

Introduction
Atherosclerosis is the most common form of vascular disease and constitutes the major cause of death, with 17.5 million related deaths annually (31% of global mortality).1 Atherosclerotic plaque represents the hallmark lesion of atherosclerosis. Most, but not all,2, 3 acute cardiac events occur in the context of plaque‐related thrombus formation, with the remainder associated with primary cardiomyopathy or arrhythmogenesis (primary myocardial electrical instability or ischemia related). Given that advances in technology allow for imaging, access, and localized treatment of atherosclerotic plaques, enormous efforts have been devoted to recognizing “vulnerable plaques,” defined as rupture‐prone (or, more teleologically relevant, event‐prone) plaques and, subsequently, treating them.4
Despite the above, results of vulnerable plaque‐oriented approaches, in terms of both personalized risk assessment and guided treatment effects, have generally been poor, thus raising doubts regarding the validity and usefulness of the vulnerable plaque concept.5, 6, 7, 8 In the present review, processes associated with decreased stability and related to the vulnerable plaque hypothesis, along with current and experimental methods for detecting vulnerable plaques, are discussed. Finally, potential alternative strategies, representing an amalgamation of differing views of atherosclerosis, will be explored.
On the Genesis of Atherosclerotic Plaque
Disruption of the endothelial cell‐mediated vessel lumen‐wall barrier is considered the earliest prodrome lesion of atherosclerosis. The molecular basis of the barrier lies in intercellular (tight and adherens junctions) and cell/extracellular matrix (ECM) adhesion, both being necessary for its integrity. Both structures are linked to the cytoskeleton, thus allowing for cellular adaptation to applied stresses.9 In fact, these stresses and consequent induced strains (deformations) are sine qua non stimuli for barrier maintenance, acting through mechanosensor‐mediated signal transduction pathways (involving actin and integrins).10 Furthermore, endothelial cells lose their polarized structure and assume a spindle‐like conformation, expressing leukocyte adhesion molecules.11 Consequently, areas of the vasculature with inherently low transmural and shear stresses (cause) and strains (effect), such as bifurcation points and the inner wall of curved vessels (such as the right coronary artery), may begin developing barrier defects early in life.12
Lipid‐Driven Mechanisms
These breaches of vascular barrier integrity allow for lipoprotein infiltration and deposition in the intima.13 Uptake by both locally present and recruited macrophages attempts to prevent their accumulation by means of reverse cholesterol transport. However, cholesterol does have deleterious cellular effects, causing apoptosis, initiating inflammation and thus preventing its removal.14 Upon further material accumulation, an amorphous lipid core (hence the component “athero,” from the Greek ἀθήρα, meaning gruel) is formed—the infamous necrotic core. Lipid particles residing in the core undergo enzymatic modifications, becoming oxidized, and trigger the release of proinflammatory mediators.15 Additionally, oxidized low‐density lipoprotein (LDL) has been found to trigger necroptosis,16 an alternate form of programmed cell death, originally a defensive infection mechanism, whereupon cells do not quietly implode, but rather release particles and messengers inducing proinflammatory activation of adjacent cells.17 Thus, lipid accumulation and modification strongly enhances both quantitative and qualitative plaque progression. Activated macrophages and apo‐/necroptotic cells within the necrotic core produce tissue factor (factor III), a major component of 1 coagulation pathway, significantly enhancing its thrombogenicity.
Lipoprotein(a) (Lp(a)) is a specialized molecule thought to have arisen recently in evolutionary history, either in the context of infection control (given its ability to amass oxidized lipids and draw inflammatory cells to injured vessel walls—normally caused by infectious agents, not atherosclerosis) or as a surrogate for ascorbic acid in vascular wall repair.18 However, in the context of atherosclerosis, it assumes 4‐fold deleterious/destabilizing effects by promoting thrombosis, inflammation, cholesterol delivery to atheromas, and smooth muscle cell (SMC) proliferation. Studies' findings have, foreseeably, associated Lp(a) levels with vulnerable plaque phenotype.19
Inflammatory‐Immune Mechanisms
Inflammation is also a major event in atherosclerosis natural history and plaque progression, usually related to decreased structural stability. Both components of immunity, innate and adaptive, have been found to be activated in atherosclerosis‐related inflammation. Postulated infectious mechanisms (Chlamydia pneumoniae) also act through this pathway, although the process soon becomes self‐sustained regardless of pathogen presence.20 Increased local thermal activity has been considered a direct effect of inflammation, stemming from presence of metabolically highly active immune cells, energy production uncoupling attributed to ischemia, and exothermic chemical reactions (such as oxidation, H2O2 dissociation).21 Moreover, given results of theoretical calculations, blood‐wall friction as well as turbulent flow at the maximal stenosis area also contribute to localized heating. Increased temperature is not only a potentially measurable result of atherosclerotic plaque presence, but has also been related to higher blood viscosity and red cell aggregation. Finally, neovascularization of advanced plaques might also play a role, on the premise of heating the metabolically more‐inert abluminal area and core of the plaque, although a cooling effect of blood‐flow–related convection should be considered.22
The Role of ECM
A second dynamic equilibrium is established regarding ECM production and degradation,2 giving rise to vessel remodeling, that is, the response of the ever changing plaque to alterations in mechanical factors.23 ECM, comprised of both cellular and acellular (usually in the form of proteoglycans) components constitutes the initial, theoretically temporary, container of the necrotic core, preventing its contact with the bloodstream. Furthermore, the mechanical strength of its luminal part (cap) determines plaque resistance to stress. SMCs and myofibroblasts are the main producers and maintainers of ECM, with inflammatory cells and necrotic core stimulating its degradation.
ECM degradation is not limited to the luminal side of the plaque; rather, abluminal layer involvement is evident, especially in plaques with larger necrotic cores (and, consequently, more macrophages and inflammation), providing a mechanistic link and interpretation of the well‐known association between positive remodeling, plaque necrotic core size, and instability.24, 25 Furthermore, direct deleterious effects of the necrotic core have been documented, in the form of cholesterol crystals that expand upon formation and with changes in temperature and pH, rupturing container (foam) cells and straining the plaque from within (not unlike ice), and pierce the fibrous cap, weakening it and providing a thrombogenic substrate to circulating coagulation components. In fact, the relationship between cap thinning and plaque propensity to rupture is not linear, given that significant increases in deformation and tensile stress, for the same load, are noted past a certain point (Young's modulus reduction >60%).26
Neoangiogenesis
Neoangiogenesis is a feature of advanced atherosclerosis, appearing late in the natural history of plaques. The main stimulus for neovessel formation comes in the form of hypoxia‐inducible factors secreted by hypoxic macrophages deep in the lesion's necrotic core27 and involves endothelial cells losing polarity and sprouting tube‐like forms as precursors to fully fledged neovessels. In fact, the anoxemia theory of atherosclerosis postulates that it is the depletion of ATP in macrophages, attributed to the extremely high rates of energy taxing cholesterol uptake, which leads to apoptosis, formation of a necrotic core, and angiogenetic stimuli. The neovessel wall lacks proper structure, in terms of elastic laminae and smooth muscle cell support, and thus they are leaky, prone to rupture, and provide the plaque with elements crucial for its continued progression,28 in the form of cholesterol (both lipoprotein and red blood cell membrane derived), cytokines, and leukocytes, straight to the necrotic core, thus establishing a vicious positive feedback loop favoring destabilization and, ultimately, rupture.29 Plaque hemorrhage may thus underlie episodes of accelerated plaque growth.30
The Many Faces of Mineralization
The final act in the course of atherosclerosis as a chronic unresolved inflammatory process lies in the permanent and secure encasement of its source area in a highly calcified structure (mineralization), replacing the temporary fibrotic “seal” produced by chemokine‐attracted intima invading SMCs. The process of vascular mineralization is now considered active, highly regulated, and akin to bone formation.31, 32 Notably, vascular calcification, as opposed to bone formation, appears to be triggered, not inhibited by, inflammation.33 However, in a process reminiscent of myocardial wall rupture risk following infarction, initial phases of calcification with formation of calcified specs and nodules (microcalcification) will actually reduce plaque mechanical strength34 by introducing contact areas between materials (calcified nodules and fibrous cap components) with different elastic moduli, different behavior under stress, and thus prone to stress‐induced debonding or caveolation.35 This effect is particularly prominent in cases of adjacent specs, interpreting central plaque rupture and high peak stresses in theoretically nonvulnerable (cap thickness >65 μm, or even >100 μm) plaques.36 The above could help explain the often contradictory findings regarding calcification effects on plaque stability,2 given that older imaging modalities could not reliably discern between spotty/nodular calcification and fused, reinforced calcified structures.
The Divergent Course of Erosion
Plaque erosion mechanisms underlie almost one quarter of all acute coronary syndrome (ACS) events,37 with rising prevalence, yet they remain largely unresolved.38 Eroded plaques tend to lack features associated with rupture prone structure, exhibiting thicker caps, large numbers of SMCs, and smaller necrotic cores, while being quiescent in terms of inflammation.39 However, significant divergence has been documented in terms of ECM properties. Both a shift toward collagen III (as opposed to collagen I usually dominating plaque ECM) and hyaluronan (a proteoglycan subtype) production has been found—in fact, hyaluronan is almost absent even at rupture sites. Given that collagen III has more‐elastic properties, compared to its type I counterpart, and that endothelial cell adhesion to hyaluronan is reduced (especially in larger‐vessel–derived endothelial cells),40 detachment of cells from their basal membrane becomes possible, even at lower stresses—or in cases of coronary spasm41 (increased membrane mobility and weaker attachment). Furthermore, not only is hyaluronan more susceptible to neutrophil‐derived protease cleavage, but also increased numbers of neutrophils have been detected near erosion‐prone plaques as well and have been shown to induce endothelial apoptosis.42 Finally, hyaluronan itself has been associated with reduced proliferation and survival of endothelial cells in cultures and possesses prothrombotic properties (increased platelet adhesion and accelerated fibrin polymerization).43 Given preponderance of women, smokers, diabetics, and the elderly among patients suffering plaque erosion, it could be inferred that certain idiosyncratic features or noxious (metabolic or exogenous) stimuli alter the usual course of events during atherosclerosis progression, leading to formation of a less‐ruptured (increased elasticity?), yet more thrombosis‐prone plaque. The lack of need for structural support with exogenous scaffolding and increased thrombogenicity may interpret the findings of the recent Effective Antithrombotic Therapy Without Stenting: Intravascular Optical Coherence Tomography‐Based Management in Plaque Erosion (EROSION) study, where antithrombotic therapy was a viable option, compared to stenting, in nonocclusive stenoses causing erosion‐related acute events.37 The enhanced role of neutrophils in plaque erosion—analogous to that of macrophages in rupture—is thought to be mediated by released strands of DNA, called neutrophil extracellular traps (NETs), originally sequestering pathogens and disarming them by means of DNA‐drawn enzymes.44 However, in the framework of plaque erosion, the process, aptly termed NETosis, leads to a prothrombotic state, by enhancing platelet aggregation and coagulation and inhibiting fibrinolysis (fibrin mesh constituting another putative pathogen trap). Indeed, myeloperoxidase‐positive cells have been found in larger quantities in the blood of patients with eroded plaques and in the overlying thrombi.45
Effects of Plaque Environment—External Stresses
Effects of abnormal exogenous stresses continue in late stages of atherosclerosis.46 Normal laminar flow causes relatively stable transverse and shear stresses, with limited oscillatory variations.11 Moreover, in addition to cell reorientation, exogenous stress leads to collagen laminae reorientation, introducing anisotropy of the fibrous cap and rendering it more prone to rupture.47 Mechanical factors may also, through mechanoreceptors, lead to a multitude of effects on other cellular processes, such as increased reactive oxygen species (ROS) production and inflammation.11 In fact, plaque tendency to rupture at the shoulders, as well as cases of fissuring in other areas of the fibrous cap, may be interpreted by integrating mechanical factors' effects on biology. As blood flow is diverged toward the remaining lumen by the upstream plaque edge, a low‐stress area occurs, prone to loss of cellular architecture and increased lipoprotein and leukocyte permeability, thus continuously providing fuel for plaque growth and inflammation. At the shoulder area, however, stresses increase considerably attributed to blood incompressibility. Thus, not only are ROS production and inflammation stimulated in the underlying tissue (as a result of high, rather than low, stresses in this area), but an abrupt transition between mildly and highly stressed areas occurs, favoring plaque rupture and neovessel formation and rupture.48 Similarly, at the downstream shoulder, flow turbulence leads to decrease of transmural pressure attributed to vortex‐formation–related energy dissipation, mirroring the events of the upstream region. Presence of a large necrotic core exaggerates this unfavorable altering of stress distribution, further straining the shoulder area.49 At intermediate plaque areas, between shoulders, highly oscillatory stresses may occur attributed to the Bernoulli effect. Similarly, this mechanism of blood acceleration through the stenotic lumen and reduction of transmural pressure exerts detrimental effects on the vasa vasorum with blood content oscillations, causing plaque expansion and dilation at every cardiac cycle, stressing and building up fatigue in the fibrous scaffold of the plaque, and potentially causing the structurally defective vasa vasorum to rupture. High shear stresses could conceivably play an analogous role in enhancing endothelial cell denudation in plaque erosion. Based on the above, it is evident that all types of abnormal stress patterns, if sustained, have discrete adverse effects on atherosclerotic lesions: Low stresses are involved in initial stages, whereas high and oscillatory stresses come into play later on in disease progression.
