) | 0.18 (0.06) | 0.22 |
MFAE indicates myocardial fatty acid esterification; MFAO, myocardial fatty acid oxidation; MFAU, myocardial fatty acid uptake.
When macroparameters were calculated by multiplying relative transfer rates by plasma FFA concentrations, we found absolute uptake, oxidation, and re‐esterification identical regardless of intervention (Figure 6).
Figure 6.
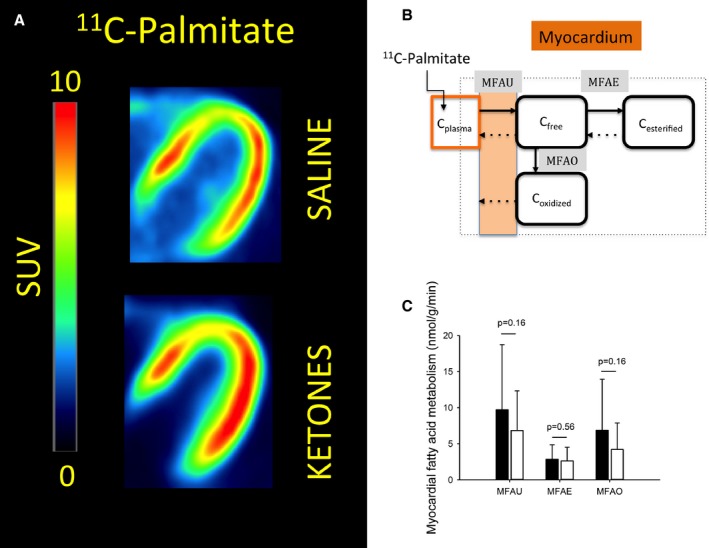
Myocardial fatty acid metabolism. A, Summed images (t=10–50 minutes) from a representative subject are presented in the left panel. There was no visible difference in accumulated 11C‐palmitate in most subjects. B, Myocardial 11C‐palmitate metabolism. The 3‐tissue compartment model has 3 free parameters (black arrows) and 3 fixed parameters (dotted arrows). C, No differences were observed in myocardial fatty acid uptake, esterification, or oxidation (black bar: Saline, white bar: Ketone). N=7, error bars are SD. MFAE indicates myocardial fatty acid esterification; MFAO, myocardial fatty acid oxidation; MFAU, myocardial fatty acid uptake; SUV, standardized uptake value.
Discussion
In line with our preliminary hypothesis, we observed a halving of myocardial glucose uptake during experimental hyperketonemia, supporting the notion that the human heart utilizes and oxidizes ketone bodies at the expense of glucose in spite of ongoing insulin stimulation. Hyperketonemia did not affect myocardial fatty acid transport or oxidative capacity in a metabolic setting of very low circulating FFAs, but increased heart rate 25% and myocardial blood flow 75%.
Myocardial Glucose Uptake Is Halved by Hyperketonemia
Several ex vivo studies have established that cardiomyocytes oxidize ketone bodies at the expense of glucose,30, 31 presumably by inhibition of pyruvate dehydrogenase32 or possibly by sequestration of coenzyme A impairing oxidative glucose flux at the entry to the tricarboxylic acid cycle.33 The ex vivo and animal studies of cardiomyocytes have not previously been extended to a human setting. However, there is substantial indirect evidence from human studies to support the notion. Ferrannini et al investigated cardiac substrate metabolism across the heart in 16‐hour‐fasted humans34 and reported that both fatty acids and ketone bodies constituted a significant proportion of cardiac fuel during basal conditions. The contribution from lipid fuels was almost completely abolished during hyperinsulinemia. Although the study indicates that the heart utilizes and oxidizes ketone bodies after fasting, it did not establish to which extent utilization of 3‐OHB, glucose, and FFA was affected by insulin, high levels of FFA, or intrinsic to the moderately high levels of 3‐OHB per se. By contrast, our data clearly show that high levels of 3‐OHB displace glucose utilization by the myocardium in the presence of well‐controlled hyperinsulinemia and suppressed FFA levels. It is of interest that the marked reduction in myocardial glucose uptake occurred in the presence of a 75% increase in myocardial blood flow. If anything, the increased myocardial blood flow would be expected to increase myocardial glucose uptake, which correlates with tissue perfusion in striated muscle.35
