

Abstract
Background
The amino acid response (AAR) is an evolutionarily conserved protective mechanism activated by amino acid deficiency through a key kinase, general control nonderepressible 2. In addition to mobilizing amino acids, the AAR broadly affects gene and protein expression in a variety of pathways and elicits antifibrotic, autophagic, and anti‐inflammatory activities. However, little is known regarding its role in cardiac stress. Our aim was to investigate the effects of halofuginone, a prolyl‐tRNA synthetase inhibitor, on the AAR pathway in cardiac fibroblasts, cardiomyocytes, and in mouse models of cardiac stress and failure.
Methods and Results
Consistent with its ability to inhibit prolyl‐tRNA synthetase, halofuginone elicited a general control nonderepressible 2–dependent activation of the AAR pathway in cardiac fibroblasts as evidenced by activation of known AAR target genes, broad regulation of the transcriptome and proteome, and reversal by l‐proline supplementation. Halofuginone was examined in 3 mouse models of cardiac stress: angiotensin II/phenylephrine, transverse aortic constriction, and acute ischemia reperfusion injury. It activated the AAR pathway in the heart, improved survival, pulmonary congestion, left ventricle remodeling/fibrosis, and left ventricular function, and rescued ischemic myocardium. In human cardiac fibroblasts, halofuginone profoundly reduced collagen deposition in a general control nonderepressible 2–dependent manner and suppressed the extracellular matrix proteome. In human induced pluripotent stem cell–derived cardiomyocytes, halofuginone blocked gene expression associated with endothelin‐1‐mediated activation of pathologic hypertrophy and restored autophagy in a general control nonderepressible 2/eIF2α‐dependent manner.
Conclusions
Halofuginone activated the AAR pathway in the heart and attenuated the structural and functional effects of cardiac stress.
Keywords: amino acid response, fibrosis, halofuginone, heart failure, hypertrophy
Subject Categories: Fibrosis, Heart Failure, Hypertrophy
Introduction
It is estimated that by 2030 the number of patients living with heart failure in the United States will increase 43% to 8.5 million. Despite recent improvements in treatment, it remains a debilitating and costly disorder with a poor prognosis and limited treatment options.1 Considerable efforts to identify new heart failure therapies across multiple modalities have focused on intrinsic cardiac mechanisms that (1) underlie pathologic processes (eg, ventricular remodeling, inflammation, fibrosis, oxidative stress, calcium dysregulation), (2) initiate regeneration (eg, cell and gene therapies), or (3) promote adaptive processes (eg, insulin/IGF‐1 signaling, AMPK, PGC1α, GSK3β). Among the various mechanisms that have been explored in heart failure, little or no attention has been given to the adaptive and protective amino acid response (AAR) pathway.
AAR is an evolutionarily conserved mechanism that is activated during periods of amino acid deprivation.2 During amino acid starvation, there is an accumulation of uncharged tRNA molecules, which are sensed by, and specifically bind to and activate, GCN2, a key kinase that mediates the AAR.3 Activated GCN2 phosphorylates itself and eIF2α, a key factor that elicits a coordinated transcriptional and translational response to restore amino acid homeostasis by affecting the utilization, acquisition, and mobilization of amino acids at the level of the cell and the organism. These responses have been shown to mediate robust antifibrotic, anti‐inflammatory, prosurvival, autophagic and metabolic effects—all actions thought to be of potential benefit in heart failure.
Halofuginone, a derivative from a Chinese herb extract, has recently been shown to be a potent inhibitor of prolyl‐tRNA synthetase, which in turn generates increased levels of uncharged prolyl‐tRNA, phosphorylation of GCN2 and eIF2α, and activation of the AAR.4 Activation of AAR by amino acid restriction or halofuginone has been shown to provide benefits in a variety of different disease models including muscular dystrophy, autoimmune diseases, metabolic syndrome, and hepatic and renal ischemia‐reperfusion injury.5, 6, 7, 8, 9 In the present study we examined the effects of halofuginone in 3 animal models of cardiac stress and heart failure and evaluated its mechanism of action in human cardiac fibroblasts and induced pluripotent stem cells (iPSC)‐derived cardiomyocytes. These data demonstrate that activation of the AAR is capable of blunting structural and functional consequences of cardiac stress and may suggest new therapeutic strategies for the treatment of heart failure.
Materials and Methods
For additional details, please refer to Data S1.
Animals
Male C57Bl/6J mice at ≈12 weeks old were used for the study. All animal studies were in compliance with the Guide for the Care and Use of Laboratory Animals as published by the US National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of GlaxoSmithKline.
In Vivo Studies
Dose Exploration Study
Mice were given chow containing halofuginone to reach a targeted dose of 0.1, 0.3, 1, and 3 mg/kg. Four days later, mice were euthanized. Liver and left ventricles were flash‐frozen in liquid nitrogen for gene expression analysis. Blood was collected for halofuginone concentration analysis.
Neurohormonal‐Induced Cardiac Stress Model
On day 0, under isoflurane (2% to 2.5%) anesthesia, osmotic pumps containing halofuginone were implanted subcutaneously in the mice based on random assignment. These pumps delivered halofuginone at the rate of 0.046 and 0.138 mg/kg per day in the low‐ and high‐dose groups, respectively. Two control groups and 1 vehicle group also received osmotic pumps that delivered saline. The high‐dose control angiotensin II/phenylephrine group was pair fed to match the food consumption of the mice dosed with 0.138 mg/kg per day halofuginone. All other groups were fed ad libitum. On day 10, both halofuginone‐treated groups and control groups received a second osmotic pump, which delivered 0.432 mg/kg per day angiotensin II and 100 mg/kg per day phenylephrine HCl. On day 24, mice were euthanized. Organ weights were recorded. Left ventricle was pulverized under liquid nitrogen and used for hydroxyproline analysis and gene expression analysis.
Hydroxyproline Liquid Chromatography–Tandem Mass Spectrometry Method
Weighed tissue samples were hydrolyzed in 6 N HCl at 110°C for 16 hours. With hydroxyproline used as standard, liquid chromatography–tandem mass spectrometry analysis was performed using positive‐mode electrospray ionization and a hydrophilic interaction liquid chromatography gradient on a silica liquid chromatography column. Quantitation was performed using Analyst mass spectrometry software and collagen concentrations calculated using Microsoft Excel. This quantitative estimate of collagen precluded further histological evaluation.
Pressure Overload Induced Heart Failure Model
Transverse aortic constriction (TAC) was performed as previously reported.10 Briefly, mice were anesthetized with pentobarbital (Nembutal) and ventilated. The transverse aorta was constricted to the size of a 27‐G needle using a 7‐0 suture between the innominate and left carotid arteries. Sham operation underwent a similar surgical procedure without constricting the aorta. Animals that survived the surgery were randomly assigned to treatment with halofuginone (0.3 mg/kg per day in chow) or a standard chow diet immediately after the TAC surgery until the end of the study (42 days). At the end of the study, echocardiographic examination was performed on surviving animals to evaluate cardiac structure and function using VEVO 2100 ultrasound system (VisualSonics, Toronto, Canada) in accordance with the American Society of Echocardiography guidelines. Left ventricular (LV) hemodynamics were measured by Millar catheter at the end of study at baseline and in the presence of 1 μg/μL saline/gram body weight/min dobutamine infusion. Plasma was collected, and proANP levels were measured by enzyme‐linked immunosorbent assay.
Fibroblast Experiments
Human cardiac fibroblasts (normal human cardiac fibroblast‐ventricle, Cat#CC2904, Lonza, Walkersville, MD) were treated with Eagle's Minimum Essential Medium (EMEM) containing ficoll for 24 to 48 hours to stimulate collagen deposition.11 At the same time of ficoll treatment, compounds such as halofuginone, borrelidin, l‐proline, or l‐threonine were added to the medium. At the end of the treatment, cells were fixed with methanol and immunostained for mature type I collagen and quantified on an Operetta High Content Imaging System (Perkin Elmer, Waltham, MA). Alternatively, cells were lysed with RIPA buffer containing a protease and phosphatase inhibitor cocktail for Western blot analysis.