Vulnerable Plaque Hypothesis
Ultimately, plaque destabilization (a general term for complication related changes) is a biomechanical phenomenon that can be thought of as depending on a complex interplay between applied stresses, structural features, and biological processes that determine mechanical strength.50, 51, 52, 53, 54, 55, 56 This approach, although not conclusive, is useful as a framework for pursuing ways to prevent, predict, and avert rupture/erosion. Obviously, focusing solely on plaque characteristics will ignore the effects that exogenous forces exert on stability. On the other hand, focusing on luminal stenosis will only assess the applied forces and disregard plaque contribution, explaining the oft‐cited finding of far greater prevalence of nonstenotic lesions in cases of destabilized (ruptured) plaques.57 Furthermore, not all cases of destabilization will lead to clinical events, given that vessel occlusion, leading to peripheral necrosis with mechanical or electrical instability, is associated with blood thrombogenicity and the ability of counter‐regulatory mechanisms to contain its sequelae—prevalence of silent ruptures in stable coronary artery disease patients has been reported to reach 58%.58
Plaque rupture is the most common form of plaque destabilization,50, 59, 60 accounting for two thirds of fatal myocardial infarctions (MIs) and sudden cardiac deaths and refers to transmural fissuring of the fibrous cap, leading to exposal of the thrombogenic and proinflammatory underlying necrotic core to circulating blood. The affected plaque usually has the features of a thin cap fibroatheroma, as described previously61 (Table 1). On the other hand, plaque erosion denotes a histologically seen thrombus superimposed on a cell‐denuded endothelial layer of a nonruptured plaque71 with different structural features, as observed in the alternative course of plaque progression. Finally, calcified nodules have been found in cases of culprit lesions, as expected by their inherent disruptive effects on plaque integrity, constituting another rupture‐prone structure, alternative to thin cap fibroatheroma.50, 59, 60
Table 1.
Thin Cap Fibroatheroma Features
| Histology61 |
| Cap thickness <65 μm (based on results by Burke et al62 acquired from ruptured coronary plaques in male cadavers) |
| Large necrotic core |
| Increased macrophage infiltration |
| Virtual histology IVUS63, 64 |
| Focal lesion, containing necrotic core (≥10% of total plaque area) in direct contact with the lumen (cap cannot be visualized) |
| Percent atheroma volume ≥40% |
| Optical coherence tomography65, 66 |
| Wide lipid arc (>90 degrees), suggesting increased lipid content. Lipid arc is defined the widest arc demarcating a signal poor region with diffuse borders65, 67 |
| Necrotic lipid pools presence (signal‐poor regions poorly delineated, underlying a signal rich cap), quantified based on number of quadrants occupied |
| Superficial microcalcifications (subtending a <90‐degree arc and with a border with lumen <100 μm thick) |
| Cap thickness <65 μm—although different limits have been proposed, with studies suggesting that OCT‐derived in vivo thin cap limit should be increased (postmortem/histological preparation—related alterations in previous studies)68, 69 |
| When virtual histology IVUS is used concomitantly to assess deeper plaque layers, OCT fibroatheromas are confirmed to have a high lipid content and low levels of fibrosis and exhibit more‐expansive remodeling.70 |
Modality‐specific definitions are necessary, given that no single approach can accurately visualize all aspects of histology, attributed to poor resolution for IVUS and poor penetrance for OCT. Given complimentary features of all methods, the best possible assessment of intrinsic instability (see Figure 1) would currently entail application of multiple modalities on a single plaque or, alternatively, combination of modalities into multifunctional systems. IVUS indicates intravascular ultrasound; OCT, optical coherence tomography.
The vulnerable plaque hypothesis was developed in an effort to better describe the unpredictability of the clinical course of atherosclerosis. Vulnerable plaques have insightfully been defined as those plaques prone to becoming culprit plaques (Table 2), causing acute vascular events or death, regardless of form, shape, stenosis, or destabilization.2 Consequently, all forms of rupture‐prone, erosion‐prone, hemorrhaging, or containing calcified nodules plaques (mainly expressing the intrinsic constituent of culpability potential), as well as significantly stenotic plaques (mainly expressing the extrinsic constituent in the form of applied stresses), can be considered vulnerable plaques as long as they have led to hard clinical events. However, this is a post‐hoc approach, of uncertain clinical significance. Thus, the term “vulnerable plaque” has, by extension, been used to denote destabilization or, even more narrowly, rupture‐prone plaques.72 Moreover, difficulties in assessing applied stresses and consequent strains attributed to requirements of advanced in silico models23, 35, 47, 51 have led to widespread adoption in the literature of the notion that structural plaque features alone can define plaque vulnerability, and, consequently, most efforts have been directed toward correlating imaging features with specific morphologies. However, this may not be the case, as evident from the pathogenesis of atherosclerosis, and visualized in Figure 1. Specific criteria, based on event‐causing plaques, have been proposed.2 Henceforth, the term “vulnerable” will be used for plaques considered unstable based on current approach, not taking into account the effect of exogenous factors, such as hydrodynamics (dynamic pressure, pressure head), flow characteristics (laminar/turbulent), and vascular anatomy and function (eg, bifurcation and tone, respectively). It is thought, based on tentative studies' results (see below), that destabilization propensity is better appreciated within the latter, rather than the former, conceptual framework.
Table 2.
Culprit Plaque‐Based “Vulnerable” Plaque Characterization Criteria
| Expressing rupture propensity |
| Active inflammation |
| Thin cap fibroatheroma morphology/yellow color on angioscopy |
| Fissured plaque |
| Calcified nodule presence |
| Intraplaque hemorrhage |
| Endothelial dysfunction |
| Positive remodeling |
| Expressing erosion propensity |
| Endothelial denudation with thrombogenic proteoglycan substrate±thrombus presence |
| Endothelial dysfunction |
| Expressing effects of extrinsic factors |
| Lumen stenosis over 90% |
Figure 1.
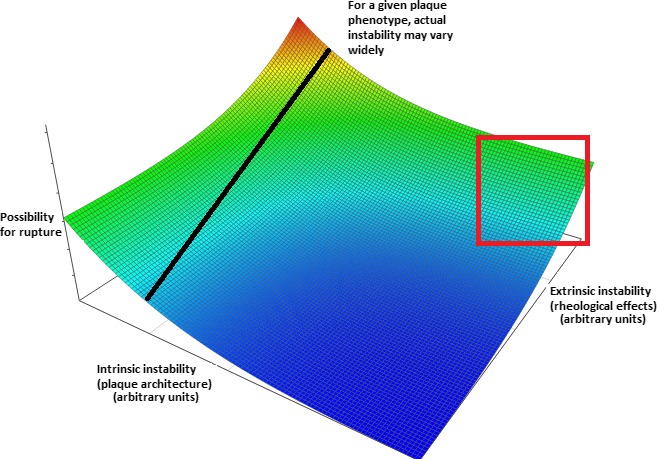
Inadequacy of modern approaches to detect plaque vulnerability. Conceptual plot visualizing the combined effects of factors leading to intrinsic and extrinsic plaque instability. Arbitrary “instability units” are used and the precise form of the relationship is purely conjectural—however, it was chosen to denote the more‐than‐additive effect of simultaneous increases in both parameters. The line drawn corresponds to plaques with a specific internal architecture and demonstrates that their actual instability (ie, possibility for rupture) is also critically dependent on external stresses applied to them. Of note, solely intrinsic features may indeed determine a truly destabilization‐prone plaque, but only at extreme values (thus clinically yielding high specificity and low sensitivity as potential criteria). Furthermore, imaging modalities rarely assess all features of vulnerability (in the current sense)—rather, they focus on specific aspects, such as lipid and calcium content, thus failing in even establishing the value in the intrinsic instability axis. Moreover, both these parameters vary with time given the (1) tendency of plaques to alternate between different structural phenotypes and (2) possibility for alterations in the rheological (dynamic pressure, pressure head, and viscosity) features of circulation (eg, following removal of an upstream lesion through successful angioplasty)—thus, a given plaque's position would not remain fixed on the plot. Clustering could be anticipated to occur in several areas of the plot, given that, often, intrinsic features affect extrinsic and vice versa (ie, a thick‐capped fibrous occlusive plaque would be subject to higher external stresses and thus usually be located in the area enclosed by the red square). Models incorporating all mentioned aspects would likely yield a much more accurate prediction regarding possibility for rupture and (should further parameters such as viscosity be added) acute events (even if silent) and thus guide treatment. Color coding: blue, red: minimal and maximal instability, respectively.
Currently, plaques are most commonly characterized as vulnerable when possessing some of the following features: large lipid cores (also laden with tissue factor); thin adluminal fibrous caps; large inflammatory infiltrates; positive remodeling; neoangiogenesis; intraplaque hemorrhage; and endothelial denudation (in vivo visible only with advance optical coherence tomography use).41, 62
Detecting the Vulnerable Plaque
Although the ability of imaging modalities to visualize atherosclerotic plaques has improved considerably during the past few years, they primarily focus, with few exceptions, on accurately depicting a still frame of the plaque structure, not the continuous and variable interaction with its surroundings (plaque‐centered approach). Thus, none of them has, beyond the context of proof‐of‐concept studies, established criteria for assessing plaque vulnerability in the broader sense, as discussed previously.
Intravascular ultrasound (IVUS) is 1 of the 2 most widely used invasive coronary atherosclerotic plaque imaging modalities, the other being optical coherence tomography. Essentially a catheter‐mounted ultrasound transducer, it follows the same principles regarding ultrasound generation, signal reception, data processing, and image presentation.63 A grayscale image is generated based on which plaques can be broadly classified as (their echogenicity compared to that of the fibrous adventitia) soft, intermediate, calcified, and mixed. Moreover, added modules can enhance tissue characterization abilities of IVUS, allowing for detection and quantification of different plaque structures—this is achieved, in broad, by analyzing, in addition to reflected signal amplitude, its frequency and power. Virtual histology IVUS, iMAP IVUS, and integrated backscatter IVUS are examples of this, with high reproducibility although they remain proprietary and use different color‐coding for the same structures.73, 74, 75
Regarding vulnerable plaques, IVUS, and its modular expansions, can reliably assess plaque burden, expansive remodeling, as well as presence and relative proportion of necrotic core, calcifications, and neoangiogenesis (contrast enhanced; penetration depth, 5 mm). Confirmation of greater strains in fibroatheromatous, as compared to fibrous plaques, has also been provided by compound ultrasound strain imaging (alternatively known as palpography/elastography).76 However, at its current form (20/40 MHz transducers), it cannot visualize plaque caps, especially thinner ones (resolution, >100 μm).63 Most prospective studies regarding ability of vulnerable plaques to predict events used IVUS‐based approaches in their definition of vulnerability.77, 78, 79, 80
Optical coherence tomography (OCT) is the second major coronary plaque invasive imaging modality that uses near‐infrared light (1.3 μm in wavelength) in a manner analogous to sonography. Light reflected form plaque structures provides image data whereas the background effect of scattered light is negated through use of interferometric techniques. Bright or dark areas occur as a result of constructive or destructive interference between reflected and reference beams. Given the much smaller wavelength of light in comparison to ultrasound, OCT resolution is 1 order of magnitude higher (≈10 μm), allowing for proper visualization of fibrous caps, collagen content (polarization‐sensitive OCT), macrophages (related to inflammation),81 neovessels (appearing as microchannels), ruptures, and thrombi. OCT performance, with regard to microcalcifications at the lower end of the spectrum (<5 μm in diameter), remains uncertain. Consequently, several, but not all, aspects of vulnerability are visualized. However, this comes at a price, considering that high scattering of light prevents imaging of plaque structures at deeper layers (penetration depth, 1.5 mm) and thus proper recognition and estimation of remodeling and necrotic core size (hence the “lipid arc” concept; Table 1). OCT has displayed high agreement with histopathology regarding both plaque characterization and cap thickness.67
OCT limitations include the need for blood displacement, attributed to the high scattering effect of its components, longer image acquisition times (although newer, frequency domain analysis‐based processing methods), frequent artifacts, inability to assess deeper plaque structures, as well as relatively poor discrimination between calcified areas and lipid core, given that both appear as signal poor areas (with clear and diffuse borders, respectively).68
A newer form of OCT, micro OCT (μOCT), is currently able to offer axial and lateral resolution in the order of 1 to 2 μm, by means of advanced frequency domain analysis and use of broad bandwidth light, approaching histology levels. Thus, events in the cellular and molecular level, crucial to atherosclerosis development and progression, are visualized, including leukocyte diapedesis, fibrin strand formation, ECM production, endothelial denudation, microcalcifications and cholesterol crystal formation, and penetration of the fibrous cap. Given that this method may, in the future, offer real‐time in vivo images (as well as 3‐dimensional reconstruction) of the vessel wall at subcellular resolution, it may prove extremely useful for the classification of atherosclerotic plaques and identification of the vulnerable ones, replacing classical histology and scanning electron microscopy, notwithstanding all inherent classical OCT limitations.
Invasive coronary thermography is an approach aiming to detect subtle temperature increases of the vessel wall in areas of heat production, usually accompanying inflamed and/or ruptured atherosclerotic plaques.82 Purpose‐built catheters include hydrofoil and balloon‐based designs, ensuring adequate apposition of the thermistor module on the vessel wall.83, 84 The latter is furthermore able to ensure temporary lumen occlusion, thus negating any cooling, convection‐based effects of blood flow, and accentuating underlying gradients.84 Indeed, studies have shown both an increase in temperature correlating with presenting disease severity (stable angina<unstable angina<acute MI), and a correlation with systemic levels of inflammatory mediators, such as C‐reactive protein.83 In addition, prognostic value of postangioplasty measured plaque temperature has been demonstrated, given that a difference of ≥0.5°C between plaque and healthy vessel wall was associated with 41% probability of adverse events (repeat infarction, recurrent angina, and death) during a follow‐up period of just 18 months.85 Good correlation with IVUS‐derived indices of vulnerability (positive remodeling) has been reported.82 Despite a sound pathophysiological basis, technical issues have, so far, precluded the use of thermography for plaque vulnerability assessment.