During a hyperinsulinemic–euglycemic clamp, the heart derives most of its ATP from oxidation of glucose or lactate36 and only a small proportion from lipids. This was also evident in this study, where glucose uptake during the control study day (in μmol/g heart tissue/min) was 30‐fold higher than fatty acid uptake as opposed to a ratio of roughly 1 observed in studies under normal fasting conditions (see eg,37). It is therefore reasonable to assume that a halving of MGU in absolute terms during the ketone body infusion implies that a significant proportion of the heart's energy needs was met by ketone body oxidation, although some of the reduction in MGU observed can also be ascribed to the increased lactate levels. On the other hand, increased lactate levels could also be a consequence of either reduced entrance of glycolytic end products in the tricarboxylic acid cycle because of substrate competition from ketones,22 or limited transfer of reducing equivalents through the malate–aspartate shuttle leading to reduced metabolism of glucose and lactate.38, 39
Myocardial Fatty Acid Kinetics Is Unaltered by Hyperketonemia in a Setting of Depressed Circulating FFAs
In theory, ketone bodies can affect fatty acid metabolism either at the level of entry into the cardiomyocytes or at the mitochondrial level. At the level of entry, active transport of fatty acids via CD36 may account for up to 60% of fatty acid uptake into the cardiomyocytes40, 41 while passive diffusion or transport via other carriers account for the rest. At the mitochondrial level, fatty acyl‐CoA is transported into the mitochondria for oxidation by carnitine palmitoyltransferase I. Carnitine palmitoyltransferase I activity has long been considered to be inhibited by malonyl‐CoA, which increases with intracellular energy substrate levels,10 but this concept of a simple substrate feedback loop has now been challenged.42, 43 In line with this, Stanley et al11 studied myocardial fatty acid oxidation both during infusion of a lipid emulsion and during infusion of lipids plus ketone bodies in pigs and found a significant decrease in fatty acid oxidation without affecting levels of malonyl‐CoA, acetyl‐CoA, or acetyl‐coA decarboxylase. In that study, ketone bodies appeared to inhibit fatty acid oxidation at the level of entry into the cell rather than via inhibition of carnitine palmitoyltransferase I. In the present study, fatty acid transport capacity into the cells was not affected by hyperketonemia as assessed by the similar MFAU rates during the saline and β‐hydroxybutyrate infusions. Ketone bodies therefore do not appear to modulate CD36 transport directly. The previously observed reduced myocardial fatty acid uptake during hyperketonemia is more likely because of reduced passive diffusion of fatty acids over the sarcolemmal membrane. However, the intact cellular membrane fatty acid transport capacity in the setting of hyperketonemia was measured during almost completely suppressed levels of circulating FFAs. For comparison, myocardial fatty acid oxidation during modest hyperketonemia (≈1 mmol/L) has been studied by others using 11C‐palmitate PET44 in human subjects and in dogs. In that study, fatty acid oxidation was significantly decreased by hyperketonemia, but this occurred in the presence of 50% reduced circulating FFAs. Most likely, their findings reflected a decreased supply of substrate rather than a direct inhibitory effect of ketone bodies on the ability of the myocardium to oxidize fatty acids.
In summary, our results from the kinetic analysis of myocardial 11C‐palmitate metabolism showed no effect of hyperketonemia on relative or absolute fatty acid uptake and oxidation rates, while relative esterification rates increased, compatible with the notion that the inhibitory effect of ketone bodies on myocardial fatty acid uptake is a consequence of substrate competition.
Myocardial Perfusion and Heart Rate Is Increased by Hyperketonemia
One of the more striking results of this study was the 75% increase in myocardial blood flow observed during hyperketonemia. There may be several explanations for this.