Real Time Polymerase Chain Reaction and Transcriptomics Analysis
For cellular experiments, total cellular RNA was isolated using the RNeasy mini kit (Qiagen, Hilden, Germany). For animal models, frozen tissues were pulverized under liquid nitrogen, and then total RNA was isolated using TRIzol reagent and RNeasy mini kit. Real‐time reverse transcription polymerase chain reaction (RT‐PCR) analysis was carried out using QuantiTect RT‐PCR (Qiagen, Hilden, Germany). For transcriptomics analysis, RNA was isolated as described above. Poly‐A mRNA enrichment was carried out using TruSeq™ RNA sample prep v2 kit (Illumina, San Diego, CA) followed by cDNA library preparation and sequencing on a HiSeq1000 by using Paired End 2×50 bp SBS sequencing kits v5 for whole‐transcriptome sequencing (Illumina, San Diego, CA). Read mapping and expression estimation were analyzed as described before.12 Pathway enrichment analysis was conducted using Ingenuity Pathway Analysis. The analysis was conducted on the differentially expressed genes from comparing halofuginone‐treated versus control samples. The significantly enriched pathways were selected using a cutoff P‐value of 0.01. Raw sequence data are available at Gene Expression Omnibus with accession ID GSE95059 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE95059).
Proteomic Analysis
NHCF‐V cells were treated with 100 nmol/L, 300 nmol/L halofuginone and 300 nmol/L halofuginone with 4 mmol/L l‐proline or DMSO for 24 hours. Cell layers were lysed in 2% SDS, peptides were labeled with 10‐plex TMT reagent (TMT10, Thermo Fisher Scientific, Waltham, MA), and fractionated on an Ultimate3000 (Dionex, Sunnyvale, CA) by using reversed‐phase chromatography at pH 12 (1 mm Xbridge column, Waters, Milford, MA). Peptide fragments were detected by liquid chromatography–tandem mass spectrometry. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE13 partner repository with the data set identifier PXD005679.
Cardiomyocyte Experiment
Human induced pluripotent stem cells (iPSC)‐derived cardiomyocytes were purchased from Cellular Dynamics International (CDI, Madison, WI). Upon defrosting, cells were allowed to mature in maintenance medium (CDI) for 10 to 14 days. Cells were placed in serum‐free medium for 24 hours before treatment. They were then treated with medium containing 1 nmol/L endothelin‐1 (ET‐1), 200 nmol/L halofuginone, 2 mmol/L l‐proline, and 2 mmol/L l‐threonine for 24 hours. Natriuretic peptide A (NPPA) mRNA expression was examined by RT‐PCR analysis. Western blot was used to determine p62 levels. microtubule‐associated protein 1 light chain 3αB level was determined by immunohistochemistry.
Statistical Analysis
Statistical analysis methods were defined in the figure legend of each figure. The comparisons between treatment groups or sample groups used 1‐way ANOVA or unpaired t tests. Based on current and historical data, the assessment of the distribution of the measurement values within treatment or sample groups identified those measurement types that had normal or log‐normal distributions. The data from measurement types with log‐normal distributions were log transformed before the ANOVA or t test comparisons. The measurements of the organ weights, the hydroxyproline, pro‐ANP, the cardiac functional measurements such as ejection fraction, the collagen intentisities, and the infarct size all follow normal distributions with similar variances by measurement type. Data from the gene expression measurements in the cellular studies and in the animal studies, follow log‐normal distributions.
Results
Halofuginone Activates the AAR Pathway
Fibroblast cell culture was used as a model system to evaluate the effects of halofuginone on the AAR pathway. Incubation of cardiac fibroblasts with halofuginone caused phosphorylation of GCN2 (a key regulator of the AAR) and upregulation of CHOP mRNA (a canonical AAR response gene), and both responses could be inhibited specifically by supplementation of l‐proline but not l‐threonine (Figure 1A through 1C). Furthermore, the effects of halofuginone on the AAR pathway were significantly attenuated in GCN2‐deficient mouse embryonic fibroblasts (Figure 1D). Similar threonine‐dependent results were observed with borrelidin, a threonyl‐tRNA synthetase inhibitor (Figure S1).
Figure 1.
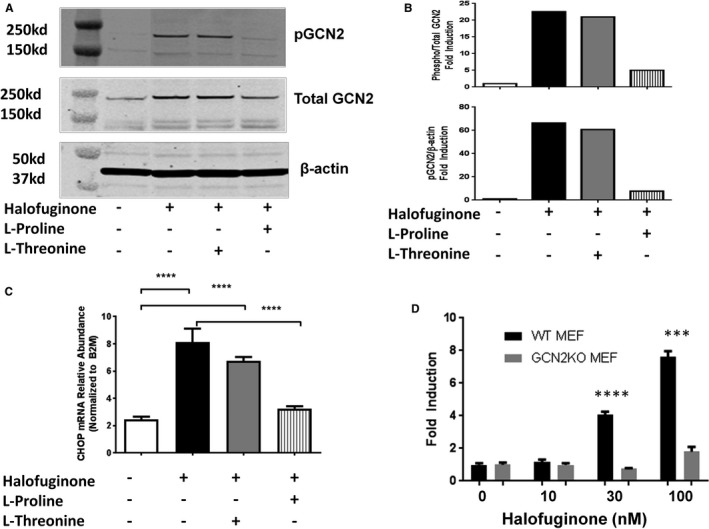
Halofuginone‐induced AAR (amino acid response) in fibroblasts. Human cardiac fibroblasts were treated with halofuginone (100 nmol/L), l‐proline (2 mmol/L), and l‐threonine (2 mmol/L) for 24 hours. Protein levels of pGCN2 (phosphorylated general control nonderepressible 2), total GCN2, and β‐actin were examined by Western blot (blot in [A]; quantitation in [B], 1 representative of 2 experiments shown) and mRNA level of CHOP (C/EBP homologous protein) was examined by RT‐PCR (reverse transcription polymerase chain reaction, [C], n=4). One way ANOVA was used for statistical analysis of log‐transformed data, ****P<0.0001. Wild‐type (WT) or GCN2 knockout (GCN2KO) mouse embryonic fibroblast (MEF) were treated with increasing concentrations of halofuginone for 24 hours. CHOP mRNA was examined by RT‐PCR (D, n=3). Unpaired t test was used on log‐transformed data: ***P<0.001, ****P<0.0001.
The effects of halofuginone are consistent with selective activation of the AAR pathway mediated by the interaction of GCN2 and uncharged tRNA generated by inhibition of prolyl‐tRNA synthetase, the molecular target of halofuginone.
Halofuginone Activates the AAR Pathway in the Heart
The ability of halofuginone to engage the AAR pathway in the mouse heart was evaluated in a dose‐exploration study. In these experiments halofuginone was administered via the dietary route by mixing it with chow at 0.8, 2.5, 8, and 25 ppm in order to achieve daily doses of 0.1, 0.3, 1, and 3 mg/kg, respectively. Blood concentrations of halofuginone were determined after 4 days of dosing, and effects on expression of canonical AAR target genes (CHOP and ASNS) were assessed in the heart and liver (Figure 2). Significant increases in AAR target genes were observed in the heart and liver at 1 and 3 mg/kg per day doses corresponding to plasma concentrations of 5 and 22 ng/mL, respectively. Lower doses of halofuginone resulted in plasma concentrations below the limits of detection. These results provided (1) in vivo evidence for AAR pathway engagement and activation in the heart and (2) guidance for potentially effective halofuginone plasma concentrations needed in subsequent studies.
Figure 2.
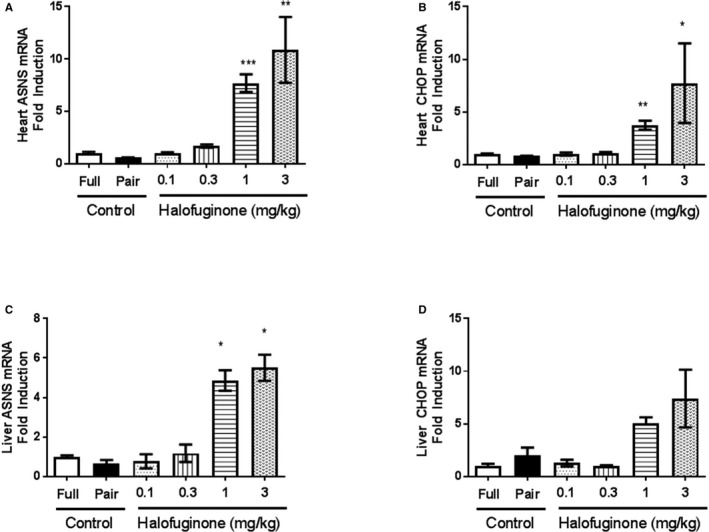
Halofuginone‐induced AAR (amino acid response) in vivo. Male C57Bl/6J mice were administered halofuginone by the dietary route to obtain targeted doses of 0.1, 0.3, 1, and 3 mg/kg for 4 days. Due to the reduction in food intake in 1 and 3 mg/kg doses (data not shown), we included 2 groups of control mice. One group was fed ad libitum (Full) and the other was pair‐fed to equal the amount of food eaten by the 3 mg/kg group (Pair). At the end of 4 days, heart (A and B) and liver (C and D) tissues were collected, and the mRNA levels of ASNS (asparagine synthetase) and CHOP (C/EBP homologous protein) were analyzed by real‐time RT‐PCR (reverse transcription polymerase chain reaction). There were 3 mice per group. Gene expression data were log transformed first. Unpaired t test of log‐transformed data were used for statistics to compare between 0.1 and 0.3 mg/kg doses and Full control, and to compare between 1, 3 mg/kg and Pair control; *, **, ***, P<0.05, 0.01, and 0.001 vs Control (Pair), respectively.