Near‐infrared spectroscopy (NIRS) utilizes characteristic emission spectra produced by plaque contents following interaction with photons (wavelength area, 700–2500 nm). Low sensitivity, in terms of induced response, and high penetration, as compared with visible light spectroscopy, render this method appropriate for assessing the lipid content of plaques, especially in cases of positive remodeling, with large, deep‐seated, lipid‐laden necrotic cores.86 Studies using NIRS have shown that large lipid content, rather than plaque burden, is associated with thin cap fibroatheroma features.25, 87 Larger lipid core burden has been shown to accurately differentiate between culprit and nonculprit lesion in ST‐elevation MI patients88 and has been associated with higher risk for periprocedural myocardial infarction.89 Interestingly, combination with IVUS may allow for concomitant appreciation of both plaque structure and composition,90 comparing favorably with OCT.87 Despite recent studies linking lipid‐rich, NIRS‐defined nonculprit plaque presence with a 4‐fold risk for adverse events (all‐cause mortality, nonfatal ACS, stroke, and unplanned revascularization—excluding those definitely related to the initial culprit lesion)91 within the first year of follow‐up, it cannot be inferred whether these were actually triggered by the detected vulnerable plaque (thus being amenable to preventive stenting) or by other, not assessed lesions (NIRS was only performed over a vessel segment). The latter could have very well been nonvulnerable lesions at the examination time, and their progression would reflect the need to reduce the atherosclerotic burden as a whole, rather than perform localized treatment. Results from the COLOR and LRP (Lipid Rich Plaque) studies are expected to shed light on the role of NIRS in guiding nonculprit vulnerable plaque stenting.92, 93
A modified form of NIRS, near‐infrared autofluorescence, involves active stimulation of lipid components to emit detectable infrared light.94 Combination with OCT has allowed for better visualization of lipid‐laden necrotic cores and, moreover, accurate localization of the area within it with the densest macrophage concentration.95
Computed tomography coronary angiography (CTCA) utilizes a computed tomography (CT) scanner in combination with iodine intravenous dye to visualize the coronary arteries. Significant advantages stem from its very nature: noninvasiveness, ability for imaging the whole coronary vasculature, and potential for assessing both vessel wall in addition to the lumen.96 Advances in technology have allowed for improved image quality with reduced scan times and radiation exposure. More specifically, multislice (or multidetector) scanners possess a 2‐dimensional detector array allowing for simultaneous acquisition of multiple planar images (slices), reducing examination time and possibility for motion artifacts—those with an array breadth of 160 mm allow for imaging of the heart in a single beat. Use of softer reconstruction kernels leads to improved soft‐tissue visualization, however at the expense of spatial resolution.97 Prospective gating can thus be used in order to synchronize image acquisition with diastole (increased coronary blood flow). Moreover, advent of dual‐energy monochromatic scanners may overcome limitations in tissue assessment caused by adjacent intense calcification, by negating the beam‐hardening effect.98 Finally, dual‐source scanners may complete the examination in only half a rotation. The related, albeit simpler, technique of calcium scoring does not use an intravenous dye and focuses primarily on coronary calcium quantification to predict the risk for subsequent events.
From a clinical standpoint, CTCA has emerged as the best noninvasive imaging modality in terms of assessing both lumen area and plaque composition,96 throughout most of the coronary vasculature (vessels >1.5 mm in diameter,96 where the majority of plaques are located99). Its accuracy in detecting lesion presence is noninferior to IVUS,100 and, consequently, it exhibits an excellent rule‐out ability with a negative prognostic value approaching 100%, even in prolonged follow‐up.101, 102 Furthermore, although unable to quantify cap thickness, it is nevertheless able to detect presence of thin cap fibroatheromas, comparing favorably with OCT.
Regarding vulnerable plaques, CTCA can reliably assess the presence, size, and thickness of the necrotic core, by grading tissue in Hounsfield units (HU; plaques with large cores will cause less attenuation and thus have lower unit values).103 Specific high‐risk plaque criteria have been developed, such as positive remodeling (remodeling index [RI], ≥1.1, also a surrogate for plaque composition104), (very) low (given that the threshold for a ≥10% necrotic core is 41 HU) attenuation plaque (LAP; <30 HU), napkin‐ring sign (a low attenuation core surrounded by a rim of relatively high attenuation—pathogenesis is still debatable), and presence of spotty calcifications (<3 mm—in accord with the role of calcium specs in destabilizing plaques; see “The Many Faces of Mineralization” subsection).96
It is noteworthy that all of its derived parameters regarding plaque characterization have emerged as significant and independent predictors of events in multiple studies. For example, a recent coronary CT study, with a follow‐up of almost 100 months,105 confirmed that, in nonobstructive disease, positive remodeling, increased plaque burden, low attenuation, and napkin‐ring sing presence are all associated with cardiac events. In addition, a plaque volume of 3 mm3/mm of vessel wall has been identified as an appropriate cutoff for prediction of adverse events.106 Presence of multiple high‐risk features has been found to confer a more‐than‐additive risk, as reported in large studies. More specifically, presence of both positive RI and LAP yielded a more than 22% probability of ACS over an average follow‐up of 27 months, as compared to <0.5% probability, should both features be absent.107 Presence of 3 high‐risk features (LAP, RI, and napkin‐ring sign) leads to a 60% survival free from ACS in the first year.108 In the latter study, the napkin‐ring sign had the highest hazard ratio (HR) for occurrence of events (HR, 5.6).
Coronary artery calcium score (CACS) is an alternative parameter measured by means of cardiac CT without the need for intravenous dye. Despite the fact that it only assesses 1 aspect of atherosclerosis, as opposed to CTCA, it has been proposed that the latter offers additional prognostic information only in patients with intermediate to high CACS.109 In another study, a combination of traditional risk factors, CACS, and CTCA yielded an area under the curve of 0.93 for the prediction of major adverse cardiovascular events (cardiac death, nonfatal infarction, and revascularization) over a follow‐up of ≈1000 days, significantly higher than that of the CACS—risk factors combination (0.82; P<0.001).110
The noninvasive nature of CTCA renders it an attractive option for population screening (primary prevention), and the ability to assess and monitor plaque burden essentially in the totality of the coronary arterial bed, with incremental improvement over conventional stratification,111 allows for the pursuit of risk‐factor–reducing strategies as an alternative to pre‐emptive stenting of high‐risk asymptomatic lesions.112 CTCA‐based 3‐dimensional reconstruction of vessel anatomy and geometry has also been used for the noninvasive assessment of fractional flow reserve in order to determine hemodynamic significance of lesions113; however a potentially more‐fruitful approach could be its integration into fully fledged computational models for the assessment of plaque stability (see below).
As pointed out in the NIRS section, although these results encourage the use of CTCA as a means to assess probability for future events, they cannot constitute, in any way, a proof for the validity of stenting nonculprit vulnerable plaques, given that they do not prove that they underlay these events.
Magnetic resonance imaging (MRI) has the best ability to visualize the soft‐tissue component of atherosclerotic plaques, as well as neovessel formation and diffusion properties (wall permeability). Major limitations include motion artifacts and use in cases of cardiac implants or devices. Importantly, MRI has been used in animal studies attempting to prospectively determine vulnerable plaque features specifically related with (pharmacologically induced) disruption.114 These were related to plaque remodeling (with positive remodeling and grater plaque area associated with future rupture) and inflammation indices (markedly increased gadolinium enhancement—denoting increased neovessel permeability and extracellular space expansion—as in intense apoptosis/necrosis). Even so, applicability to humans, the time frame between feature presence and (nontriggered) rupture and relation with clinical events remains unknown.
Molecular imaging involving targeting and visualizing specific components of biological processes, has been extensively used in attempts to visualize the vulnerable plaque. Theoretically, any modality may be used for molecular imaging as long as a proper tracer can be developed, for example, photon‐emitting or possessing paramagnetic properties. Any substance of interest may act as a target provided that it can be either attached to or modified as a tracer.
Positron emission tomography (PET) and single‐photon emission computed tomography (SPECT) are the 2 modalities mostly associated with molecular imaging, often in conjunction with CT/MRI imaging,115 to provide anatomic background and resolution on top of the mainly functional results of molecular imaging.
Virtually every atherosclerosis‐related process has been studied by means of molecular imaging. More important, vulnerability‐related processes have proven amenable to imaging, including leukocyte adhesion (through involved proteins, such as selectins and vascular cellular adhesion molecule 1), macrophage content (osteopontin),115 collagen degradation (labeled matrix metalloproteinase [MMP] inhibitors), cell apoptosis (use of annexin that binds to lipids exclusively present in the outer layer of apoptotic cell membrane) or necroptosis (radiolabeled necrostatin, a preferential inhibitor of necroptosis),16 and neoangiogenesis (use of labeled anti–vascular endothelial growth factor antibodies as tracers). PET has also been experimentally evaluated for the study of intraplaque hypoxia27 and assessment of macrophage content. Significance of tracer choice can be perceived in the case of plaque calcification, where PET‐CT use of radioactive sodium fluoride as a tracer may reveal sites of active calcification, thus differentiating between stabilizing (encasing) and destabilizing (ongoing) calcification (see above). This is achieved because of preferential binding of the tracer on hydroxyapatite crystals, whose exposed surface is much larger in areas of ongoing crystal formation.116
PET is generally favored over SPECT because of lower artifact rates and improved spatial resolution as a result of detection of 2, as opposed to a single, oppositely moving photons.
The advent of nanotechnology has further widened the repertoire of imaging probes, allowing the construction of complex nanostructures (nanoparticles and magnetic nanoparticles), engineered according to the desired targets.117 For instance, nanoparticles composed of cross‐linked iron oxide molecules and 19F perfluorocarbon are used to detect macrophages and in MRI spectroscopy, respectively (the latter attributed to excellent background noise suppression—no fluoride isotopes exist in tissues). Many of these probes may double as treatment vectors (theranostics), inasmuch as under the influence of infrared lasers from a catheter they overheat and release energy to the surrounding tissue, leading to (localized and targeted) irreparable damage (nanobombs).118
Interestingly, different approaches to optical microscopy (Brillouin microscopy) could allow for direct estimation of plaque stiffness without need for computational models (see below). More specifically, they exploit differential energy shift of emitted photons following interaction with different areas of an anisotropically deformed (strained) structure (such as a plaque cap). Proof‐of‐applicability studies have been undertaken in an ex vivo setting, providing encouraging results regarding potential mounting of Brillouin scattering systems on coronary catheters.
Computational models will allow for simulation of plaque behavior under (patho)physiological conditions. These include finite element analysis models, assessing forces applied on a surface though numerical approximation of the solution by dividing the surface into multiple (>10 000) elementary components with homogeneous properties,52 and fluid‐structure interaction simulations, also integrating rheological effects on plaque structural stress. They are the only means able to actually assess true vulnerability by integrating the plaque structure with its surroundings; however, all of them are critically dependent on an accurate imaging technique to provide vessel geometry and plaque structural data as input.23 Intrinsic parameters classically associated with vulnerability (cap thickness, necrotic core size, vessel anisotropy, and all forms of calcification) are combined with the anticipated effects of applied stresses, fatigue attributed to vessel motion during the cardiac cycle, and blood pressure/flow effects to yield meaningful results regarding conditions leading to rupture/erosion. Should more parameters be available as inputs and not need to be inferred (such as stain by use of Brillouin microscopy), results will accordingly be more precise. A major limitation of current computational models lies in the perceived difference of structural properties between plaque components in vivo and purified materials used to determine inserted parameters.119 Despite the above, advances in algorithms have led to reduction of necessary time to construct the model and obtain convergent solutions (ie, functions describing plaque behavior) from weeks (!) to less than 2 hours.51 The latter is comparable to offline time needed for 3‐dimensional reconstruction of IVUS and OCT data; thus, it may herald active application of models to clinical practice in the near future so as to evaluate the atherosclerotic plaque not as an isolated structure, but as part of a living vessel.
Although relatively scarce, some clinical data exist advocating the added prognostic value of integrating computational models in clinical practice. More specifically, even in the negative Prediction of Progression of Coronary Artery Disease and Clinical Outcome Using Vascular Profiling of Shear Stress and Wall Morphology (PREDICTION) study, wall shear stress was found predictive of a future decrease in luminal area and clinically significant obstruction,80 whereas balloon‐induced rupture was noted to occur at the site of the maximal structural stress in a 2001 study using IVUS to generate a finite‐element model.53 More important, in patients presenting with ACSs, increased stress values were noted in high‐risk areas, suggesting a potential cause‐and‐effect relation.54 Obviously, large, well‐designed prospective studies and, subsequently, randomized, clinical trials are currently lacking and sorely needed to verify or refute the validity of the above integrative approach.
Questioning the Vulnerable Plaque Hypothesis
Considerable issues are currently being raised with regard to the vulnerable plaque hypothesis context, especially its clinical significance.6, 7, 120 Despite vulnerable plaque features presence having high sensitivity in cases of acute events, prospective studies, such as Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT),79 VH‐IVUS in Vulnerable Atherosclerosis (VIVA),77 and the European Collaborative Project on Inflammation and Vascular Wall Remodeling in Atherosclerosis (ATHEROREMO‐IVUS),78 have consistently shown that their positive predictive value is low, implying low prognostic value on an individual basis, used in addition to classical risk‐stratification factors. In the same framework, the PREDICTION study only demonstrated an ability for IVUS and shear stress features to predict plaque enlargement and lumen narrowing, not acute events.80 Failure of a shear‐stress inclusive approach to define plaque vulnerability may be related to IVUS resolution, considerably inferior to that of OCT. Moreover, in the recently presented 2‐year follow‐up of the COLOR registry,93 NIRS‐determined culprit plaque vulnerable phenotype was not associated with a difference in major adverse cardiovascular events occurrence (5.4% vs 6.3% in the nonvulnerable plaques; P=0.41), suggesting that a stented lesion has the same possibility to cause a new clinical event regardless of its type. In fact, adverse events during follow‐up were more commonly related to nonculprit lesions; however, outcomes for the NIRS‐defined vulnerable nonculprit plaques have not yet been analyzed. In conjunction, the full results from the COLOR and LRP studies (NCT02033694)92 are expected to definitely answer whether NIRS fares any better in identifying plaques prone to causing future events.