First, it seems obvious to relate this to the concomitant 25% increase in heart rate. Numerous studies have shown a linear relationship between resting heart rate and MBF on a population level (reviewed by45). However, on the level of the individual, heart rate and myocardial perfusion do not necessarily increase in tandem in the 60 to 80 beats/min range, because any increase in heart rate shortens the duration of the diastole whereby coronary blood flow is inhibited. Only when heart rate is increased to such an extent that metabolic factors become more prominent, does significant vasodilation occur. The relationship between increasing heart rate and myocardial blood flow is therefore sigmoidal.46 The modest average ketone‐induced increase in heart rate of 18 beats/min observed in this study should therefore not translate into a 75% increase in MBF.
Second, it is possible that increased blood pH caused by infusion of a β‐hydroxybutyrate salt might affect myocardial blood flow. Thus, acutely elevated levels of ketone bodies result in a loss of potassium from the kidney tubules as the cation acts as a partner to the negatively charged β‐hydroxybutyrate. Decreasing levels of potassium in the blood spark an exchange of H+ from the blood with intracellular potassium, and the resulting decrease in blood H+ constitutes a metabolic alkalosis. In line with this, 4 patients in whom we measured blood pH during the ketone body infusion had pH values ≈7.5 and potassium just above 3 mmol/L. Since a discrete 0.08 increase in blood pH has been shown to increase myocardial blood flow up to 50% in mongrel dogs,47 it is conceivable that the mild metabolic alkalosis our subjects suffered during the ketone body infusion may contribute to the increased MBF.
Third, hyperketonemia seems to increase perfusion or blood flow in other tissues independently of changes in pH or heart rate. Forearm blood increases 15% in human volunteers during infusion of β‐hydroxybutyrate48 in amounts sufficient to obtain plateau values of as little as 0.5 mmol/L, a fraction of what we observed in this study. More interesting from a physiological point of view are the numerous studies that report a stimulatory effect of ketone bodies on cerebral blood flow. Hasselbalch et al observed a 39% increase in cerebral blood flow in human volunteers during short‐term infusion of β‐hydroxybutyrate,20 a finding that could not be ascribed to either blood glucose levels or pH, perhaps reflecting that ketone bodies act directly on the endothelium to produce vasodilation. Another study reported that 3‐OHB infusion increased renal plasma flow measured with hippurate by 20% in humans,21 thus supporting the concept of general vasodilatory effects of ketone bodies.
As outlined above, several factors may combine to explain how infusion of ketone bodies results in a significantly increased MBF, whereas the mechanisms leading to increased heart rate are more uncertain. If anything, β‐hydroxybutyrate decreases sympathetic outflow in HEK293 cells by antagonizing the G‐Protein Coupled Receptor 41,49 which in theory should translate into a decreased heart rate. Also, the modest sodium loading associated with infusion of the ketone body salt should contribute to a lowering of heart rate rather than an increment.50 On the other hand, in studies in athletes fed a ketogenic diet, resting heart rate has been demonstrated to be slightly increased51 despite comparable levels of norepinephrine. Unfortunately, measurements of heart rate have not been reported in previous studies using exogenous ketone body infusions to study the effects of hyperketonemia on diverse segments of physiology and pathophysiology.20, 52, 53
It should be noted that many patients with the metabolic syndrome and type 2 diabetes have endothelial dysfunction and that increased myocardial perfusion in these patients may have beneficial effects.54 It thus remains a possibility that the sharp reduction in cardiovascular mortality following treatment with SGLT‐2 inhibitors in clinical trials15 could be caused by a combination of increased myocardial blood flow and increased myocardial oxygen efficiency driven by high levels of ketone bodies.
Experimental Design
In vivo studies of substrate metabolism are inherently challenging. For instance, ketone bodies bind to nicotinic acid receptors55 and inhibit peripheral lipolysis and reduce circulating FFAs. Reduced levels of FFAs simultaneously increase insulin sensitivity, lower insulin levels, and reduce blood glucose56 and endogenous ketogenesis. As a result, any intervention involving ketone bodies is bound to affect the most prominent circulating energy substrates as well as their regulatory hormones.