Halofuginone in the Neurohormonal Induced Cardiac Stress Model
To determine the effects of activating the AAR pathway in a model of cardiac stress, we first evaluated the effects of halofuginone treatment in mice receiving a continuous infusion of angiotensin II (AngII) and phenylephrine (PE). In this model, mice were treated with either low (0.046 mg/kg per day) or high (0.138 mg/kg per day) doses of halofuginone via osmotic minipumps, commencing 10 days before introducing the AngII/PE infusion pumps and continued throughout the subsequent 14 days of AngII/PE infusion. Based on the dose exploration study above, these low‐ and high‐dose infusions would provide plasma concentrations similar to 0.3 and 1.0 mg/kg per day (chow dosing), causing modest or robust AAR pathway activation, respectively. The high dose of halofuginone reduced food intake and required a separate pair‐fed control group to normalize any potential effects of caloric restriction. Food consumption was unaffected in the low‐dose group and comparable to a control group fed ad libitum. We also included a vehicle group that only received saline via osmotic pumps throughout the study.
There was no mortality in the saline vehicle group; however, mortality was ≈55% in both control groups receiving AngII/PE alone (Figure 3A). In contrast, AngII/PE mice receiving halofuginone had significantly improved survival using log‐rank survival test; ie, 21% and 9% mortality in the low‐ and high‐dose groups, respectively. As exemplified by reductions in hydroxyproline and organ weights, halofuginone had a modest antifibrotic effect in the LV (Figure 3B) and dose‐dependently attenuated the cardiac hypertrophy and lung congestion induced by AngII/PE infusion (Figure 3C and 3D). AngII/PE treatment caused a ≈10% reduction in gastrocnemius muscle weight/tibia length ratio, and halofuginone did not further reduce gastrocnemius muscle weight in this model (data not shown). The infusion of the AngII/PE combination elicited a gene expression pattern indicative of pathologic left ventricular remodeling, ie, induction of NPPA and type I collagen α1 and decreased α myosin heavy chain and microtubule‐associated protein 1 light chain 3α (LC3) (Figure 3E through 3H). Halofuginone treatment dose‐dependently attenuated the expression of NPPA, type I collagen α1 and increased the expression of α myosin heavy chain and LC3, suggesting attenuation of the neurohormonal induced heart failure phenotype associated with hypertrophy, fibrosis, and deficient autophagy.
Figure 3.
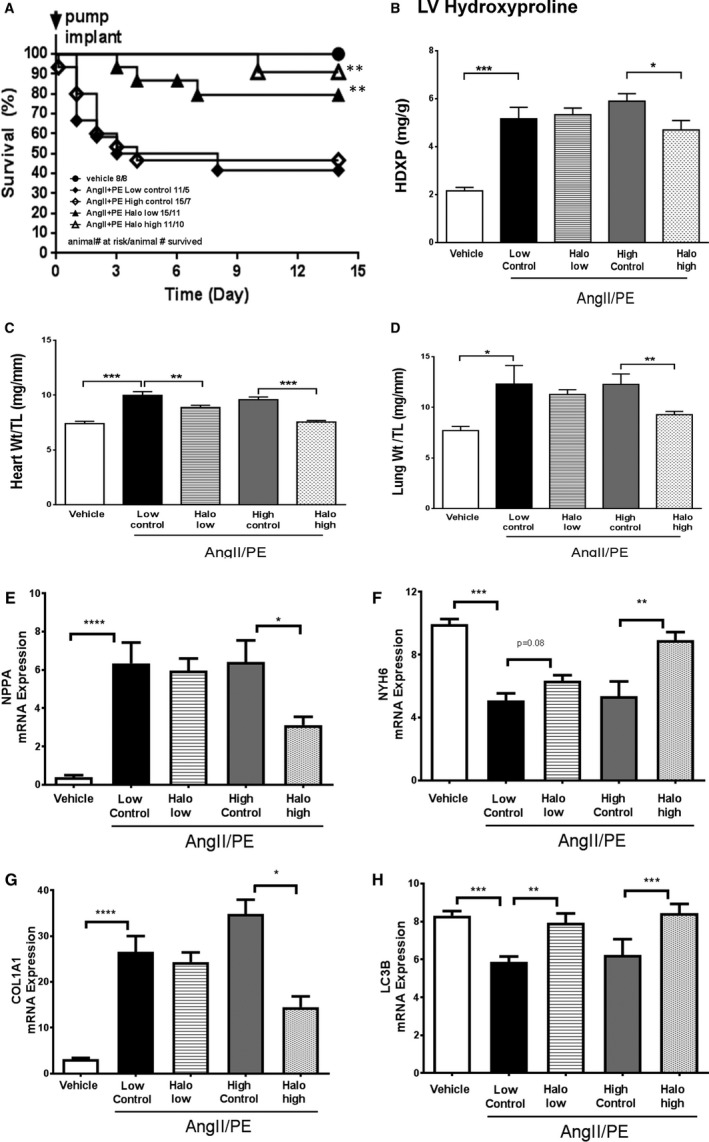
Halofuginone in AngII/PE (angiotensin II/phenylephrine) mouse model. Male C57Bl/6J mice were treated with halofuginone via minipumps that delivered 0.3 or 1 mg/kg doses (AngII/PE Halo low and AngII/PE Halo high) throughout the study (beginning 10 days before the 2‐week AngII/PE infusion). The vehicle group received neither halofuginone nor AngII/PE treatment. The AngII/PE low control group was fed ad libitum. The AngII/PE high control group was pair‐fed to match food consumption of the AngII/PE Halo high group. Survival was monitored following AngII/PE treatment (A). Log‐rank (Mantel‐Cox) test was used for survival analysis: **P<0.01 vs AngII/PE low and high control groups. At the end of tge study, heart hydroxyproline content (HDXP, as a readout for fibrosis) was measured by LC‐MS (liquid chromatography–mass spectrometry, [B]). Heart weight, lung weight, and tibia length (TL) were recorded, and ratios were calculated (C and D). Cardiac gene expression was conducted by real‐time RT‐PCR (reverse transcription polymerase chain reaction) and plotted (E through H). There were 8 (8) mice in the vehicle, 11 (5) in the low control, 15 (7) in high control, 15 (11) in the Halo low, and 11 (10) in the Halo high groups at the beginning of AngII/PE infusion. Numbers in the parentheses reflect the number of animals that survived until the end of the study, which were used for analysis in (B through H). Unpaired t test was used for statistics on raw data in B and C. Unpaired t test was used for statistics on log‐transformed data (E through H): *, **, ***, ****, P<0.05, 0.01, 0.001, and 0.0001, respectively.
Halofuginone in the TAC Model
Halofuginone was also evaluated in the TAC pressure overload model to determine effects on cardiac function. Immediately after the TAC procedure, a subset of the mice were fed a diet containing 2.5 ppm halofuginone in order to attain plasma concentrations modeled on the basis of previous studies (see above). At the end of the study (6 weeks), mortality was 33% in the TAC group, and no mortality was observed in the sham group or in the TAC group treated with halofuginone (log‐rank survival test, TAC versus TAC+halofuginone, P=0.1, Figure 4A). TAC also induced cardiac hypertrophy and increased lung weight (indicative of pulmonary congestion), which were both attenuated by halofuginone treatment (Figure 4B and 4C). Circulating pro‐ANP levels were correspondingly elevated by TAC and again decreased by halofuginone treatment (Figure 4D). Ejection fraction (Figure 4E) and fractional area change (Figure S2) were reduced in TAC animals compared with sham, and there was a trend toward improvement in the halofuginone‐treated group. Consistent with these effects, LV end‐diastolic pressure and cardiac reserve (dobutamine stress test, ΔdP/dt) were significantly improved in the halofuginone group (Figure 4F and 4G). These results suggest that halofuginone prevents LV dysfunction induced by pressure overload.
Figure 4.