Recent findings appear to suggest that the notion of nonstenotic plaques underlying ACSs is a misconception, given that, when having been assessed close to the episode of which they were the culprit, plaques appear severely stenotic, having undergone a period of accelerated growth.79, 121 Although vulnerable plaques may carry high lipid burdens, masked by their expansive remodeling, 1 of the major tenets of the vulnerable plaque hypothesis is that stenosis degree has little impact on the plaque's fate, compared to its structural features (although nothing actually precludes it from being severely stenotic).2 However, this controversy can be reconciled by considering that plaques with accelerated progression are, in fact, vulnerable plaques, fueled by repeated episodes of hemorrhage and subclinical (healed) rupture and erosion.122
Studies suggesting plaques may alternate between vulnerable and nonvulnerable phenotypes123 cast doubt on the significance of diagnosing plaque vulnerability and, conversely, suggest that nonvulnerable plaques may transition into an unstable morphology and suffer rupture or erosion. The stochastic (by current knowledge), rather than determinate, nature of plaque progression is indirectly supported by the finding that extensive nonobstructive atherosclerosis confers similar risk for MI and sudden death as obstructive, but less‐extensive, disease.124 Additionally, occurrence of clinical events heavily depends on blood and myocardial features (Figure 2); thus, significance of treating rupture prone plaques becomes unclear. However, it is not inconceivable that plaque interdependence may extend well beyond an observed domino‐like effect (a destabilized plaque releasing mediators knocking others—even in remote vascular beds—out of balance).125 More specifically, flow alterations following a silent event or even a successful coronary intervention could inevitably, and currently unpredictably, alter the course of even distant lesions. Finally, the number of nonculprit, asymptomatic, potentially type‐switching plaques may be such, especially in cases of high atherosclerotic burden5 that renders, in combination with the above, all efforts at localized “vulnerability‐guided” treatment, without the ability to accurately assess implications futile, if not dangerous.126
Figure 2.
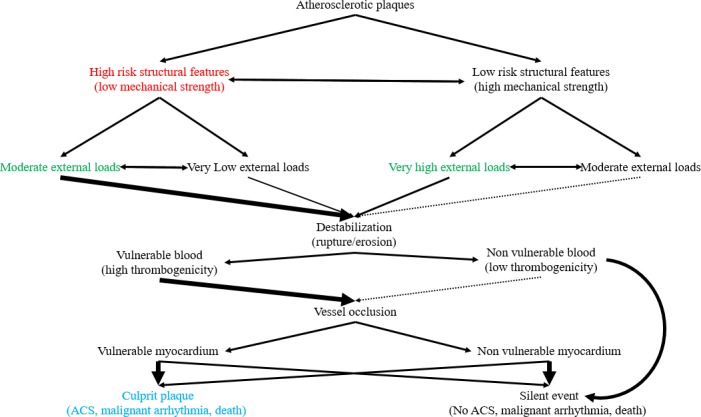
Projected natural course of atherosclerotic plaques based on their intrinsic and extrinsic features. This figure depicts potential evolution of plaques based on an integrative assessment, including external and internal factors, and underlies the necessary paradigm shift in the vulnerable plaque definition. Size and thickness of arrows implies relative probability for a course between the 2 listed in every step. Obviously, the default state, that is, maintenance of the status quo with plaques remaining quiescent, is far more likely in all cases—no arrow denotes certainty for an event. As seen in the diagram, even stable, by all accounts, plaques can rupture/erode. This can be attributed to a phenotype/external stressor shift, an erroneous assessment, or simply an unlikely event given that plaque behavior is, as everything in medicine, the sum of probabilistic, not determinate, events. Additional levels of uncertainty are inserted through the, currently unpredictable, blood and myocardium response to a destabilizing event. Nonvulnerable blood and plaque destabilization combination may also lead to events, albeit rarely. “Silent” events are considered, for the purposes of this figure, both those resolving previous to thrombus formation and those with thrombosis yet remaining clinically undetected. “High” and “low” risk structural features can only be defined relatively, that is, how would the plaque behave in cases of applied loads that can be considered moderate. Obviously, further research is necessary to elucidate these parameters. Color coding: blue—initial definition; red—current perception; green—integrative approach. Double‐headed arrows denote bidirectional processes. ACS indicates acute coronary syndrome.
Despite the above, the apparent shortcomings of the vulnerable plaque hypothesis are not attributed to it having been superseded by our ability to prevent, diagnose, and treat coronary atherosclerosis. Rather, current widespread interpretation and focus on imaging and still‐frame structural features have significantly diverged from the original concept, with regard to which we have only covered half the necessary path.46 More specifically, renewed focus on extrinsic effects on plaques and development of accurate and usable computational models127 will allow for improved assessment of plaques prone to destabilization of any form. Large, prospective, clinical trials will be mandated to acquire relevant data to be used in the development of appropriate vulnerability criteria. Although this will only apply to plaque destabilization, not hard clinical event prognosis, an accurate model incorporating all additional blood, myocardium, and patient‐related parameters (indeed replacing the plaque vulnerability concept) is not expected to be available in the foreseeable future.
Regarding treatment options, it is true that numerous vulnerable plaques usually coexist in patients at risk for cardiovascular events.128 This, in combination with the domino effect during an ACS, potential for plaque morphology alteration with regression of the vulnerable phenotype and presence of further determinants of clinical outcome (vulnerable blood—vulnerable myocardium) argue against isolating and locally treating vulnerable plaques, and in favor for implementing systemic atherosclerosis treatment approaches,129, 130, 131 interpreting presence of these plaques to signify the degree of its progression in terms of natural history. Exceptions to the above may arise in cases of nonculprit destabilized plaque detection during an acute event (potential for domino effect, as suggested by findings of the CvLPRIT [Complete versus Lesion‐only Primary PCI trial] trial132), presence of accelerated plaque progression with increasing stenosis (nonetheless requiring close patient follow‐up), and establishment of clinically applicable criteria for imminent plaque destabilization.114 Thus, in this aspect, more judicious use of targeted localized therapies is warranted, as indeed suggested by vulnerable plaque skeptics.
Current and Future Perspectives
In addition to the classical armamentarium, several newer conceptual and systemic treatment approaches for reducing atherosclerosis burden and preventing its progression are now being actively pursued.
Autophagy is a cellular process affecting both lipid accumulation and inflammation. In its kernel, autophagy describes the process through which cells are able to digest some of their components (ranging in size from signal molecules and lipids to organelles),133 and reclaim usable core materials, such as aminoacids.134 The biological role of autophagy, somewhat counterintuitive, appears to lie not only in ensuring recycling of damaged or degraded molecules, especially long‐lasting ones, but also in allowing cells to regulate processes, such as inflammation, by degrading triggering stimuli, thus ending their effects.135, 136 Master regulators of autophagy include the mammalian target of rapamycin complex and AMP‐activated protein kinase.137 Additional autophagy activating stimuli include endoplasmic reticulum stress (caused by misfolded proteins), ROS, and modified lipids, such as oxidized LDL.138, 139 Reliance of cellular protein renewal on autophagy and inhibitory effects of energy and nutrient abundance on the latter have given rise to the hypothesis that autophagy status plays a crucial role in determining vascular age (vessels being under continuous stress and in need of replacing worn components). Research findings have confirmed the above and consequently provided a plausible basis for the well‐known adage relating reduced caloric intake to longevity. On the other hand, overstimulation of autophagy ultimately leads to cell death and plaque destabilization attributed to destruction of necessary structures to ensure survival.140
In the context of atherosclerosis, dysregulation of autophagy has been described to affect endothelial cells, SMCs, and macrophages.135 More specifically, endothelial cells depend critically on autophagy for maintenance of proper function, and the nitric oxide (NO) system has been shown to rely on presence of normal autophagy levels (increases in endothelial NO synthase).141 Contact of endothelial cells with components of the ECM induces autophagy, and the latter may inhibit neoangiogenesis.142 Effects of autophagy on smooth muscle include inhibition of cell death following exposure to lipid peroxides, thus stabilizing lesions.138 The most significant effects of autophagy on atherosclerosis, however, are related to macrophages. In addition to the established cytoplasmic cholesterol efflux, formation of autophagosomes encircling lipid droplets allows for a second pathway contributing to the removal of lipid material from plaques.143 Interestingly, inflammatory and lipid mediators (interleukin‐6 and oxidized LDL), have been shown to inhibit macrophage autophagy in a vicious circle.144 Indeed, autophagy inhibition and stimulation, often in tandem with apoptosis stimulation and inhibition, have been shown to promote and arrest, respectively, atherosclerotic plaque progression.145
microRNAs (miRNAs) are small, noncoding RNA molecules that act as post‐transcriptional regulators of gene expression.146 In atherosclerotic plaques, alterations in miRNA signatures have been found in all major involved cell types, with macrophage and endothelial‐cell–derived miRNAs promoting plaque destabilization (through promotion of inflammation and angiogenesis, respectively).147 Conversely, SMC miRNA profile may be modulated to increase plaque cap integrity, and thus stability, by either switching them to a less‐synthetic, more‐contractile phenotype, improving plaque mechanical strength, or inducing increased production of collagen to increase fibrous cap thickness.148 Modulation involves both upregulation (by administration of miRNA mimics) and downregulation (through use of antagomirs and miRNA sponges). Notable, the miRNA approach is the only one able to address all aspects of plaque destabilization attributed to miRNA involvement in all phases of atherosclerosis, efficiency of the miRNA gene expression control mechanism, and the potential to include multiple miRNAs in a single vector. Finally, yet other miRNAs have been shown to act by affecting autophagy149 and be upregulated in areas of increased endothelial shear stress. Despite several more‐promising in vitro and animal studies,150 clinical application of miRNA manipulation is currently impeded by lack of adequate vectors and off‐target effects.
The notion of cell senescence is also being strongly implicated in atherosclerosis in current studies. In principle, senescence is a ubiquitous biological process underlying not only biological ageing and tissue degeneration, but tissue remodeling as well (removal of differentiated cells with limited functional potential and replacement by other from the progenitor cell pool).151 Thus, the established fact of (biological) age being the strongest predictor and risk factor for vascular disease is put into perspective. Broadly, cell senescence can be classified as replicative (associated with telomere shortening), stress‐induced premature senescence (not associated with telomeres per se, but exhibiting the same biochemical changes arresting cell‐cycle progression), and oncogene induced, with only the former 2 involved in atherosclerosis progression. In addition to changes in telomeres, chromatin structure (switch to heterochromatin—transcriptionally quiescent), and inhibition of cell‐cycle completion promoting proteins (mainly cyclin‐dependent kinases), senescent cells display a characteristic secretory profile, including specific cytokines.152, 153 Although, in principle, a method to promote senescent cell clearance by phagocytosis (efferocytosis) and replacement, long‐term persistence of this secretory phenotype leads to disproportional proliferation, angiogenesis, endothelial dysfunction, and inflammatory cell recruitment.151
In general, the role of senescence in atherosclerosis and plaque vulnerability appears to be related to both exhaustion of replicative potential of cells attributed to constant need for replenishment of disease‐related losses and premature induction of functional handicaps (cell‐cycle arrest) not only precluding self‐renewal (autophagy and synthesis of components), but also inducing a noxious secretory phenotype. Senescence has been found to occur in both SMCs and endothelial cells, rendering them dysfunctional. Verily, presence of senescent SMCs has been linked to atherogenesis and presence of unstable plaque features, such as large necrotic core and thinner fibrous cap (potentially mediated by senescence‐related cytokines),154 whereas senescent endothelial cells are characterized by both NO synthase uncoupling and over‐response to inflammatory stimuli.155
Significant advances in the understanding of inflammation have led to the notion that inflammation abatement is an active, rather than passive, process.156 Specialized proresolving mediators (SPMs) are a group of structurally related signaling molecules derived from polyunsaturated fatty acids acted upon by 1 or a combination of lipoxygenase, cyclooxygenase, and cytochrome P450 monooxygenase enzymes (thus nonselective inhibition of lipoxygenase may not always be beneficial). Main subclasses are resolvins, maresins, protectins, and lipoxins. More broadly, they are considered autacoids (αὑτός+ἀκω, “self‐heal”), hormones produced, acting, and catabolized locally. It appears that they constitute the second part of a negative feedback loop, being produced by inflammatory cells and acting on them and other cells in the tissue vicinity to promote healing, including efferocytosis stimulation,157 and quench proinflammatory stimuli. Moreover, SPMs initiate a positive feedback loop for the firm induction and progression of the reparatory process. In atherosclerotic plaques, in accord with the concept of chronic inflammatory activation, plaque progression and assumption of a vulnerable phenotype are both related to increased levels of proinflammatory lipid mediators (such as leukotriene B4) and decreased levels of SPMs,158 evidencing that, in chronicity, inflammation has gone awry. Thus, inflammation control should involve not only suppression of promoting pathways, as is the current treatment paradigm in atherosclerosis, by means of medications (eg, statins), but also reinforcement of those promoting healing and dampening of inflammatory responses. Recent research has established that exogenous administration of a cocktail of SPMs does indeed lead to plaque stabilization by halting further engorgement of the necrotic core, leading to increases in the numbers of SMCs and cap thickness and altering macrophage phenotype to a reparatory one (M2).158
Further pathways of interest for research and putative significance for the natural history of atherosclerosis have been identified. Pyroptosis is among them and refers to a form of proinflammatory cell death of immune cells, following detection of foreign antigens within, whereupon cells commit suicide while releasing immune‐stimulatory molecules.159 Differences from apoptosis include dependence on caspase 1 activation and formation of the inflammasome (as opposed to caspases 8 and 9 and apoptosome, respectively). The difference from necroptosis lies in the occurrence in immune (as opposed to tissue) cells. Studies have shown that intracellular oxidized LDL may act as an inductor of macrophage pyroptosis, through a mitochondrial‐DNA–dependent pathway. Although apoptosis, necroptosis, and autophagy have been reported in atherosclerosis, it is entirely possible that pyroptosis of immune cells is also involved (and thus apoptotic foam cells are, at least in part, actually pyroptotic cells), more dynamically contributing to plaque destabilization.160 On more‐conventional grounds, generalized immunosuppressive treatment has been implemented with favorable results whereas therapies targeted to specific components (eg, inhibition of MMPs) have yielded encouraging results.