This study was designed in an attempt to control these particular challenges: The modest (0.3 mIE/kg per minute) hyperinsulinemic–euglycemic clamp ensured comparable insulin and glucose levels on both study days while still allowing for myocardial oxidation of other fuel sources. In addition, the HE‐clamp also suppressed adipose tissue lipolysis and thus circulating fatty acids. Our subjects only differed significantly in terms of ketone body levels. Metabolic conditions with ample supplies of glucose, high insulin, and suppressed circulating fatty acid levels most closely mimic the fed state. It can be argued that ketone body levels only rarely are increased during feeding, but as previously mentioned, this may be the case in patients with heart failure57 or in diabetic patients treated with SGLT‐2 inhibitors.15 Furthermore, a very recent study has shown that co‐administration of glucose and 3‐hydroxybutyrate esters, creating a similar metabolically favorable scenario with high insulin levels and suppressed FFAs, improves exercise performance in humans.58
Some major drawbacks to the study design should be mentioned. By design, FFA levels were clamped at comparable low levels on both study days, which does not reflect the classical hyperketonemic setting of increased circulating FFAs as seen during fasting, metabolic stress, or exercise. It is possible that this may have influenced our estimates of 11C‐palmitate kinetics. Also, blood ketone body levels were experimentally rapidly elevated and sustained for only 6.5 hours, which precludes us from studying the metabolic effects of putative negative feedback mechanisms on key ketolytic enzymes in the heart.59 The proof‐of‐concept nature of the study design called for a significant increase in circulating ketone bodies in order to obtain robust primary end point results (PET‐based MGU). The ketone body concentrations obtained in our volunteers are therefore much greater than what could be expected from treatment with SGLT‐2 inhibitors or even several days of fasting. Finally, our study design did not include direct measurements of cardiac ketone body uptake, and we can therefore only conclude by inference that the reduced myocardial glucose uptake was replaced by ketone bodies.
Conclusion
We used dynamic 11C‐palmitate, 15O‐H2O, and 18F‐FDG PET/CT to demonstrate for the first time in humans that hyperketonemia in the setting of concomitant hyperinsulinemia significantly reduces myocardial glucose uptake and increases myocardial blood flow. The striking ability of 3‐OHB to replace myocardial glucose utilization and to increase myocardial blood flow warrants further investigations of the potential of ketone bodies to increase myocardial oxygen efficiency (eg, during exercise and in patients with cardiac disease), in line with the marked reduction of cardiovascular mortality in patients rendered hyperketonemic by treatment with SGLT‐2 inhibitors.
Sources of Funding
Møller has an unrestricted grant from The Danish Council for Strategic Research—DSF 0603‐00479B.
Disclosures
Dr Wiggers has been a principal or subinvestigator in studies involving the following pharmaceutical companies: MSD, Bayer, Daiichi‐Sankyo, Novartis, Novo Nordisk, Sanofi‐Aventis and Pfizer, outside of the submitted work; Dr Jessen reported grants from The Danish Council for Independent Research and The Novo Nordisk Foundation, outside of the submitted work; Dr Møller reports grants from the Danish Council for Strategic Research (grant no. 0603‐00479B) during the conduct of this study. All other authors have nothing to disclose.
(J Am Heart Assoc. 2017;6:e005066. DOI: 10.1161/JAHA.116.005066.)