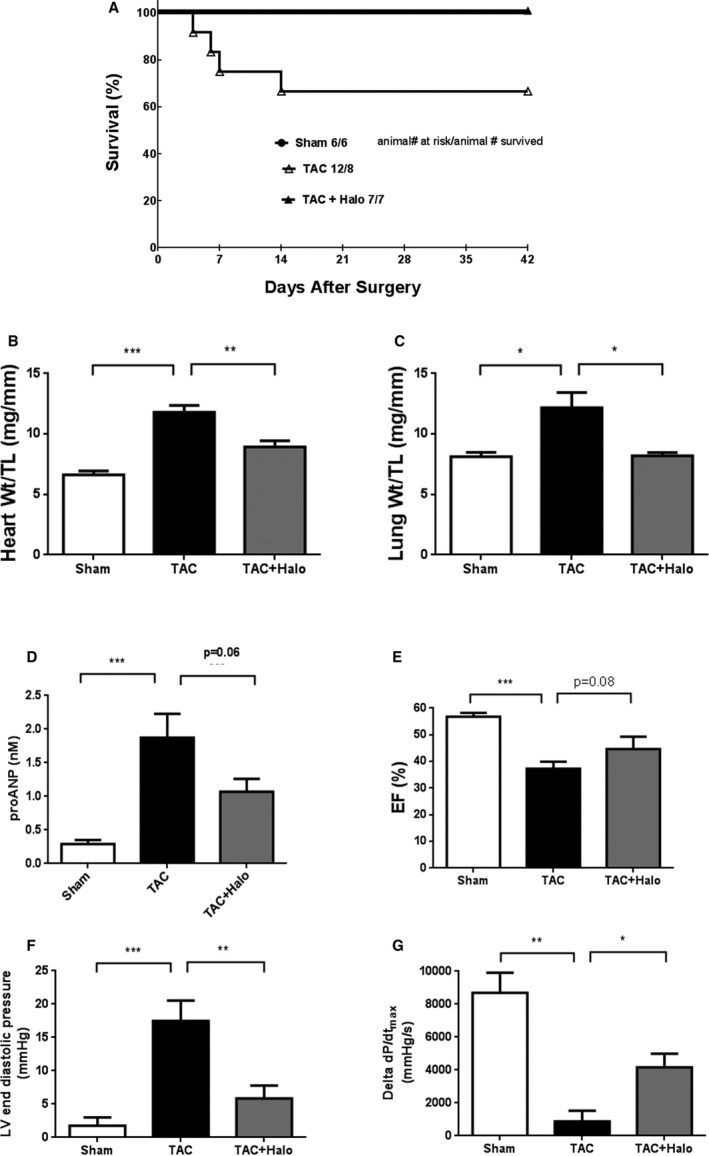
Halofuginone in TAC (transverse aortic constriction) mouse model. Male C57Bl/6J mice were subjected to TAC to induce heart failure. Halofuginone was delivered in chow (0.3 mg/kg dose) starting on the same day that TAC was performed. Survival was monitored in each group. A, Log rank survival test, TAC vs TAC+Halo, P=0.1. At end of study (6 weeks), heart weight, lung weight, and tibia length were recorded, and ratios were calculated (B and C). Circulating proANP (pro–atrial natriuretic peptide) level was measure by ELISA (enzyme‐linked immunosorbent assay, [D]). Ejection fraction (EF) was measured by echocardiography at the end of study (E). Left ventricle end‐diastolic pressure at baseline and the change in cardiac contractility (ΔdP/dT) following dobutamine challenge were measured at the end of study (F and G). There were 6 (6), 12 (8) and 7 (7) mice in the sham, TAC, and TAC+halofuginone groups at the beginning of the study, respectively. Numbers in the parentheses reflect the number of animals that survived until the end of the study, which were used for analysis (B through G). Unpaired t test was used for statistics: *, **, ***, P<0.05, 0.01, and 0.001, respectively.
In addition to nonischemic cardiac stress models such as AngII/PE and TAC, our recent preliminary studies suggest that GCN2 may also be playing a protective role in ischemic cardiac stress (Figure S3). We found that halofuginone treatment in mice reduced infarct size from 41% to 28% of area at risk after a 30‐minute occlusion of the left anterior descending coronary artery and 24 hours of reperfusion without affecting the area at risk (as a percentage of LV).
Halofuginone and AAR in Human Cardiac Fibroblasts
In vitro cellular models were used to explore the protective mechanisms of halofuginone in the heart. The antifibrotic effects of halofuginone were evaluated in human cardiac fibroblasts by assessing effects on collagen production, deposition, and regulation. In a collagen deposition assay,11 halofuginone reduced type I collagen deposition (pIC50=100 nmol/L) as well as type I collagen α1 gene and protein expression at concentrations that had no adverse effects on cell health (Figure 5A and 5B). These effects were blocked by supplementation with excess l‐proline but not l‐threonine (Figure 5C and 5D). Similar results were observed with borrelidin, a threonyl‐tRNA synthetase inhibitor, and its effect was blocked by l‐threonine but not l‐proline (Figure S4). In addition, amino acid–deficient media and l‐leucinol (mimic of l‐leucine and inhibitor of Leucyl‐tRNA synthetase) also inhibited collagen deposition (Figure S5). These results provide evidence that the antifibrotic effects of halofuginone in the heart are related to direct reduction of stress‐induced collagen formation by cardiac fibroblasts and that these effects are mediated by activation of the AAR pathway.
Figure 5.
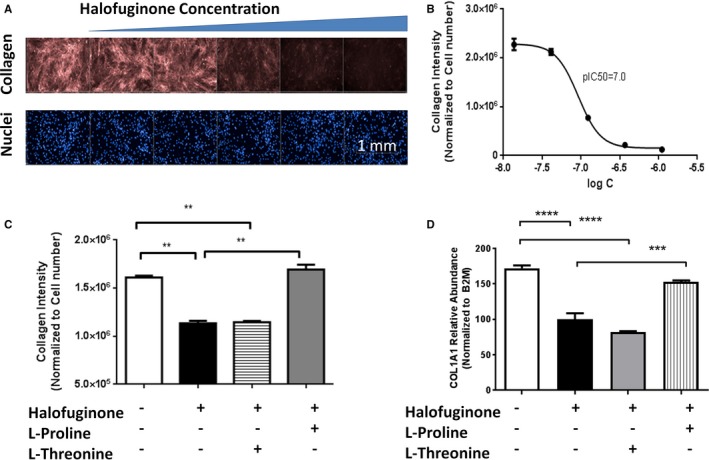
Halofuginone inhibits collagen deposition in cardiac fibroblasts. Normal human cardiac fibroblasts were incubated with media containing ficoll (to stimulate collagen deposition) in the presence of increasing concentrations of halofuginone. The deposition of mature collagen by these fibroblasts was examined by immunohistochemistry (A and B). Fibroblasts were treated with halofuginone 100 nmol/L, l‐proline 2 mmol/L, and l‐threonine 2 mmol/L for 24 hours. At the end of the treatment, collagen deposition and COL1A1 mRNA (Type I collagen 1α1 messenger ribonucleic acid [C and D]) were measured by immunohistochemistry and reverse transcription polymerase chain reaction, respectively. N=5 for B and C, N=4 for D. One‐way ANOVA with Tukey post hoc analysis, *, **, ***, P<0.05, 0.001 and 0.0001, respectively.
Proteomic and Transcriptomic Effects of Halofuginone in Human Cardiac Fibroblasts
To gain a more complete understanding of the effects of halofuginone on the extracellular matrix (ECM) we performed a proteomic analysis of cardiac fibroblasts.
Cardiac fibroblasts were treated with 100 nmol/L or 300 nmol/L halofuginone or with 300 nmol/L halofuginone with 4 mmol/L l‐proline for 24 hours. By use of a mass spectrometry–based method, more than 7000 proteins were identified and quantified. Halofuginone dose‐dependently altered protein expression in these cells (Figure 6A and 6B, Table S1). At 300 nmol/L, halofuginone significantly altered the expression of 355 proteins (4.6% of the total). The majority of differentially regulated proteins were decreased (322 decreased versus 33 increased), and nearly all were reversed, by l‐proline supplementation, suggesting that halofuginone‐induced changes were mediated through inhibition of prolyl‐tRNA synthetase (Figure 6C). A REACTOME proteomic pathway analysis confirmed that halofuginone significantly downregulated half of the proteins assigned to the ECM pathway (Figure 6D).
Figure 6.