Conclusions
The concept of vulnerable plaque was developed with the intent of formally defining and describing atherosclerotic plaques having caused acute clinical events (a posteriori). Need for clinical applicability led to definition shifting toward plaques prone to causing events and by extension prone to destabilization (rupture, erosion). Most subsequent clinical studies further narrowed the scope by focusing on intrinsic (structural) determinants of instability, such as quantitative and qualitative composition and remodeling. Attributed to the neglect accorded to effects of extrinsic stressors (eg, dynamic pressure/pressure head, turbulent/laminar flow; see Figure 1), and the routine use of a single imaging modality in plaque assessment, this led to plaques considered nonvulnerable, under the current concept, to be found causing events.
Despite the above, the vulnerable plaque concept has led to tremendous advances in our understanding of pathogenesis and treatment of atherosclerosis, along with substantial improvement of the relevant technological armamentarium. Results from newer studies and trials appear to suggest that a change of approach to the vulnerable plaque concept is mandated to further improve patient diagnosis and treatment. However, a return to the fundamentals of the vulnerable plaque hypothesis appears more appropriate (Table 3).
Table 3.
Vulnerable Plaque Conundrum
| What it originally meant |
| A plaque that is the culprit lesion for an acute coronary event |
| What it should mean |
| A plaque that is prone to rupture when all intrinsic and extrinsic effects are taken into account (regardless of structure) |
| How it is currently interpreted in the majority of literature |
| A plaque with specific morphological features—usually referring exclusively to thin cap fibroatheromas |
This table attempts to summarize the different connotations inherent in the term “vulnerable plaque.” It can be appreciated that a teleological approach has shifted toward a utilitarian one, allowing for easier classification of plaques as “vulnerable” or not, yet depriving this characterization of prognostic implications.
In particular, improvement in the assessment of the complex interplay between intrinsic and extrinsic factors mentioned above (Figure 1) by use of advanced computational models is crucial in more accurately determining which plaques are truly vulnerable161 and thus allow for a more‐accurate estimate of their “natural future” (Figure 2). Their detection should be interpreted as a marker of advanced atherosclerosis progression, in the context of such systemic alterations as deranged autophagy, SPM and noncoding RNA regulation, and treated both aggressively and systematically.
The fundamental question whether and under what circumstances localized treatment of plaques should be pursued is further complicated by two factors: The ability of plaques to alternate between different structural phenotypes (thus changing their location in the Figure 1 curve) and plaque interdependence, as described above. It could be safely said, based on large trials' evidence, that treating current (nonculprit) vulnerable plaques does not affect disease progression and patient course. However, whether this will hold true in the future, with integrative approaches, incorporating advanced computational models, and changing the working definition of vulnerability, cannot be at present determined, pending development of such tools and related prospective trials.
The vulnerable blood and vulnerable myocardium concepts should be brought back into focus to allow for assessment of the vulnerable patient by determining which plaque‐blood‐myocardium combinations are prone to leading to catastrophic events (that may actually be arrhythmic rather than ischemic). On the other hand, not all silent events are innocent: It is conceivable that they might alter, by mechanisms discussed previously, the course of other plaques, in the long term—although at present this is merely conjectural and hypothesis generating.
In conclusion, although the vulnerable plaque is a valid concept in principle, holding great promise for future research, current interpretation of (incomplete) data for vulnerability assessment has rendered it inaccurate. Resources could be more effectively redirected toward large‐scale efforts to reduce the epidemiological atherosclerosis burden, for which there is solid evidence for improving life expectancy and quality, and also toward developing more‐sophisticated models for an integrative approach to plaque assessment. In any case, it should be kept in mind that “preventing is better than treating.” Thus, guided localized treatment of plaques, rather than systemic approach to patients, is, in the great majority of cases and given lack of reliable simulation models, not advisable at present for reasons of effectiveness and safety.
Disclosures
None.
J Am Heart Assoc. 2017;6:e005543. DOI: 10.1161/JAHA.117.005543.
References
- 1. WHO . Cardiovascular diseases (CVDs). Fact sheet ‐ Reviewed June. 2016.
- 2. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P, Pasterkamp G, Fayad Z, Stone PH, Waxman S, Raggi P, Madjid M, Zarrabi A, Burke A, Yuan C, Fitzgerald PJ, Siscovick DS, de Korte CL, Aikawa M, Airaksinen KE, Assmann G, Becker CR, Chesebro JH, Farb A, Galis ZS, Jackson C, Jang IK, Koenig W, Lodder RA, March K, Demirovic J, Navab M, Priori SG, Rekhter MD, Bahr R, Grundy SM, Mehran R, Colombo A, Boerwinkle E, Ballantyne C, Insull W Jr, Schwartz RS, Vogel R, Serruys PW, Hansson GK, Faxon DP, Kaul S, Drexler H, Greenland P, Muller JE, Virmani R, Ridker PM, Zipes DP, Shah PK, Willerson JT. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part II. Circulation. 2003;108:1772–1778. [DOI] [PubMed] [Google Scholar]
- 3. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P, Pasterkamp G, Fayad Z, Stone PH, Waxman S, Raggi P, Madjid M, Zarrabi A, Burke A, Yuan C, Fitzgerald PJ, Siscovick DS, de Korte CL, Aikawa M, Juhani Airaksinen KE, Assmann G, Becker CR, Chesebro JH, Farb A, Galis ZS, Jackson C, Jang IK, Koenig W, Lodder RA, March K, Demirovic J, Navab M, Priori SG, Rekhter MD, Bahr R, Grundy SM, Mehran R, Colombo A, Boerwinkle E, Ballantyne C, Insull W Jr, Schwartz RS, Vogel R, Serruys PW, Hansson GK, Faxon DP, Kaul S, Drexler H, Greenland P, Muller JE, Virmani R, Ridker PM, Zipes DP, Shah PK, Willerson JT. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circulation. 2003;108:1664–1672. [DOI] [PubMed] [Google Scholar]
- 4. Bourantas CV, Garcia‐Garcia HM, Farooq V, Maehara A, Xu K, Genereux P, Diletti R, Muramatsu T, Fahy M, Weisz G, Stone GW, Serruys PW. Clinical and angiographic characteristics of patients likely to have vulnerable plaques: analysis from the PROSPECT study. JACC Cardiovasc Imaging. 2013;6:1263–1272. [DOI] [PubMed] [Google Scholar]
- 5. Arbab‐Zadeh A, Fuster V. The myth of the “vulnerable plaque”: transitioning from a focus on individual lesions to atherosclerotic disease burden for coronary artery disease risk assessment. J Am Coll Cardiol. 2015;65:846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bourantas CV, Garcia‐Garcia HM, Torii R, Zhang YJ, Westwood M, Crake T, Serruys PW. Vulnerable plaque detection: an unrealistic quest or a feasible objective with a clinical value? Heart. 2016;102:581–589. [DOI] [PubMed] [Google Scholar]
- 7. Libby P, Pasterkamp G. Requiem for the ‘vulnerable plaque’. Eur Heart J. 2015;36:2984–2987. [DOI] [PubMed] [Google Scholar]
- 8. Zheng B, Mintz GS, McPherson JA, De Bruyne B, Farhat NZ, Marso SP, Serruys PW, Stone GW, Maehara A. Predictors of plaque rupture within nonculprit fibroatheromas in patients with acute coronary syndromes: the prospect study. JACC Cardiovasc Imaging. 2015;8:1180–1187. [DOI] [PubMed] [Google Scholar]
- 9. Tominaga J, Fukunaga Y, Abelardo E, Nagafuchi A. Defining the function of beta‐catenin tyrosine phosphorylation in cadherin‐mediated cell‐cell adhesion. Genes Cells. 2008;13:67–77. [DOI] [PubMed] [Google Scholar]
- 10. Collins NT, Cummins PM, Colgan OC, Ferguson G, Birney YA, Murphy RP, Meade G, Cahill PA. Cyclic strain‐mediated regulation of vascular endothelial occludin and ZO‐1: influence on intercellular tight junction assembly and function. Arterioscler Thromb Vasc Biol. 2006;26:62–68. [DOI] [PubMed] [Google Scholar]
- 11. Jufri NF, Mohamedali A, Avolio A, Baker MS. Mechanical stretch: physiological and pathological implications for human vascular endothelial cells. Vasc Cell. 2015;7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheruvu PK, Finn AV, Gardner C, Caplan J, Goldstein J, Stone GW, Virmani R, Muller JE. Frequency and distribution of thin‐cap fibroatheroma and ruptured plaques in human coronary arteries: a pathologic study. J Am Coll Cardiol. 2007;50:940–949. [DOI] [PubMed] [Google Scholar]
- 13. Saybolt MD, Lilly SM, Patel D, Hamamdzic D, Llano R, Fenning RS, Madden S, Wilensky RL. The vulnerable artery: early and rapid deposition of lipid in coronary arteries is associated with subsequent development of thin‐cap fibroatheromas. EuroIntervention. 2016;11:e1612–e1618. [DOI] [PubMed] [Google Scholar]
- 14. Janoudi A, Shamoun FE, Kalavakunta JK, Abela GS. Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque. Eur Heart J. 2016;37:1959–1967. [DOI] [PubMed] [Google Scholar]
- 15. Valente AJ, Irimpen AM, Siebenlist U, Chandrasekar B. OxLDL induces endothelial dysfunction and death via TRAF3IP2: inhibition by HDL3 and AMPK activators. Free Radic Biol Med. 2014;70:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karunakaran D, Geoffrion M, Wei L, Gan W, Richards L, Shangari P, DeKemp EM, Beanlands RA, Perisic L, Maegdefessel L, Hedin U, Sad S, Guo L, Kolodgie FD, Virmani R, Ruddy T, Rayner KJ. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci Adv. 2016;2:e1600224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. [DOI] [PubMed] [Google Scholar]
- 18. Banach M. Lipoprotein (a)‐we know so much yet still have much to learn. J Am Heart Assoc. 2016;5:e003597 DOI: 10.1161/jaha.116.003597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Niccoli G, Cin D, Scalone G, Panebianco M, Abbolito S, Cosentino N, Jacoangeli F, Refaat H, Gallo G, Salerno G, Volpe M, Crea F, De Biase L. Lipoprotein (a) is related to coronary atherosclerotic burden and a vulnerable plaque phenotype in angiographically obstructive coronary artery disease. Atherosclerosis. 2016;246:214–220. [DOI] [PubMed] [Google Scholar]
- 20. Sander D, Winbeck K, Klingelhofer J, Etgen T, Conrad B. Progression of early carotid atherosclerosis is only temporarily reduced after antibiotic treatment of Chlamydia pneumoniae seropositivity. Circulation. 2004;109:1010–1015. [DOI] [PubMed] [Google Scholar]
- 21. Madjid M, Willerson JT, Casscells SW. Intracoronary thermography for detection of high‐risk vulnerable plaques. J Am Coll Cardiol. 2006;47:C80–C85. [DOI] [PubMed] [Google Scholar]
- 22. Tenaglia AN, Peters KG, Sketch MH Jr, Annex BH. Neovascularization in atherectomy specimens from patients with unstable angina: implications for pathogenesis of unstable angina. Am Heart J. 1998;135:10–14. [DOI] [PubMed] [Google Scholar]
- 23. Ohayon J, Finet G, Le Floc'h S, Cloutier G, Gharib AM, Heroux J, Pettigrew RI. Biomechanics of atherosclerotic coronary plaque: site, stability and in vivo elasticity modeling. Ann Biomed Eng. 2014;42:269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Varnava AM, Mills PG, Davies MJ. Relationship between coronary artery remodeling and plaque vulnerability. Circulation. 2002;105:939–943. [DOI] [PubMed] [Google Scholar]
- 25. Ota H, Magalhaes MA, Torguson R, Negi S, Kollmer MR, Spad MA, Gai J, Satler LF, Suddath WO, Pichard AD, Waksman R. The influence of lipid‐containing plaque composition assessed by near‐infrared spectroscopy on coronary lesion remodelling. Eur Heart J Cardiovasc Imaging. 2016;17:821–831. [DOI] [PubMed] [Google Scholar]