References
- 1. Taegtmeyer H. Failing heart and starving brain: ketone bodies to the rescue. Circulation. 2016;134:265–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA‐REG Outcome Trial: a “thrifty substrate” hypothesis. Diabetes Care. 2016;39:1108–1114. [DOI] [PubMed] [Google Scholar]
- 3. Cahill GF Jr. Fuel metabolism in starvation. Annu Rev Nutr. 2006;26:1–22. [DOI] [PubMed] [Google Scholar]
- 4. Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, Veech RL, Passonneau JV. Control of glucose utilization in working perfused rat heart. J Biol Chem. 1994;269:25502–25514. [PubMed] [Google Scholar]
- 5. Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. 2004;70:309–319. [DOI] [PubMed] [Google Scholar]
- 6. Boehm EA, Jones BE, Radda GK, Veech RL, Clarke K. Increased uncoupling proteins and decreased efficiency in palmitate‐perfused hyperthyroid rat heart. Am J Physiol Heart Circ Physiol. 2001;280:H977–H983. [DOI] [PubMed] [Google Scholar]
- 7. Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009;59:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reger MA, Henderson ST, Hale C, Cholerton B, Baker LD, Watson GS, Hyde K, Chapman D, Craft S. Effects of beta‐hydroxybutyrate on cognition in memory‐impaired adults. Neurobiol Aging. 2004;25:311–314. [DOI] [PubMed] [Google Scholar]
- 9. White H, Venkatesh B. Clinical review: ketones and brain injury. Crit Care. 2011;15:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. [DOI] [PubMed] [Google Scholar]
- 11. Stanley WC, Meadows SR, Kivilo KM, Roth BA, Lopaschuk GD. Beta‐hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl‐CoA content. Am J Physiol Heart Circ Physiol. 2003;285:H1626–H1631. [DOI] [PubMed] [Google Scholar]
- 12. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E. Suppression of oxidative stress by beta‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, Rame JE. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE; Investigators E‐RO . Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–2128. [DOI] [PubMed] [Google Scholar]
- 16. Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA‐REG OUTCOME study? A unifying hypothesis. Diabetes Care. 2016;39:1115–1122. [DOI] [PubMed] [Google Scholar]
- 17. Mather KJ, Steinberg HO, Baron AD. Insulin resistance in the vasculature. J Clin Invest. 2013;123:1003–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crea F, Camici PG, Bairey Merz CN. Coronary microvascular dysfunction: an update. Eur Heart J. 2014;35:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res. 2016;118:1808–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hasselbalch SG, Madsen PL, Hageman LP, Olsen KS, Justesen N, Holm S, Paulson OB. Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. Am J Physiol. 1996;270:E746–E751. [DOI] [PubMed] [Google Scholar]
- 21. Fioretto P, Trevisan R, Velussi M, Cernigoi A, De Riva C, Bressan M, Doria A, Pauletto N, Angeli P, De Dona C, Nosadini R. Glomerular filtration rate is increased in man by the infusion of both D, L‐3‐hydroxybutyric acid and sodium D, L‐3‐hydroxybutyrate. J Clin Endocrinol Metab. 1987;65:331–338. [DOI] [PubMed] [Google Scholar]
- 22. Bing RJ, Siegel A, Ungar I, Gilbert M. Metabolism of the human heart. II. Studies on fat, ketone and amino acid metabolism. Am J Med. 1954;16:504–515. [DOI] [PubMed] [Google Scholar]
- 23. Landau BR, Wahren J. Nonproductive exchanges: the use of isotopes gone astray. Metabolism. 1992;41:457–459. [DOI] [PubMed] [Google Scholar]
- 24. Sorensen LK, Rittig NF, Holmquist EF, Jorgensen KA, Jorgensen JO, Moller N, Johannsen M. Simultaneous determination of beta‐hydroxybutyrate and beta‐hydroxy‐beta‐methylbutyrate in human whole blood using hydrophilic interaction liquid chromatography electrospray tandem mass spectrometry. Clin Biochem. 2013;46:1877–1883. [DOI] [PubMed] [Google Scholar]