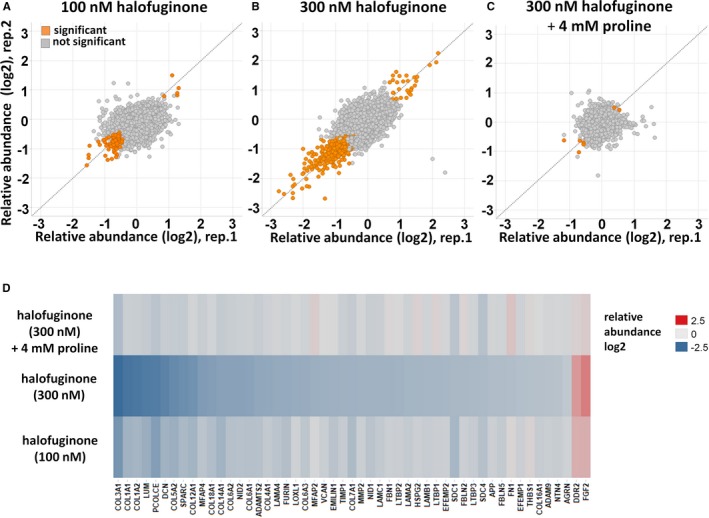
Halofuginone‐mediated proteomic changes in cardiac fibroblasts. Normal human cardiac fibroblasts were incubated with 100 nmol/L and 300 nmol/L halofuginone and 4 mmol/L l‐proline for 24 hours. Proteins were extracted and analyzed with a mass spectrometry–based quantitative proteomics approach. A through C, Scatter plot representation of relative protein abundances obtained for different treatment conditions compared to vehicle‐treated cells. The 2 axes are relative abundance (log 2‐fold change) from 2 different replicates in this experiment. Proteins with significantly altered abundance (P<0.05) compared with untreated samples are displayed in orange. D, A heat‐map representation of extracellular matrix proteins displaying significant abundance changes from cells treated with halofuginone compared to vehicle (average of n=2). Upregulated proteins are shown in red; downregulated proteins are shown in blue.
Transcriptomic analysis in cardiac fibroblasts showed that 200 nmol/L halofuginone upregulated 569 mRNAs and downregulated 608 mRNAs (Tables S2 through S4); this is a markedly different pattern relative to the profound downregulation observed in the proteome. It is noteworthy that Ingenuity analysis of the proteome and transcriptome demonstrated only a partial overlap of differentially regulated canonical pathways (Tables S4 and S5).
Additional analysis of the top 100 downregulated proteins showed significant regulation of heart failure–related proteins, TGFβ, and Wnt‐related proteins (Figure S6A and S6B).
Halofuginone in Cardiomyocytes
Human iPSC‐derived cardiomyocytes were also used to explore the protective mechanisms of halofuginone in the heart. The cardiac hypertrophy observed in vivo was modeled in cardiomyocytes by activating the pathologic hypertrophy pathway with ET‐114 (Figure 7A). As expected, ET‐1 significantly increased NPPA mRNA expression associated with Gαq‐mediated pathologic hypertrophy. Incubation with 200 nmol/L halofuginone abolished the ET‐1–mediated increases in NPPA. These effects were attenuated by l‐proline supplementation, consistent with its action to inhibit prolyl‐tRNA synthetase.
Figure 7.
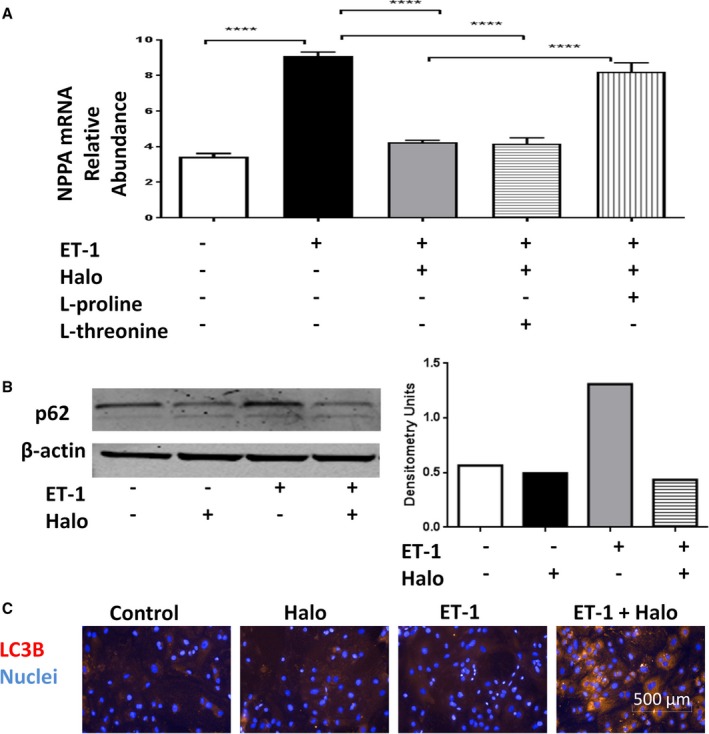
Halofuginone effects in cardiomyocytes. Human induced pluripotent stem cell–derived cardiomyocytes were treated with 1 nmol/L endothelin‐1 (ET‐1), 200 nmol/L halofuginone (Halo), 2 mmol/L l‐threonine, and/or 2 mmol/L l‐proline for 24 hours. Total cellular RNA was harvested, and NPPA (proatrial natriuretic peptide) mRNA expression was examined by real‐time reverse transcription polymerase chain reaction analysis (A), N=3 for each treatment condition. One‐way ANOVA with Tukey post hoc analysis: ****P<0.0001. The protein level of p62 was detected by Western blot (B). The level of LC3B was detected by immunohistochemistry (C). One representative of 2 experiments was shown for B and C.
Impaired autophagy is associated with cardiac stress and is believed to contribute to cardiac dysfunction.15 We found that halofuginone treatment alone or in combination with ET‐1 was able to reduce the expression of p62, a protein that is degraded during autophagy (Figure 7B)16 and increased LC3B‐positive vesicles (Figure 7C)—a response pattern consistent with an increased autophagic flux.
Overall, our studies demonstrate that halofuginone activated the AAR in vitro (cardiomyocytes and fibroblast) and in vivo (heart and liver) through an action consistent with inhibition of prolyl‐tRNA synthetase, generation of uncharged tRNA, activation of GCN2, and inhibition of cardiac hypertrophy, fibrosis, dysfunction, and autophagic deficits.
Discussion
In the present study we used halofuginone to activate the AAR in the heart and evaluate its effects on the cardiac response to stress. Dose‐related activation of the AAR with halofuginone improved survival and blunted LV pathological gene expression, cardiac hypertrophy, pulmonary congestion, cardiac fibrosis, and ventricular dysfunction in murine models of neurohormonal stress and pressure overload. These results suggest that activation of the AAR pathway in the heart may be a novel mechanism to combat the deleterious effects of cardiac stress in the failing myocardium.
Consistent with its reported action as a prolyl‐tRNA synthetase inhibitor, halofuginone activated key signaling elements of the AAR response pathway (GCN2 and elF2α) and AAR response genes (ASNS and CHOP) and broadly regulated the transcriptome and proteome in cardiac fibroblasts. The critical role of GCN2 in mediating the downstream effects of halofuginone was demonstrated in GCN2‐null MEFs in which halofuginone failed to activate CHOP transcription. We also examined the converse by creating a constitutively active mammalian GCN2 with S808G and F855L mutations that have been demonstrated in a yeast homologue to prevent autoinhibition of the C‐terminal domain3, 17 (Figure S7). The overexpression of the constitutively active GCN2 mutant mimicked the effects of halofuginone by robustly enhancing pGCN2 and p‐eIF2α and inhibiting collagen deposition. Taken together, these results demonstrate the necessary and sufficient role of GCN2 in regulating the AAR when activated by halofuginone. In addition, the ability of l‐proline (but not l‐threonine) supplementation to block virtually all of the effects of halofuginone evaluated in this study suggest a highly selective and reversible cellular action of halofuginone consistent with competitive inhibition of prolyl‐tRNA synthetase.
Reducing amino acid intake, especially essential amino acids, has been shown to activate the AAR and reduces food consumption, leading to caloric restriction and decreased body weight.18, 19 In our in vivo studies, halofuginone at high does (1‐3 mg/kg) activated the AAR and reduced food intake and body weight. However, the cardioprotective effects of halofuginone could not be attributed to caloric restriction because pair‐fed controls exhibited similar pathology to a control AngII/PE group that was fed ad libitum. In addition, halofuginone treatment also reduced the effects of cardiac stress at lower doses that did not affect food intake or body weight. These results rule out potentially confounding effects of caloric restriction and support activation of the AAR as a principal action of halofuginone in these studies.