- 26. Pei X, Nadkarni S, Li ZY. A parametric study of inflammatory effects on plaque mechanical stress. Int J Cardiol. 2016;205:157–159. [DOI] [PubMed] [Google Scholar]
- 27. Nie X, Laforest R, Elvington A, Randolph GJ, Zheng J, Voller T, Abendschein DR, Lapi S, Woodard PK. PET/MR imaging of hypoxic atherosclerosis using 64Cu‐ATSM in a rabbit model. J Nucl Med. 2016;57:2006–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Taruya A, Tanaka A, Nishiguchi T, Matsuo Y, Ozaki Y, Kashiwagi M, Shiono Y, Orii M, Yamano T, Ino Y, Hirata K, Kubo T, Akasaka T. Vasa vasorum restructuring in human atherosclerotic plaque vulnerability: a clinical optical coherence tomography study. J Am Coll Cardiol. 2015;65:2469–2477. [DOI] [PubMed] [Google Scholar]
- 29. Doyle B, Caplice N. Plaque neovascularization and antiangiogenic therapy for atherosclerosis. J Am Coll Cardiol. 2007;49:2073–2080. [DOI] [PubMed] [Google Scholar]
- 30. Yokoya K, Takatsu H, Suzuki T, Hosokawa H, Ojio S, Matsubara T, Tanaka T, Watanabe S, Morita N, Nishigaki K, Takemura G, Noda T, Minatoguchi S, Fujiwara H. Process of progression of coronary artery lesions from mild or moderate stenosis to moderate or severe stenosis: a study based on four serial coronary arteriograms per year. Circulation. 1999;100:903–909. [DOI] [PubMed] [Google Scholar]
- 31. Rajamannan NM, Nealis TB, Subramaniam M, Pandya S, Stock SR, Ignatiev CI, Sebo TJ, Rosengart TK, Edwards WD, McCarthy PM, Bonow RO, Spelsberg TC. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast‐like bone formation. Circulation. 2005;111:3296–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–386. [DOI] [PubMed] [Google Scholar]
- 33. Hjortnaes J, Butcher J, Figueiredo JL, Riccio M, Kohler RH, Kozloff KM, Weissleder R, Aikawa E. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur Heart J. 2010;31:1975–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Collin J, Gossl M, Matsuo Y, Cilluffo RR, Flammer AJ, Loeffler D, Lennon RJ, Simari RD, Spoon DB, Erbel R, Lerman LO, Khosla S, Lerman A. Osteogenic monocytes within the coronary circulation and their association with plaque vulnerability in patients with early atherosclerosis. Int J Cardiol. 2015;181:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maldonado N, Kelly‐Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, Cardoso L, Weinbaum S. A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: potential implications for plaque rupture. Am J Physiol Heart Circ Physiol. 2012;303:H619–H628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tanaka A, Imanishi T, Kitabata H, Kubo T, Takarada S, Tanimoto T, Kuroi A, Tsujioka H, Ikejima H, Ueno S, Kataiwa H, Okouchi K, Kashiwaghi M, Matsumoto H, Takemoto K, Nakamura N, Hirata K, Mizukoshi M, Akasaka T. Morphology of exertion‐triggered plaque rupture in patients with acute coronary syndrome: an optical coherence tomography study. Circulation. 2008;118:2368–2373. [DOI] [PubMed] [Google Scholar]
- 37. Jia H, Dai J, Hou J, Xing L, Ma L, Liu H, Xu M, Yao Y, Hu S, Yamamoto E, Lee H, Zhang S, Yu B, Jang IK. Effective anti‐thrombotic therapy without stenting: intravascular optical coherence tomography‐based management in plaque erosion (the EROSION study). Eur Heart J. 2016; DOI: 10.1093/eurheartj/ehw381. Available at: https://academic.oup.com/eurheartj/article-abstract/doi/10.1093/eurheartj/ehw381/2661754/Effective-anti-thrombotic-therapy-without-stenting?redirectedFrom=fulltext. Accessed February 11, 2017. [DOI] [PubMed] [Google Scholar]
- 38. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852–1866. [DOI] [PubMed] [Google Scholar]
- 39. Kramer MC, Rittersma SZ, de Winter RJ, Ladich ER, Fowler DR, Liang YH, Kutys R, Carter‐Monroe N, Kolodgie FD, van der Wal AC, Virmani R. Relationship of thrombus healing to underlying plaque morphology in sudden coronary death. J Am Coll Cardiol. 2010;55:122–132. [DOI] [PubMed] [Google Scholar]
- 40. Lokeshwar VB, Selzer MG. Differences in hyaluronic acid‐mediated functions and signaling in arterial, microvessel, and vein‐derived human endothelial cells. J Biol Chem. 2000;275:27641–27649. [DOI] [PubMed] [Google Scholar]
- 41. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–C18. [DOI] [PubMed] [Google Scholar]
- 42. Quillard T, Araujo HA, Franck G, Shvartz E, Sukhova G, Libby P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J. 2015;36:1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koshiishi I, Shizari M, Underhill CB. CD44 can mediate the adhesion of platelets to hyaluronan. Blood. 1994;84:390–396. [PubMed] [Google Scholar]
- 44. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. [DOI] [PubMed] [Google Scholar]
- 45. Ferrante G, Nakano M, Prati F, Niccoli G, Mallus MT, Ramazzotti V, Montone RA, Kolodgie FD, Virmani R, Crea F. High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: a clinicopathological study. Circulation. 2010;122:2505–2513. [DOI] [PubMed] [Google Scholar]
- 46. Toutouzas K, Benetos G, Karanasos A, Chatzizisis YS, Giannopoulos AA, Tousoulis D. Vulnerable plaque imaging: updates on new pathobiological mechanisms. Eur Heart J. 2015;36:3147–3154. [DOI] [PubMed] [Google Scholar]
- 47. Liang X, Xenos M, Alemu Y, Rambhia SH, Lavi I, Kornowski R, Gruberg L, Fuchs S, Einav S, Bluestein D. Biomechanical factors in coronary vulnerable plaque risk of rupture: intravascular ultrasound‐based patient‐specific fluid‐structure interaction studies. Coron Artery Dis. 2013;24:75–87. [DOI] [PubMed] [Google Scholar]
- 48. Tuenter A, Selwaness M, Arias Lorza A, Schuurbiers JC, Speelman L, Cibis M, van der Lugt A, de Bruijne M, van der Steen AF, Franco OH, Vernooij MW, Wentzel JJ. High shear stress relates to intraplaque haemorrhage in asymptomatic carotid plaques. Atherosclerosis. 2016;251:348–354. [DOI] [PubMed] [Google Scholar]
- 49. Wentzel JJ, Schuurbiers JC, Gonzalo Lopez N, Gijsen FJ, van der Giessen AG, Groen HC, Dijkstra J, Garcia‐Garcia HM, Serruys PW. In vivo assessment of the relationship between shear stress and necrotic core in early and advanced coronary artery disease. EuroIntervention. 2013;9:989–995; discussion 995. [DOI] [PubMed] [Google Scholar]
- 50. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. [DOI] [PubMed] [Google Scholar]
- 51. Huang X, Yang C, Zheng J, Bach R, Muccigrosso D, Woodard PK, Tang D. 3D MRI‐based multicomponent thin layer structure only plaque models for atherosclerotic plaques. J Biomech. 2016;49:2726–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brown AJ, Teng Z, Evans PC, Gillard JH, Samady H, Bennett MR. Role of biomechanical forces in the natural history of coronary atherosclerosis. Nat Rev Cardiol. 2016;13:210–220. [DOI] [PubMed] [Google Scholar]
- 53. Ohayon J, Teppaz P, Finet G, Rioufol G. In‐vivo prediction of human coronary plaque rupture location using intravascular ultrasound and the finite element method. Coron Artery Dis. 2001;12:655–663. [DOI] [PubMed] [Google Scholar]
- 54. Teng Z, Brown AJ, Calvert PA, Parker RA, Obaid DR, Huang Y, Hoole SP, West NE, Gillard JH, Bennett MR. Coronary plaque structural stress is associated with plaque composition and subtype and higher in acute coronary syndrome: the BEACON I (biomechanical evaluation of atheromatous coronary arteries) study. Circ Cardiovasc Imaging. 2014;7:461–470. [DOI] [PubMed] [Google Scholar]
- 55. Finet G, Ohayon J, Rioufol G. Biomechanical interaction between cap thickness, lipid core composition and blood pressure in vulnerable coronary plaque: impact on stability or instability. Coron Artery Dis. 2004;15:13–20. [DOI] [PubMed] [Google Scholar]
- 56. Ohayon J, Finet G, Gharib AM, Herzka DA, Tracqui P, Heroux J, Rioufol G, Kotys MS, Elagha A, Pettigrew RI. Necrotic core thickness and positive arterial remodeling index: emergent biomechanical factors for evaluating the risk of plaque rupture. Am J Physiol Heart Circ Physiol. 2008;295:H717–H727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ambrose JA, Tannenbaum MA, Alexopoulos D, Hjemdahl‐Monsen CE, Leavy J, Weiss M, Borrico S, Gorlin R, Fuster V. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol. 1988;12:56–62. [DOI] [PubMed] [Google Scholar]
- 58. Arbustini E, Grasso M, Diegoli M, Pucci A, Bramerio M, Ardissino D, Angoli L, de Servi S, Bramucci E, Mussini A, Minzioni G, Vigano M, Specchia G. Coronary atherosclerotic plaques with and without thrombus in ischemic heart syndromes: a morphologic, immunohistochemical, and biochemical study. Am J Cardiol. 1991;68:36B–50B. [DOI] [PubMed] [Google Scholar]
- 59. Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–671. [DOI] [PubMed] [Google Scholar]
- 60. Davies MJ. A macro and micro view of coronary vascular insult in ischemic heart disease. Circulation. 1990;82:II38–II46. [PubMed] [Google Scholar]
- 61. Kolodgie FD, Burke AP, Farb A, Gold HK, Yuan J, Narula J, Finn AV, Virmani R. The thin‐cap fibroatheroma: a type of vulnerable plaque: the major precursor lesion to acute coronary syndromes. Curr Opin Cardiol. 2001;16:285–292. [DOI] [PubMed] [Google Scholar]
- 62. Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336:1276–1282. [DOI] [PubMed] [Google Scholar]
- 63. Garcia‐Garcia HM, Gogas BD, Serruys PW, Bruining N. IVUS‐based imaging modalities for tissue characterization: similarities and differences. Int J Cardiovasc Imaging. 2011;27:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rodriguez‐Granillo GA, Garcia‐Garcia HM, Mc Fadden EP, Valgimigli M, Aoki J, de Feyter P, Serruys PW. In vivo intravascular ultrasound‐derived thin‐cap fibroatheroma detection using ultrasound radiofrequency data analysis. J Am Coll Cardiol. 2005;46:2038–2042. [DOI] [PubMed] [Google Scholar]
- 65. Kume T, Okura H, Yamada R, Kawamoto T, Watanabe N, Neishi Y, Sadahira Y, Akasaka T, Yoshida K. Frequency and spatial distribution of thin‐cap fibroatheroma assessed by 3‐vessel intravascular ultrasound and optical coherence tomography: an ex vivo validation and an initial in vivo feasibility study. Circ J. 2009;73:1086–1091. [DOI] [PubMed] [Google Scholar]
- 66. Prati F, Regar E, Mintz GS, Arbustini E, Di Mario C, Jang IK, Akasaka T, Costa M, Guagliumi G, Grube E, Ozaki Y, Pinto F, Serruys PW. Expert review document on methodology, terminology, and clinical applications of optical coherence tomography: physical principles, methodology of image acquisition, and clinical application for assessment of coronary arteries and atherosclerosis. Eur Heart J. 2010;31:401–415. [DOI] [PubMed] [Google Scholar]
- 67. Yabushita H, Bouma BE, Houser SL, Aretz HT, Jang IK, Schlendorf KH, Kauffman CR, Shishkov M, Kang DH, Halpern EF, Tearney GJ. Characterization of human atherosclerosis by optical coherence tomography. Circulation. 2002;106:1640–1645. [DOI] [PubMed] [Google Scholar]
- 68. Sinclair H, Bourantas C, Bagnall A, Mintz GS, Kunadian V. OCT for the identification of vulnerable plaque in acute coronary syndrome. JACC Cardiovasc Imaging. 2015;8:198–209. [DOI] [PubMed] [Google Scholar]
- 69. Yonetsu T, Kakuta T, Lee T, Takahashi K, Kawaguchi N, Yamamoto G, Koura K, Hishikari K, Iesaka Y, Fujiwara H, Isobe M. In vivo critical fibrous cap thickness for rupture‐prone coronary plaques assessed by optical coherence tomography. Eur Heart J. 2011;32:1251–1259. [DOI] [PubMed] [Google Scholar]
- 70. Miyamoto Y, Okura H, Kume T, Kawamoto T, Neishi Y, Hayashida A, Yamada R, Imai K, Saito K, Yoshida K. Plaque characteristics of thin‐cap fibroatheroma evaluated by OCT and IVUS. JACC Cardiovasc Imaging. 2011;4:638–646. [DOI] [PubMed] [Google Scholar]
- 71. Farb A, Burke AP, Tang AL, Liang TY, Mannan P, Smialek J, Virmani R. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93:1354–1363. [DOI] [PubMed] [Google Scholar]
- 72. Muller JE, Abela GS, Nesto RW, Tofler GH. Triggers, acute risk factors and vulnerable plaques: the lexicon of a new frontier. J Am Coll Cardiol. 1994;23:809–813. [DOI] [PubMed] [Google Scholar]
- 73. Garcia‐Garcia HM, Mintz GS, Lerman A, Vince DG, Margolis MP, van Es GA, Morel MA, Nair A, Virmani R, Burke AP, Stone GW, Serruys PW. Tissue characterisation using intravascular radiofrequency data analysis: recommendations for acquisition, analysis, interpretation and reporting. EuroIntervention. 2009;5:177–189. [DOI] [PubMed] [Google Scholar]