- 25. Harms HJ, Knaapen P, de Haan S, Halbmeijer R, Lammertsma AA, Lubberink M. Automatic generation of absolute myocardial blood flow images using [15O]H2O and a clinical PET/CT scanner. Eur J Nucl Med Mol Imaging. 2011;38:930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bergmann SR, Weinheimer CJ, Markham J, Herrero P. Quantitation of myocardial fatty acid metabolism using PET. J Nucl Med. 1996;37:1723–1730. [PubMed] [Google Scholar]
- 27. Guiducci L, Jarvisalo M, Kiss J, Nagren K, Viljanen A, Naum AG, Gastaldelli A, Savunen T, Knuuti J, Salvadori PA, Ferrannini E, Nuutila P, Iozzo P. [11C]palmitate kinetics across the splanchnic bed in arterial, portal and hepatic venous plasma during fasting and euglycemic hyperinsulinemia. Nucl Med Biol. 2006;33:521–528. [DOI] [PubMed] [Google Scholar]
- 28. Patlak CS, Blasberg RG. Graphical evaluation of blood‐to‐brain transfer constants from multiple‐time uptake data. Generalizations. J Cereb Blood Flow Metab. 1985;5:584–590. [DOI] [PubMed] [Google Scholar]
- 29. Botker HE, Goodwin GW, Holden JE, Doenst T, Gjedde A, Taegtmeyer H. Myocardial glucose uptake measured with fluorodeoxyglucose: a proposed method to account for variable lumped constants. J Nucl Med. 1999;40:1186–1196. [PubMed] [Google Scholar]
- 30. Crawford PA, Crowley JR, Sambandam N, Muegge BD, Costello EK, Hamady M, Knight R, Gordon JI. Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc Natl Acad Sci USA. 2009;106:11276–11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pelletier A, Tardif A, Gingras MH, Chiasson JL, Coderre L. Chronic exposure to ketone bodies impairs glucose uptake in adult cardiomyocytes in response to insulin but not vanadate: the role of PI3‐K. Mol Cell Biochem. 2007;296:97–108. [DOI] [PubMed] [Google Scholar]
- 32. Garland PB, Newsholme EA, Randle PJ. Regulation of glucose uptake by muscle. 9. Effects of fatty acids and ketone bodies, and of alloxan‐diabetes and starvation, on pyruvate metabolism and on lactate‐pyruvate and L‐glycerol 3‐phosphate‐dihydroxyacetone phosphate concentration ratios in rat heart and rat diaphragm muscles. Biochem J. 1964;93:665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russell RR III, Taegtmeyer H. Coenzyme A sequestration in rat hearts oxidizing ketone bodies. J Clin Invest. 1992;89:968–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ferrannini E, Santoro D, Bonadonna R, Natali A, Parodi O, Camici PG. Metabolic and hemodynamic effects of insulin on human hearts. Am J Physiol. 1993;264:E308–E315. [DOI] [PubMed] [Google Scholar]
- 35. Baron AD. Vascular reactivity. Am J Cardiol. 1999;84:25J–27J. [DOI] [PubMed] [Google Scholar]
- 36. Depre C, Vanoverschelde JL, Taegtmeyer H. Glucose for the heart. Circulation. 1999;99:578–588. [DOI] [PubMed] [Google Scholar]
- 37. Taylor M, Wallhaus TR, Degrado TR, Russell DC, Stanko P, Nickles RJ, Stone CK. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F]fluoro‐6‐thia‐heptadecanoic acid and [18F]FDG in patients with congestive heart failure. J Nucl Med. 2001;42:55–62. [PubMed] [Google Scholar]
- 38. O'Donnell JM, Kudej RK, LaNoue KF, Vatner SF, Lewandowski ED. Limited transfer of cytosolic NADH into mitochondria at high cardiac workload. Am J Physiol Heart Circ Physiol. 2004;286:H2237–H2242. [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi K, Neely JR. Control of maximum rates of glycolysis in rat cardiac muscle. Circ Res. 1979;44:166–175. [DOI] [PubMed] [Google Scholar]
- 40. Kuang M, Febbraio M, Wagg C, Lopaschuk GD, Dyck JR. Fatty acid translocase/CD36 deficiency does not energetically or functionally compromise hearts before or after ischemia. Circulation. 2004;109:1550–1557. [DOI] [PubMed] [Google Scholar]
- 41. Luiken JJ, Coort SL, Koonen DP, van der Horst DJ, Bonen A, Zorzano A, Glatz JF. Regulation of cardiac long‐chain fatty acid and glucose uptake by translocation of substrate transporters. Pflugers Arch. 2004;448:1–15. [DOI] [PubMed] [Google Scholar]
- 42. van der Vusse GJ. The fascinating and elusive life of cardiac fatty acids. Cardiovasc Res. 2011;92:363–364. [DOI] [PubMed] [Google Scholar]