Potential mechanisms underlying the cardioprotective actions of halofuginone were evaluated in human cardiac fibroblasts and iPSC‐derived cardiomyocytes. In a cardiac fibroblast collagen deposition assay, halofuginone and amino acid deficiency abolished spontaneous collagen production and deposition by limiting collagen gene transcription at concentrations that had no effect on cell count or cell health. In addition to type I collagen, halofuginone (100‐300 nmol/L) significantly downregulated 40 to 50 ECM‐related proteins, suggesting a powerful effect on extracellular matrix remodeling consistent with the reduction in hydroxyproline content observed in vivo. This powerful effect on the ECM occurs at both the transcriptional level (mRNA) and translational level (protein) and may be related to recently described attenuation of TGFβ‐mediated ALK5/SMAD2/3 signaling.20 In addition, halofuginone could also impact myofibroblast differentiation shown by the 6‐fold downregulation of αSMA mRNA (Table S2). However, differential effects of ALK5 inhibitors and halofuginone on collagen deposition (data not shown) suggest that further work is needed to understand the role of TGFβ signaling in mediating halofuginone actions.
The effects of halofuginone on pathologic hypertrophic signaling were evaluated in human iPSC‐derived cardiomyocytes. Specifically, ET‐1 was used to activate the Gαq hypertrophic pathway and regulation of the pathological gene program, as exemplified by NPPA mRNA upregulation. Halofuginone abolished ET‐1 mediated upregulation of NPPA mRNA, representing the first description of blockade of Gαq‐mediated cardiac hypertrophic gene expression by tRNA synthetase inhibition. However, it is not known precisely how tRNA synthetase inhibition abolishes ET‐1–mediated Gαq activation and subsequent Ca2+/calcineurin/NFAT‐ dependent and/or MAPK‐dependent regulation of gene transcription (for review see Mudd and Kass21). This is an important area for future investigation.
Halofuginone also significantly enhanced indices (LC3B, p62) of autophagy in cardiomyocytes similar to amino acid deprivation, which also activates autophagy through a GCN2‐dependent mechanism (reviewed in Carroll et al22). In addition, overexpression of the constitutively active GCN2 mutant mimicked the effects of halofuginone by robustly increasing p‐GCN2 and p‐eIF2α and increasing the LC3B II/I ratio, suggesting enhanced autophagic flux (Figure S8). Halofuginone regulation of autophagy may be related to GCN2/eIF2α‐dependent expression of critical autophagosome components23 and/or inhibition of amino acid activation of mTOR (a critical regulator of autophagy). Notably there was no regulation of E3 ubiquitin ligase mRNA levels such as Murf (SMURF1), Atrogin (FBXO32), or Parkin (PARK2) with the exception of SMURF2 (up 2.2‐fold) by halofuginone (Table S2). Of note, it was recently reported that amino acid starvation leads to induction of Sestrin2 in a GCN2/ATF‐dependent manner, which sustains the repression of mTORC1.24
The robust effects of halofuginone at the level of the cardiac fibroblast and cardiomyocyte are likely to play important roles in mitigating the effects of cardiac stress. However, we cannot rule out important known systemic actions of halofuginone related to tRNA synthetase inhibition. Specifically, halofuginone has powerful anti‐inflammatory effects mediated by inhibition of Th17 signaling that are protective in a number of disease models such as autoimmune encephalomyelitis and graft‐versus‐host disease.9, 25, 26 It is noteworthy that an imbalance of Th17 cells has been reported in patients with chronic heart failure27 and is correlated with the severity of myocardial dysfunction in a rat model of heart failure.28 Therefore, halofuginone may also limit cardiac stress through inhibition of Th17 cells.
A recently reported study conducted in GCN2 knockout mice does not appear to support the beneficial effects of halofuginone‐mediated GCN2 activation described in our present study.29 In fact, these investigators found that GCN2‐knockout mice exhibited reduced contractile dysfunction, lung congestion, and inflammation following cardiac stress induced by TAC. The explanation for this discrepancy most likely lies in the fundamental differences between genetic and pharmacologic manipulation of the pathway. The episodic modulation of a specific activity within a pathway as expected by pharmacologic manipulation may cause effects very different from a permanent ablation of a protein having multiple actions and interactions. This is analogous to reported phenotypic differences observed between genetically engineered knockout and catalytically dead knockin mouse models.30 Nonetheless, the precise mechanism that account for these differences remain to be determined.
As mentioned above, CHOP expression was used in the present study as a marker of eIF2α activation by the AAR. However, CHOP is an important integrated stress response mediator regulating oxidative stress and apoptosis and is likely to play a role in the protective effects of halofuginone.31, 32 However, the effects of CHOP appear to be complex, demonstrating protection when knocked out in the setting of cardiac pressure overload and in ischemia and reperfusion33, 34 and deleterious when knocked out in a mouse model of myocardial infarction caused by permanent coronary artery ligation.35 Further studies are required to determine the role of CHOP in mediating the cardioprotective effects of halofuginone/AAR.
Despite the compelling effects to reduce cardiac stress, activation of the AAR by inhibiting tRNA synthetase is likely to be problematic if prolonged. In this regard, a reduction of the intracellular pool of charged tRNA and inhibition of protein translation through p‐eIF2α would be expected to significantly reduce synthesis of essential proteins. In fact, halofuginone exhibits significant cellular toxicity at concentrations >1 μmol/L in cardiac fibroblasts (data not shown) and has a limited safety window in mice.36 These liabilities are further compounded by the significant accumulation of halofuginone in liver, kidney, and other organs.36 Thus, the limitations of activating the AAR by inhibition of tRNA synthetase may be mitigated by intermittent dosing regimens and/or by inhibitors with improved distribution and clearance properties. Furthermore, future studies will be needed to determine if strategic deprivation of dietary amino acids could be used to blunt the effects of cardiac stress.
In summary, our studies demonstrated that halofuginone acts selectively to activate the AAR pathway, and its actions in vivo blunt the effects of cardiac stress in the failing myocardium. Activation of AAR appears to mediate the antihypertrophic, antifibrotic, and autophagic effects of halofuginone and represents a compelling and novel approach to the treatment of heart failure (Figure 8).
Figure 8.
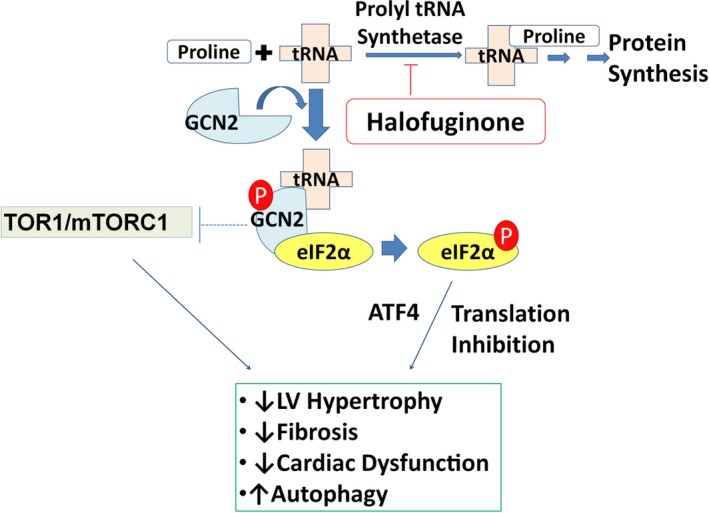
Mechanisms of halofuginone action. Under normal conditions, prolyl‐tRNA is aminoacylated with l‐proline by prolyl‐tRNA synthetase and made available for protein synthesis. Halofuginone inhibits the action of prolyl‐tRNA synthetase and in turn generates elevated levels of uncharged prolyl‐tRNA that can bind GCN2 (general control nonderepressible 2). Once bound by uncharged tRNA, GCN2 is activated by autophosphorylation and then phosphorylates eIF2α (eukaryotic translation initiation factor 2α), which leads to gene transcription and activation of the AAR (amino acid response) pathway.
Sources of Funding
The study conducted in Figure S3 was supported by grants from the National Heart, Lung, and Blood Institute to Force (HL061688, HL119234) and American Heart Association Scientist Development Grant (13SDG16930103) to Lal. All the rest of the studies were funded by GlaxoSmithKline.
Disclosures
None.
Supporting information
Data S1. Material and Methods
Table S1. Differentially Regulated Protein by 300 nmol/L Halofuginone
Table S2. Differentially Regulated mRNA by 200 nmol/L Halofuginone
Table S3. FPKM Values From RNAseq Analysis
Table S4. Ingenuity pathway analysis of differentially regulated transcriptome.
Table S5. Ingenuity pathway analysis of differentially regulated proteome.