- 74. Sathyanarayana S, Carlier S, Li W, Thomas L. Characterisation of atherosclerotic plaque by spectral similarity of radiofrequency intravascular ultrasound signals. EuroIntervention. 2009;5:133–139. [DOI] [PubMed] [Google Scholar]
- 75. Sinclair H, Veerasamy M, Bourantas C, Egred M, Nair A, Calvert PA, Brugaletta S, Mintz GS, Kunadian V. The role of virtual histology intravascular ultrasound in the identification of coronary artery plaque vulnerability in acute coronary syndromes. Cardiol Rev. 2016;24:303–309. [DOI] [PubMed] [Google Scholar]
- 76. Hansen HH, de Borst GJ, Bots ML, Moll FL, Pasterkamp G, de Korte CL. Validation of noninvasive in vivo compound ultrasound strain imaging using histologic plaque vulnerability features. Stroke. 2016;47:2770–2775. [DOI] [PubMed] [Google Scholar]
- 77. Calvert PA, Obaid DR, O'Sullivan M, Shapiro LM, McNab D, Densem CG, Schofield PM, Braganza D, Clarke SC, Ray KK, West NE, Bennett MR. Association between IVUS findings and adverse outcomes in patients with coronary artery disease: the VIVA (VH‐IVUS in Vulnerable Atherosclerosis) Study. JACC Cardiovasc Imaging. 2011;4:894–901. [DOI] [PubMed] [Google Scholar]
- 78. Cheng JM, Garcia‐Garcia HM, de Boer SP, Kardys I, Heo JH, Akkerhuis KM, Oemrawsingh RM, van Domburg RT, Ligthart J, Witberg KT, Regar E, Serruys PW, van Geuns RJ, Boersma E. In vivo detection of high‐risk coronary plaques by radiofrequency intravascular ultrasound and cardiovascular outcome: results of the ATHEROREMO‐IVUS study. Eur Heart J. 2014;35:639–647. [DOI] [PubMed] [Google Scholar]
- 79. Stone GW, Maehara A, Lansky AJ, de Bruyne B, Cristea E, Mintz GS, Mehran R, McPherson J, Farhat N, Marso SP, Parise H, Templin B, White R, Zhang Z, Serruys PW. A prospective natural‐history study of coronary atherosclerosis. N Engl J Med. 2011;364:226–235. [DOI] [PubMed] [Google Scholar]
- 80. Stone PH, Saito S, Takahashi S, Makita Y, Nakamura S, Kawasaki T, Takahashi A, Katsuki T, Nakamura S, Namiki A, Hirohata A, Matsumura T, Yamazaki S, Yokoi H, Tanaka S, Otsuji S, Yoshimachi F, Honye J, Harwood D, Reitman M, Coskun AU, Papafaklis MI, Feldman CL. Prediction of progression of coronary artery disease and clinical outcomes using vascular profiling of endothelial shear stress and arterial plaque characteristics: the PREDICTION Study. Circulation. 2012;126:172–181. [DOI] [PubMed] [Google Scholar]
- 81. Tearney GJ, Yabushita H, Houser SL, Aretz HT, Jang IK, Schlendorf KH, Kauffman CR, Shishkov M, Halpern EF, Bouma BE. Quantification of macrophage content in atherosclerotic plaques by optical coherence tomography. Circulation. 2003;107:113–119. [DOI] [PubMed] [Google Scholar]
- 82. Toutouzas K, Synetos A, Stefanadi E, Vaina S, Markou V, Vavuranakis M, Tsiamis E, Tousoulis D, Stefanadis C. Correlation between morphologic characteristics and local temperature differences in culprit lesions of patients with symptomatic coronary artery disease. J Am Coll Cardiol. 2007;49:2264–2271. [DOI] [PubMed] [Google Scholar]
- 83. Stefanadis C, Diamantopoulos L, Vlachopoulos C, Tsiamis E, Dernellis J, Toutouzas K, Stefanadi E, Toutouzas P. Thermal heterogeneity within human atherosclerotic coronary arteries detected in vivo: a new method of detection by application of a special thermography catheter. Circulation. 1999;99:1965–1971. [DOI] [PubMed] [Google Scholar]
- 84. Stefanadis C, Toutouzas K, Vavuranakis M, Tsiamis E, Vaina S, Toutouzas P. New balloon‐thermography catheter for in vivo temperature measurements in human coronary atherosclerotic plaques: a novel approach for thermography? Catheter Cardiovasc Interv. 2003;58:344–350. [DOI] [PubMed] [Google Scholar]
- 85. Stefanadis C, Toutouzas K, Tsiamis E, Stratos C, Vavuranakis M, Kallikazaros I, Panagiotakos D, Toutouzas P. Increased local temperature in human coronary atherosclerotic plaques: an independent predictor of clinical outcome in patients undergoing a percutaneous coronary intervention. J Am Coll Cardiol. 2001;37:1277–1283. [DOI] [PubMed] [Google Scholar]
- 86. Madder RD, Goldstein JA, Madden SP, Puri R, Wolski K, Hendricks M, Sum ST, Kini A, Sharma S, Rizik D, Brilakis ES, Shunk KA, Petersen J, Weisz G, Virmani R, Nicholls SJ, Maehara A, Mintz GS, Stone GW, Muller JE. Detection by near‐infrared spectroscopy of large lipid core plaques at culprit sites in patients with acute ST‐segment elevation myocardial infarction. JACC Cardiovasc Interv. 2013;6:838–846. [DOI] [PubMed] [Google Scholar]
- 87. Roleder T, Kovacic JC, Ali Z, Sharma R, Cristea E, Moreno P, Sharma SK, Narula J, Kini AS. Combined NIRS and IVUS imaging detects vulnerable plaque using a single catheter system: a head‐to‐head comparison with OCT. EuroIntervention. 2014;10:303–311. [DOI] [PubMed] [Google Scholar]
- 88. Madder RD, Puri R, Muller JE, Harnek J, Gotberg M, VanOosterhout S, Chi M, Wohns D, McNamara R, Wolski K, Madden S, Sidharta S, Andrews J, Nicholls SJ, Erlinge D. Confirmation of the intracoronary near‐infrared spectroscopy threshold of lipid‐rich plaques that underlie ST‐segment‐elevation myocardial infarction. Arterioscler Thromb Vasc Biol. 2016;36:1010–1015. [DOI] [PubMed] [Google Scholar]
- 89. Goldstein JA, Maini B, Dixon SR, Brilakis ES, Grines CL, Rizik DG, Powers ER, Steinberg DH, Shunk KA, Weisz G, Moreno PR, Kini A, Sharma SK, Hendricks MJ, Sum ST, Madden SP, Muller JE, Stone GW, Kern MJ. Detection of lipid‐core plaques by intracoronary near‐infrared spectroscopy identifies high risk of periprocedural myocardial infarction. Circ Cardiovasc Interv. 2011;4:429–437. [DOI] [PubMed] [Google Scholar]
- 90. Pu J, Mintz GS, Brilakis ES, Banerjee S, Abdel‐Karim AR, Maini B, Biro S, Lee JB, Stone GW, Weisz G, Maehara A. In vivo characterization of coronary plaques: novel findings from comparing greyscale and virtual histology intravascular ultrasound and near‐infrared spectroscopy. Eur Heart J. 2012;33:372–383. [DOI] [PubMed] [Google Scholar]
- 91. Oemrawsingh RM, Cheng JM, Garcia‐Garcia HM, van Geuns RJ, de Boer SP, Simsek C, Kardys I, Lenzen MJ, van Domburg RT, Regar E, Serruys PW, Akkerhuis KM, Boersma E. Near‐infrared spectroscopy predicts cardiovascular outcome in patients with coronary artery disease. J Am Coll Cardiol. 2014;64:2510–2518. [DOI] [PubMed] [Google Scholar]
- 92. The Lipid‐Rich Plaque Study (LRP) (NCT02033694) ‐ Principal Investigator: Waksman, R Available at: https://clinicaltrials.gov/ct2/show/NCT02033694?term=NCT02033694&rank=1. Accessed February 11, 2017.
- 93. Weisz G. Color: A prospective, multicenter registry evaluating the relationship between lipid‐rich plaque and two‐year outcomes after stent implantation in patients with coronary artery disease. Presented at the 2016 Transcatheter Cardiovascular Therapeutics (TCT) Congress, Washington, DC. [Google Scholar]
- 94. Psaltis PJ, Nicholls SJ. Imaging: focusing light on the vulnerable plaque. Nat Rev Cardiol. 2016;13:253–255. [DOI] [PubMed] [Google Scholar]
- 95. Ughi GJ, Wang H, Gerbaud E, Gardecki JA, Fard AM, Hamidi E, Vacas‐Jacques P, Rosenberg M, Jaffer FA, Tearney GJ. Clinical characterization of coronary atherosclerosis with dual‐modality OCT and near‐infrared autofluorescence imaging. JACC Cardiovasc Imaging. 2016;9:1304–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rodriguez‐Granillo GA, Carrascosa P, Bruining N, Waksman R, Garcia‐Garcia HM. Defining the non‐vulnerable and vulnerable patients with computed tomography coronary angiography: evaluation of atherosclerotic plaque burden and composition. Eur Heart J Cardiovasc Imaging. 2016;17:481–491. [DOI] [PubMed] [Google Scholar]
- 97. Achenbach S, Boehmer K, Pflederer T, Ropers D, Seltmann M, Lell M, Anders K, Kuettner A, Uder M, Daniel WG, Marwan M. Influence of slice thickness and reconstruction kernel on the computed tomographic attenuation of coronary atherosclerotic plaque. J Cardiovasc Comput Tomogr. 2010;4:110–115. [DOI] [PubMed] [Google Scholar]
- 98. Yu L, Leng S, McCollough CH. Dual‐energy CT‐based monochromatic imaging. AJR Am J Roentgenol. 2012;199:S9–S15. [DOI] [PubMed] [Google Scholar]
- 99. Wang JC, Normand SL, Mauri L, Kuntz RE. Coronary artery spatial distribution of acute myocardial infarction occlusions. Circulation. 2004;110:278–284. [DOI] [PubMed] [Google Scholar]
- 100. Voros S, Rinehart S, Qian Z, Joshi P, Vazquez G, Fischer C, Belur P, Hulten E, Villines TC. Coronary atherosclerosis imaging by coronary CT angiography: current status, correlation with intravascular interrogation and meta‐analysis. JACC Cardiovasc Imaging. 2011;4:537–548. [DOI] [PubMed] [Google Scholar]
- 101. Andreini D, Pontone G, Mushtaq S, Bartorelli AL, Bertella E, Antonioli L, Formenti A, Cortinovis S, Veglia F, Annoni A, Agostoni P, Montorsi P, Ballerini G, Fiorentini C, Pepi M. A long‐term prognostic value of coronary CT angiography in suspected coronary artery disease. JACC Cardiovasc Imaging. 2012;5:690–701. [DOI] [PubMed] [Google Scholar]
- 102. Motoyama S, Ito H, Sarai M, Kondo T, Kawai H, Nagahara Y, Harigaya H, Kan S, Anno H, Takahashi H, Naruse H, Ishii J, Hecht H, Shaw LJ, Ozaki Y, Narula J. Plaque characterization by coronary computed tomography angiography and the likelihood of acute coronary events in mid‐term follow‐up. J Am Coll Cardiol. 2015;66:337–346. [DOI] [PubMed] [Google Scholar]
- 103. Choi BJ, Kang DK, Tahk SJ, Choi SY, Yoon MH, Lim HS, Kang SJ, Yang HM, Park JS, Zheng M, Hwang GS, Shin JH. Comparison of 64‐slice multidetector computed tomography with spectral analysis of intravascular ultrasound backscatter signals for characterizations of noncalcified coronary arterial plaques. Am J Cardiol. 2008;102:988–993. [DOI] [PubMed] [Google Scholar]
- 104. Kashiwagi M, Tanaka A, Kitabata H, Tsujioka H, Kataiwa H, Komukai K, Tanimoto T, Takemoto K, Takarada S, Kubo T, Hirata K, Nakamura N, Mizukoshi M, Imanishi T, Akasaka T. Feasibility of noninvasive assessment of thin‐cap fibroatheroma by multidetector computed tomography. JACC Cardiovasc Imaging. 2009;2:1412–1419. [DOI] [PubMed] [Google Scholar]
- 105. Conte E, Annoni A, Pontone G, Mushtaq S, Guglielmo M, Baggiano A, Volpato V, Agalbato C, Bonomi A, Veglia F, Formenti A, Fiorentini C, Bartorelli AL, Pepi M, Andreini D. Evaluation of coronary plaque characteristics with coronary computed tomography angiography in patients with non‐obstructive coronary artery disease: a long‐term follow‐up study. Eur Heart J Cardiovasc Imaging. 2016; DOI: 10.1093/ehjci/jew200. Available at: https://academic.oup.com/ehjcimaging/articleabstract/doi/10.1093/ehjci/jew200/3060682/Evaluation-of-coronary-plaque-characteristics-with?redirectedFrom=fulltext. Accessed February 11, 2017. [DOI] [PubMed] [Google Scholar]
- 106. Dwivedi G, Liu Y, Tewari S, Inacio J, Pelletier‐Galarneau M, Chow BJ. Incremental prognostic value of quantified vulnerable plaque by cardiac computed tomography: a pilot study. J Thorac Imaging. 2016;31:373–379. [DOI] [PubMed] [Google Scholar]
- 107. Motoyama S, Sarai M, Harigaya H, Anno H, Inoue K, Hara T, Naruse H, Ishii J, Hishida H, Wong ND, Virmani R, Kondo T, Ozaki Y, Narula J. Computed tomographic angiography characteristics of atherosclerotic plaques subsequently resulting in acute coronary syndrome. J Am Coll Cardiol. 2009;54:49–57. [DOI] [PubMed] [Google Scholar]
- 108. Otsuka K, Fukuda S, Tanaka A, Nakanishi K, Taguchi H, Yoshikawa J, Shimada K, Yoshiyama M. Napkin‐ring sign on coronary CT angiography for the prediction of acute coronary syndrome. JACC Cardiovasc Imaging. 2013;6:448–457. [DOI] [PubMed] [Google Scholar]
- 109. Detrano R, Guerci AD, Carr JJ, Bild DE, Burke G, Folsom AR, Liu K, Shea S, Szklo M, Bluemke DA, O'Leary DH, Tracy R, Watson K, Wong ND, Kronmal RA. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358:1336–1345. [DOI] [PubMed] [Google Scholar]