- 43. Kudej RK, Fasano M, Zhao X, Lopaschuk GD, Fischer SK, Vatner DE, Vatner SF, Lewandowski ED. Second window of preconditioning normalizes palmitate use for oxidation and improves function during low‐flow ischaemia. Cardiovasc Res. 2011;92:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vanoverschelde JL, Wijns W, Kolanowski J, Bol A, Decoster PM, Michel C, Cogneau M, Heyndrickx GR, Essamri B, Melin JA. Competition between palmitate and ketone bodies as fuels for the heart: study with positron emission tomography. Am J Physiol. 1993;264:H701–H707. [DOI] [PubMed] [Google Scholar]
- 45. Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88:1009–1086. [DOI] [PubMed] [Google Scholar]
- 46. Heusch G. Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol. 2008;153:1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang HH, Katz RL. Effects of changes in coronary blood pH on the heart. Circ Res. 1965;17:114–122. [DOI] [PubMed] [Google Scholar]
- 48. Walker M, Fulcher GR, Marsiaj H, Orskov H, Alberti KG. The independent effect of ketone bodies on forearm glucose metabolism in normal man. Scand J Clin Lab Invest. 1991;51:605–613. [DOI] [PubMed] [Google Scholar]
- 49. Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, Kobayashi M, Hirasawa A, Tsujimoto G. Short‐chain fatty acids and ketones directly regulate sympathetic nervous system via G protein‐coupled receptor 41 (GPR41). Proc Natl Acad Sci USA. 2011;108:8030–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McNeely JD, Windham BG, Anderson DE. Dietary sodium effects on heart rate variability in salt sensitivity of blood pressure. Psychophysiology. 2008;45:405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zajac A, Poprzecki S, Maszczyk A, Czuba M, Michalczyk M, Zydek G. The effects of a ketogenic diet on exercise metabolism and physical performance in off‐road cyclists. Nutrients. 2014;6:2493–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miles JM, Nissen SL, Rizza RA, Gerich JE, Haymond MW. Failure of infused beta‐hydroxybutyrate to decrease proteolysis in man. Diabetes. 1983;32:197–205. [DOI] [PubMed] [Google Scholar]
- 53. Blomqvist G, Alvarsson M, Grill V, Von Heijne G, Ingvar M, Thorell JO, Stone‐Elander S, Widen L, Ekberg K. Effect of acute hyperketonemia on the cerebral uptake of ketone bodies in nondiabetic subjects and IDDM patients. Am J Physiol Endocrinol Metab. 2002;283:E20–E28. [DOI] [PubMed] [Google Scholar]
- 54. Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. [DOI] [PubMed] [Google Scholar]
- 55. Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, Ren N, Kaplan R, Wu K, Wu TJ, Jin L, Liaw C, Chen R, Richman J, Connolly D, Offermanns S, Wright SD, Waters MG. (D)‐beta‐hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA‐G. J Biol Chem. 2005;280:26649–26652. [DOI] [PubMed] [Google Scholar]
- 56. Gormsen LC, Jessen N, Gjedsted J, Gjedde S, Norrelund H, Lund S, Christiansen JS, Nielsen S, Schmitz O, Moller N. Dose‐response effects of free fatty acids on glucose and lipid metabolism during somatostatin blockade of growth hormone and insulin in humans. J Clin Endocrinol Metab. 2007;92:1834–1842. [DOI] [PubMed] [Google Scholar]
- 57. Du Z, Shen A, Huang Y, Su L, Lai W, Wang P, Xie Z, Xie Z, Zeng Q, Ren H, Xu D. 1H‐NMR‐based metabolic analysis of human serum reveals novel markers of myocardial energy expenditure in heart failure patients. PLoS One. 2014;9:e88102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, Murray AJ, Stubbs B, West J, McLure SW, King MT, Dodd MS, Holloway C, Neubauer S, Drawer S, Veech RL, Griffin JL, Clarke K. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 2016;24:256–268. [DOI] [PubMed] [Google Scholar]
- 59. Wentz AE, d'Avignon DA, Weber ML, Cotter DG, Doherty JM, Kerns R, Nagarajan R, Reddy N, Sambandam N, Crawford PA. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J Biol Chem. 2010;285:24447–24456. [DOI] [PMC free article] [PubMed] [Google Scholar]