Figure S1. Borrelidin induced amino acid response (AAR) in fibroblasts. Human cardiac fibroblasts were treated with Borrelidin (100 nmol/L), l‐proline (2 mmol/L), and l‐threonine (2 mmol/L) for 24 hours. Protein levels of phosphorylated general control nonderepressible 2 (pGCN2), total GCN2, and β‐actin were examined by Western blot (A), and mRNA level of C/EBP homologous protein (CHOP) was examined by reverse transcription polymerase chain reaction (RT‐PCR, [B], n=4). One‐way ANOVA was used for statistical analysis of log‐transformed data. ***P<0.001, ****P<0.0001. Wild‐type (WT) or GCN2 knockout (GCN2KO) mouse embryonic fibroblasts (MEF) were treated with increasing concentrations of Halofuginone for 24 hours. CHOP mRNA was examined by RT‐PCR. Unpaired t test was used for statistical analysis of log‐transformed data, ****P<0.0001 (C, n=4).
Figure S2. Halofuginone improves fractional area change (FAC) in transverse aortic constriction (TAC) mouse model. C57Bl/6J mice were subjected to TAC to induce heart failure and treated with halofuginone as described in Figure 6. FAC was measured by echocardiography. There were 6, 9, and 7 mice in the sham, TAC, and TAC+halofuginone groups. Unpaired t test. ***P<0.001.
Figure S3. Halofuginone reduces infarct size in cardiac ischemia/reperfusion (I/R) injury. C57BL/6 mice (11 mice in each group) were treated with 6 μg of halofuginone daily (≈2 mg/kg) for 3 days. I/R injury was then induced by ligation of coronary artery for 30 minutes and reperfusion for 24 hours. The heart was harvested with staining with Evans blue and triphenyl tetrazolium chloride (TTC) dye. Representative heart images from control (A) and halofuginone‐treated animals (B) were shown. Infarct size (as percetage of area at risk) and area at risk (as percentage of left ventricle) were plotted (C). Unpaired t test, **P<0.01.
Figure S4. Borrelidin inhibits collagen deposition in cardiac fibroblast. Normal human cardiac fibroblasts were incubated with media containing ficoll to stimulate collagen deposition. Borrelidin was added at the time of ficoll treatment. The deposition of mature collagen by fibroblasts was examined by immunohistochemistry (A, n=5). Fibroblasts were treated with 10 nmol/L borrelidin, 2 mmol/L l‐proline, or 2 mmol/L l‐threonine for 24 hours. At the end of the treatment, collagen deposition and Type I collagen 1α1 (COL1A1) mRNA (B, n=4) were measured by immunohistochemistry and reverse transcription polymerase chain reaction (RT‐PCR), respectively. One‐way ANOVA comparisons were used to analyze raw collagen deposition data and log‐transformed gene expression data, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Figure S5. Amino acid starvation inhibits collagen deposition. WI38 fibroblast cells were stimulated with either complete Dulbecco's modified Eagle medium (DMEM) with ficoll or DMEM deficient in l‐lysine, l‐leucine, l‐arginine, and l‐gulatmine with ficoll (A, n=5). Twenty‐four hours later, cells were fixed with methanol and immunostained with anti–mature type I collagen antibody. Normal human cardiac fibroblasts were stimulated with ficoll medium and treated with l‐leucinol, an l‐leucine analog15,16 and inhibitor of Leucyl‐tRNA synthetase (B, n=5). Twenty‐four hours later, cells were fixed with methanol and immunostained with anti–mature type I collagen antibody. Unpaired t test ***P<0.001.
Figure S6. Two key differentially regulated pathways. The top 100 downregulated proteins (from proteomic study) in cardiac fibroblasts were grouped according to their functions or properties based on Ingenuity description and the published literature. Differentially expressed heart failure–related proteins are shown (A). Differentially expressed transforming growth factor β (TGFβ) and Wnt‐related proteins are shown (B).
Figure S7. Overexpression of general control nonderepressible 2 (GCN2) activates amino acid response (AAR) and reduces collagen deposition. Normal human cardiac fibroblasts were transfected with BacMam virus encoding a constitively active form of GCN2 or treated with halofuginone in the presence of ficoll medium. The expression of total GCN2, phosphorylated GCN2 (p‐GCN2), total eukaryotic translation initiation factor 2α (eIF2α), phosphorylated eIF2α (peIF2α), and tubulin were detected by Western blot (A). Deposition of collagen was determined by immunohistochemistry (B) and quantitated by image analysis (C, n=3). A BacMam virus that does not carry any protein was used as negative control and had no effect on AAR activation or collagen deposition at similar multiplicity of transfection (MOT, data not shown).
Figure S8. Overexpression of general control nonderepressible 2 (GCN2) activates amino acid response (AAR) and enhance autophagy in cardiomyocytes. Human induced pluripotent stem cell (iPSC)‐derived cardiomyocytes were transfected with a BacMam virus encoding a constitutively active form of GCN2. The expression of total GCN2, phosphorylated GCN2 (p‐GCN2), total eukaryotic translation initiation factor 2α (eIF2α), phosphorylated‐eIF2α (p‐eIF2α), microtubule‐associated proteins 1A/1B light chain (LC3BI and LC3BII) and tubulin were detected by Western blot (A). Band intensity values normalized by the housekeeper tubulin were used to compare relative expression levels. LC3B ratio is shown as the normalized value of LC3BII over LC3BI (B).
Acknowledgments
We would like to thank Mark E. Burgert for his recommendation and review of the statistics analyses. We would also like to thank Christopher Traini and Shanker Kalyana Sundaram for their help in submitting the RNAseq data to GEO.
(J Am Heart Assoc. 2017;6:e004453 DOI: 10.1161/JAHA.116.004453.)28487390
References
- 1. Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sikalidis AK. Cellular and animal indispensable amino acid limitation responses and health promotion. Can the two be linked? A critical review. Int J Food Sci Nutr. 2013;64:300–311. [DOI] [PubMed] [Google Scholar]
- 3. Dong J, Qiu H, Garcia‐Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA‐binding domain. Mol Cell. 2000;6:269–279. [DOI] [PubMed] [Google Scholar]
- 4. Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim YJ, Lee HK, Cortese JF, Wirth DF, Dignam JD, Rao A, Yeo CY, Mazitschek R, Whitman M. Halofuginone and other febrifugine derivatives inhibit prolyl‐tRNA synthetase. Nat Chem Biol. 2012;8:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guo F, Cavener DR. The GCN2 eIF2α kinase regulates fatty‐acid homeostasis in the liver during deprivation of an essential amino acid. Cell Metab. 2007;5:103–114. [DOI] [PubMed] [Google Scholar]
- 6. Huebner KD, Jassal DS, Halevy O, Pines M, Anderson JE. Functional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginone. Am J Physiol Heart Circ Physiol. 2008;294:H1550–H1561. [DOI] [PubMed] [Google Scholar]
- 7. Turgeman T, Hagai Y, Huebner K, Jassal DS, Anderson JE, Genin O, Nagler A, Halevy O, Pines M. Prevention of muscle fibrosis and improvement in muscle performance in the mdx mouse by halofuginone. Neuromusc Disord. 2008;18:857–868. [DOI] [PubMed] [Google Scholar]
- 8. Peng W, Robertson L, Gallinetti J, Mejia P, Vose S, Charlip A, Chu T, Mitchell JR. Surgical stress resistance induced by single amino acid deprivation requires Gcn2 in mice. Sci Transl Med. 2012;4:118ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule‐Smith A, Lefebvre RE, Unutmaz D, Mazitschek R, Waldner H, Whitman M, Keller T, Rao A. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324:1334–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richards DA, Bao W, Rambo MV, Burgert M, Jucker BM, Lenhard SC. Examining the relationship between exercise tolerance and isoproterenol‐based cardiac reserve in murine models of heart failure. J Appl Physiol (1985). 2013;114:1202–1210. [DOI] [PubMed] [Google Scholar]
- 11. Chen CZ, Peng YX, Wang ZB, Fish PV, Kaar JL, Koepsel RR, Russell AJ, Lareu RR, Raghunath M. The Scar‐in‐a‐Jar: studying potential antifibrotic compounds from the epigenetic to extracellular level in a single well. Br J Pharmacol. 2009;158:1196–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA‐Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vizcaino JA, Csordas A, Del‐Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dorn GW, Brown JH. Gq signaling in cardiac adaptation and maladaptation. Trends Cardiovasc Med. 1999;9:26–34. [DOI] [PubMed] [Google Scholar]
- 15. Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rusten TE, Stenmark H. p62, an autophagy hero or culprit? Nat Cell Biol. 2010;12:207–209. [DOI] [PubMed] [Google Scholar]