- 110. Hou ZH, Lu B, Gao Y, Jiang SL, Wang Y, Li W, Budoff MJ. Prognostic value of coronary CT angiography and calcium score for major adverse cardiac events in outpatients. JACC Cardiovasc Imaging. 2012;5:990–999. [DOI] [PubMed] [Google Scholar]
- 111. Versteylen MO, Kietselaer BL, Dagnelie PC, Joosen IA, Dedic A, Raaijmakers RH, Wildberger JE, Nieman K, Crijns HJ, Niessen WJ, Daemen MJ, Hofstra L. Additive value of semiautomated quantification of coronary artery disease using cardiac computed tomographic angiography to predict future acute coronary syndrome. J Am Coll Cardiol. 2013;61:2296–2305. [DOI] [PubMed] [Google Scholar]
- 112. Cheezum MK, Hulten EA, Smith RM, Taylor AJ, Kircher J, Surry L, York M, Villines TC. Changes in preventive medical therapies and CV risk factors after CT angiography. JACC Cardiovasc Imaging. 2013;6:574–581. [DOI] [PubMed] [Google Scholar]
- 113. Norgaard BL, Leipsic J, Gaur S, Seneviratne S, Ko BS, Ito H, Jensen JM, Mauri L, De Bruyne B, Bezerra H, Osawa K, Marwan M, Naber C, Erglis A, Park SJ, Christiansen EH, Kaltoft A, Lassen JF, Botker HE, Achenbach S. Diagnostic performance of noninvasive fractional flow reserve derived from coronary computed tomography angiography in suspected coronary artery disease: the NXT trial (analysis of coronary blood flow using CT angiography: next steps). J Am Coll Cardiol. 2014;63:1145–1155. [DOI] [PubMed] [Google Scholar]
- 114. Pham TA, Hua N, Phinikaridou A, Killiany R, Hamilton J. Early in vivo discrimination of vulnerable atherosclerotic plaques that disrupt: a serial MRI study. Atherosclerosis. 2016;244:101–107. [DOI] [PubMed] [Google Scholar]
- 115. Qiao H, Wang Y, Zhang R, Gao Q, Liang X, Gao L, Jiang Z, Qiao R, Han D, Zhang Y, Qiu Y, Tian J, Gao M, Cao F. MRI/optical dual‐modality imaging of vulnerable atherosclerotic plaque with an osteopontin‐targeted probe based on Fe3O4 nanoparticles. Biomaterials. 2016;112:336–345. [DOI] [PubMed] [Google Scholar]
- 116. Quirce R, Martinez‐Rodriguez I, Banzo I, Jimenez‐Bonilla J, Martinez‐Amador N, Ibanez‐Bravo S, Lopez‐Defillo J, Jimenez‐Alonso M, Revilla MA, Carril JM. New insight of functional molecular imaging into the atheroma biology: 18F‐NaF and 18F‐FDG in symptomatic and asymptomatic carotid plaques after recent CVA. Preliminary results. Clin Physiol Funct Imaging. 2016;36:499–503. [DOI] [PubMed] [Google Scholar]
- 117. Karimi M, Zare H, Bakhshian Nik A, Yazdani N, Hamrang M, Mohamed E, Sahandi Zangabad P, Moosavi Basri SM, Bakhtiari L, Hamblin MR. Nanotechnology in diagnosis and treatment of coronary artery disease. Nanomedicine (Lond). 2016;11:513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kharlamov AN. Plasmonic photothermal therapy for atheroregression below Glagov threshold. Future Cardiol. 2013;9:405–425. [DOI] [PubMed] [Google Scholar]
- 119. Walsh MT, Cunnane EM, Mulvihill JJ, Akyildiz AC, Gijsen FJ, Holzapfel GA. Uniaxial tensile testing approaches for characterisation of atherosclerotic plaques. J Biomech. 2014;47:793–804. [DOI] [PubMed] [Google Scholar]
- 120. Pasterkamp G, den Ruijter HM, Libby P. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat Rev Cardiol. 2017;14:21–29. [DOI] [PubMed] [Google Scholar]
- 121. Glaser R, Selzer F, Faxon DP, Laskey WK, Cohen HA, Slater J, Detre KM, Wilensky RL. Clinical progression of incidental, asymptomatic lesions discovered during culprit vessel coronary intervention. Circulation. 2005;111:143–149. [DOI] [PubMed] [Google Scholar]
- 122. Sun J, Underhill HR, Hippe DS, Xue Y, Yuan C, Hatsukami TS. Sustained acceleration in carotid atherosclerotic plaque progression with intraplaque hemorrhage: a long‐term time course study. JACC Cardiovasc Imaging. 2012;5:798–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kubo T, Maehara A, Mintz GS, Doi H, Tsujita K, Choi SY, Katoh O, Nasu K, Koenig A, Pieper M, Rogers JH, Wijns W, Bose D, Margolis MP, Moses JW, Stone GW, Leon MB. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol. 2010;55:1590–1597. [DOI] [PubMed] [Google Scholar]
- 124. Bittencourt MS, Hulten E, Ghoshhajra B, O'Leary D, Christman MP, Montana P, Truong QA, Steigner M, Murthy VL, Rybicki FJ, Nasir K, Gowdak LH, Hainer J, Brady TJ, Di Carli MF, Hoffmann U, Abbara S, Blankstein R. Prognostic value of nonobstructive and obstructive coronary artery disease detected by coronary computed tomography angiography to identify cardiovascular events. Circ Cardiovasc Imaging. 2014;7:282–291. [DOI] [PubMed] [Google Scholar]
- 125. Chung JW, Bang OY, Lee MJ, Hwang J, Cha J, Choi JH, Choe YH. Echoing plaque activity of the coronary and intracranial arteries in patients with stroke. Stroke. 2016;47:1527–1533. [DOI] [PubMed] [Google Scholar]
- 126. Iniguez A, Brener SJ, Jimenez VA, Maehara A, Mintz GS, Xu K, Weisz G, Lansky AJ, De Bruyne B, Serruys PW, Stone GW. Significance of prior percutaneous revascularisation in patients with acute coronary syndromes: insights from the prospective PROSPECT registry. EuroIntervention. 2016;11:1468–1474. [DOI] [PubMed] [Google Scholar]
- 127. Guo X, Zhu J, Maehara A, Monoly D, Samady H, Wang L, Billiar KL, Zheng J, Yang C, Mintz GS, Giddens DP, Tang D. Quantify patient‐specific coronary material property and its impact on stress/strain calculations using in vivo IVUS data and 3D FSI models: a pilot study. Biomech Model Mechanobiol. 2017;16:333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Goldstein JA, Demetriou D, Grines CL, Pica M, Shoukfeh M, O'Neill WW. Multiple complex coronary plaques in patients with acute myocardial infarction. N Engl J Med. 2000;343:915–922. [DOI] [PubMed] [Google Scholar]
- 129. Casscells W, Naghavi M, Willerson JT. Vulnerable atherosclerotic plaque: a multifocal disease. Circulation. 2003;107:2072–2075. [DOI] [PubMed] [Google Scholar]
- 130. Maseri A, Fuster V. Is there a vulnerable plaque? Circulation. 2003;107:2068–2071. [DOI] [PubMed] [Google Scholar]
- 131. Hannan EL, Samadashvili Z, Walford G, Jacobs AK, Stamato NJ, Venditti FJ, Holmes DR Jr, Sharma S, King SB III. Staged versus one‐time complete revascularization with percutaneous coronary intervention for multivessel coronary artery disease patients without ST‐elevation myocardial infarction. Circ Cardiovasc Interv. 2013;6:12–20. [DOI] [PubMed] [Google Scholar]
- 132. Gershlick AH, Khan JN, Kelly DJ, Greenwood JP, Sasikaran T, Curzen N, Blackman DJ, Dalby M, Fairbrother KL, Banya W, Wang D, Flather M, Hetherington SL, Kelion AD, Talwar S, Gunning M, Hall R, Swanton H, McCann GP. Randomized trial of complete versus lesion‐only revascularization in patients undergoing primary percutaneous coronary intervention for STEMI and multivessel disease: the CVLPRIT trial. J Am Coll Cardiol. 2015;65:963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. [DOI] [PubMed] [Google Scholar]
- 134. Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–743. [DOI] [PubMed] [Google Scholar]
- 135. Nussenzweig SC, Verma S, Finkel T. The role of autophagy in vascular biology. Circ Res. 2015;116:480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Emanuel R, Sergin I, Bhattacharya S, Turner JN, Epelman S, Settembre C, Diwan A, Ballabio A, Razani B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid‐induced lysosomal dysfunction and downstream sequelae. Arterioscler Thromb Vasc Biol. 2014;34:1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ryter SW, Lee SJ, Smith A, Choi AM. Autophagy in vascular disease. Proc Am Thorac Soc. 2010;7:40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hill BG, Haberzettl P, Ahmed Y, Srivastava S, Bhatnagar A. Unsaturated lipid peroxidation‐derived aldehydes activate autophagy in vascular smooth‐muscle cells. Biochem J. 2008;410:525–534. [DOI] [PubMed] [Google Scholar]
- 139. Perrotta I, Aquila S. The role of oxidative stress and autophagy in atherosclerosis. Oxid Med Cell Longev. 2015;2015:130315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Martinet W, De Meyer GR. Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. 2009;104:304–317. [DOI] [PubMed] [Google Scholar]
- 141. Guo F, Li X, Peng J, Tang Y, Yang Q, Liu L, Wang Z, Jiang Z, Xiao M, Ni C, Chen R, Wei D, Wang GX. Autophagy regulates vascular endothelial cell eNOS and ET‐1 expression induced by laminar shear stress in an ex vivo perfused system. Ann Biomed Eng. 2014;42:1978–1988. [DOI] [PubMed] [Google Scholar]
- 142. Tuloup‐Minguez V, Hamai A, Greffard A, Nicolas V, Codogno P, Botti J. Autophagy modulates cell migration and beta1 integrin membrane recycling. Cell Cycle. 2013;12:3317–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang T, Zhang L, Hu J, Duan Y, Zhang M, Lin J, Man W, Pan X, Jiang Z, Zhang G, Gao B, Wang H, Sun D. Mst1 participates in the atherosclerosis progression through macrophage autophagy inhibition and macrophage apoptosis enhancement. J Mol Cell Cardiol. 2016;98:108–116. [DOI] [PubMed] [Google Scholar]
- 145. Mueller MA, Beutner F, Teupser D, Ceglarek U, Thiery J. Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR‐/‐ mice despite severe hypercholesterolemia. Atherosclerosis. 2008;198:39–48. [DOI] [PubMed] [Google Scholar]
- 146. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. [DOI] [PubMed] [Google Scholar]
- 147. Martin K, O'Sullivan JF, Caplice NM. New therapeutic potential of microrna treatment to target vulnerable atherosclerotic lesions and plaque rupture. Curr Opin Cardiol. 2011;26:569–575. [DOI] [PubMed] [Google Scholar]
- 148. Ulrich V, Rotllan N, Araldi E, Luciano A, Skroblin P, Abonnenc M, Perrotta P, Yin X, Bauer A, Leslie KL, Zhang P, Aryal B, Montgomery RL, Thum T, Martin K, Suarez Y, Mayr M, Fernandez‐Hernando C, Sessa WC. Chronic miR‐29 antagonism promotes favorable plaque remodeling in atherosclerotic mice. EMBO Mol Med. 2016;8:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Menghini R, Casagrande V, Marino A, Marchetti V, Cardellini M, Stoehr R, Rizza S, Martelli E, Greco S, Mauriello A, Ippoliti A, Martelli F, Lauro R, Federici M. MiR‐216a: a link between endothelial dysfunction and autophagy. Cell Death Dis. 2014;5:e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR‐221 and miR‐222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Garrido AM, Bennett M. Assessment and consequences of cell senescence in atherosclerosis. Curr Opin Lipidol. 2016;27:431–438. [DOI] [PubMed] [Google Scholar]
- 152. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. [DOI] [PubMed] [Google Scholar]
- 153. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring p16(Ink4a)‐positive cells shorten healthy lifespan. Nature. 2016;530:184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, Gray K, Kumar S, Clarke M, Bennett M. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132:1909–1919. [DOI] [PubMed] [Google Scholar]
- 155. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544. [DOI] [PubMed] [Google Scholar]
- 156. Thorp EB. Proresolving lipid mediators restore balance to the vulnerable plaque. Circ Res. 2016;119:972–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, Tabas I. An imbalance between specialized pro‐resolving lipid mediators and pro‐inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, Silvestre‐Roig C, Dittmar G, Doring Y, Drechsler M, Weber C, Zimmer R, Cenac N, Soehnlein O. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res. 2016;119:1030–1038. [DOI] [PubMed] [Google Scholar]
- 159. Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P. Major cell death pathways at a glance. Microbes Infect. 2009;11:1050–1062. [DOI] [PubMed] [Google Scholar]
- 160. Chang W, Lin J, Dong J, Li D. Pyroptosis: an inflammatory cell death implicates in atherosclerosis. Med Hypotheses. 2013;81:484–486. [DOI] [PubMed] [Google Scholar]
- 161. Corban MT, Eshtehardi P, Suo J, McDaniel MC, Timmins LH, Rassoul‐Arzrumly E, Maynard C, Mekonnen G, King S III, Quyyumi AA, Giddens DP, Samady H. Combination of plaque burden, wall shear stress, and plaque phenotype has incremental value for prediction of coronary atherosclerotic plaque progression and vulnerability. Atherosclerosis. 2014;232:271–276. [DOI] [PubMed] [Google Scholar]