- 17. Qiu H, Hu C, Dong J, Hinnebusch AG. Mutations that bypass tRNA binding activate the intrinsically defective kinase domain in GCN2. Genes Dev. 2002;16:1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harper AE, Peters JC. Protein intake, brain amino acid and serotonin concentrations and protein self‐selection. J Nutr. 1989;119:677–689. [DOI] [PubMed] [Google Scholar]
- 19. Morrison CD, Reed SD, Henagan TM. Homeostatic regulation of protein intake: in search of a mechanism. Am J Physiol Regul Integr Comp Physiol. 2012;302:R917–R928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cui Z, Crane J, Xie H, Jin X, Zhen G, Li C, Xie L, Wang L, Bian Q, Qiu T, Wan M, Ding S, Yu B, Cao X. Halofuginone attenuates osteoarthritis by inhibition of TGF‐β activity and H‐type vessel formation in subchondral bone. Ann Rheum Dis. 2015;75:1714–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mudd JO, Kass DA. Tackling heart failure in the twenty‐first century. Nature. 2008;451:919–928. [DOI] [PubMed] [Google Scholar]
- 22. Carroll B, Korolchuk VI, Sarkar S. Amino acids and autophagy: cross‐talk and co‐operation to control cellular homeostasis. Amino Acids. 2015;47:2065–2088. [DOI] [PubMed] [Google Scholar]
- 23. B'chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2α/ATF4 pathway is essential for stress‐induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ye J, Palm W, Peng M, King B, Lindsten T, Li MO, Koumenis C, Thompson CB. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev. 2015;29:2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng H, Tian J, Zeng L, Pan B, Li Z, Song G, Chen W, Xu K. Halofugine prevents cutaneous graft versus host disease by suppression of Th17 differentiation. Hematology. 2012;17:261–267. [DOI] [PubMed] [Google Scholar]
- 26. Carlson TJ, Pellerin A, Djuretic IM, Trivigno C, Koralov SB, Rao A, Sundrud MS. Halofuginone‐induced amino acid starvation regulates Stat3‐dependent Th17 effector function and reduces established autoimmune inflammation. J Immunol. 2014;192:2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li N, Bian H, Zhang J, Li X, Ji X, Zhang Y. The Th17/Treg imbalance exists in patients with heart failure with normal ejection fraction and heart failure with reduced ejection fraction. Clin Chim Acta. 2010;411:1963–1968. [DOI] [PubMed] [Google Scholar]
- 28. Zhang Q, Hu LQ, Yin CS, Chen P, Li HQ, Sun X, Yan G. Catechin ameliorates cardiac dysfunction in rats with chronic heart failure by regulating the balance between Th17 and Treg cells. Inflamm Res. 2014;63:619–628. [DOI] [PubMed] [Google Scholar]
- 29. Lu Z, Xu X, Fassett J, Kwak D, Liu X, Hu X, Wang H, Guo H, Xu D, Yan S, McFalls EO, Lu F, Bache RJ, Chen Y. Loss of the eukaryotic initiation factor 2α kinase general control nonderepressible 2 protects mice from pressure overload‐induced congestive heart failure without affecting ventricular hypertrophy. Hypertension. 2014;63:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rommel C, Vanhaesebroeck B, Vogt PK. More than just kinases: the scaffolding functions of PI3K . In: Costa C, Hirsch E, eds. Phosphoinositide 3‐kinase in health and disease. Springer; 2011:171–181. [Google Scholar]
- 31. Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. 2008;118:3378–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Silva RM, Ries V, Oo TF, Yarygina O, Jackson‐Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005;95:974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K, Hokimoto S, Yamamuro M, Iwawaki T, Gotoh T, Ogawa H, Oike Y. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1124–1132. [DOI] [PubMed] [Google Scholar]
- 34. Fu HY, Okada K, Liao Y, Tsukamoto O, Isomura T, Asai M, Sawada T, Okuda K, Asano Y, Sanada S, Asanuma H, Asakura M, Takashima S, Komuro I, Kitakaze M, Minamino T. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum‐mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122:361–369. [DOI] [PubMed] [Google Scholar]
- 35. Luo G, Li Q, Zhang X, Shen L, Xie J, Zhang J, Kitakaze M, Huang X, Liao Y. Ablation of C/EBP homologous protein increases the acute phase mortality and doesn't attenuate cardiac remodeling in mice with myocardial infarction. Biochem Biophys Res Commun. 2015;464:201–207. [DOI] [PubMed] [Google Scholar]
- 36. Stecklair KP, Hamburger DR, Egorin MJ, Parise RA, Covey JM, Eiseman JL. Pharmacokinetics and tissue distribution of halofuginone (NSC 713205) in CD2F1 mice and Fischer 344 rats. Cancer Chemother Pharmacol. 2001;48:375–382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Material and Methods
Table S1. Differentially Regulated Protein by 300 nmol/L Halofuginone
Table S2. Differentially Regulated mRNA by 200 nmol/L Halofuginone
Table S3. FPKM Values From RNAseq Analysis
Table S4. Ingenuity pathway analysis of differentially regulated transcriptome.
Table S5. Ingenuity pathway analysis of differentially regulated proteome.
Figure S1. Borrelidin induced amino acid response (AAR) in fibroblasts. Human cardiac fibroblasts were treated with Borrelidin (100 nmol/L), l‐proline (2 mmol/L), and l‐threonine (2 mmol/L) for 24 hours. Protein levels of phosphorylated general control nonderepressible 2 (pGCN2), total GCN2, and β‐actin were examined by Western blot (A), and mRNA level of C/EBP homologous protein (CHOP) was examined by reverse transcription polymerase chain reaction (RT‐PCR, [B], n=4). One‐way ANOVA was used for statistical analysis of log‐transformed data. ***P<0.001, ****P<0.0001. Wild‐type (WT) or GCN2 knockout (GCN2KO) mouse embryonic fibroblasts (MEF) were treated with increasing concentrations of Halofuginone for 24 hours. CHOP mRNA was examined by RT‐PCR. Unpaired t test was used for statistical analysis of log‐transformed data, ****P<0.0001 (C, n=4).
Figure S2. Halofuginone improves fractional area change (FAC) in transverse aortic constriction (TAC) mouse model. C57Bl/6J mice were subjected to TAC to induce heart failure and treated with halofuginone as described in Figure 6. FAC was measured by echocardiography. There were 6, 9, and 7 mice in the sham, TAC, and TAC+halofuginone groups. Unpaired t test. ***P<0.001.
Figure S3. Halofuginone reduces infarct size in cardiac ischemia/reperfusion (I/R) injury. C57BL/6 mice (11 mice in each group) were treated with 6 μg of halofuginone daily (≈2 mg/kg) for 3 days. I/R injury was then induced by ligation of coronary artery for 30 minutes and reperfusion for 24 hours. The heart was harvested with staining with Evans blue and triphenyl tetrazolium chloride (TTC) dye. Representative heart images from control (A) and halofuginone‐treated animals (B) were shown. Infarct size (as percetage of area at risk) and area at risk (as percentage of left ventricle) were plotted (C). Unpaired t test, **P<0.01.
Figure S4. Borrelidin inhibits collagen deposition in cardiac fibroblast. Normal human cardiac fibroblasts were incubated with media containing ficoll to stimulate collagen deposition. Borrelidin was added at the time of ficoll treatment. The deposition of mature collagen by fibroblasts was examined by immunohistochemistry (A, n=5). Fibroblasts were treated with 10 nmol/L borrelidin, 2 mmol/L l‐proline, or 2 mmol/L l‐threonine for 24 hours. At the end of the treatment, collagen deposition and Type I collagen 1α1 (COL1A1) mRNA (B, n=4) were measured by immunohistochemistry and reverse transcription polymerase chain reaction (RT‐PCR), respectively. One‐way ANOVA comparisons were used to analyze raw collagen deposition data and log‐transformed gene expression data, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Figure S5. Amino acid starvation inhibits collagen deposition. WI38 fibroblast cells were stimulated with either complete Dulbecco's modified Eagle medium (DMEM) with ficoll or DMEM deficient in l‐lysine, l‐leucine, l‐arginine, and l‐gulatmine with ficoll (A, n=5). Twenty‐four hours later, cells were fixed with methanol and immunostained with anti–mature type I collagen antibody. Normal human cardiac fibroblasts were stimulated with ficoll medium and treated with l‐leucinol, an l‐leucine analog15,16 and inhibitor of Leucyl‐tRNA synthetase (B, n=5). Twenty‐four hours later, cells were fixed with methanol and immunostained with anti–mature type I collagen antibody. Unpaired t test ***P<0.001.
Figure S6. Two key differentially regulated pathways. The top 100 downregulated proteins (from proteomic study) in cardiac fibroblasts were grouped according to their functions or properties based on Ingenuity description and the published literature. Differentially expressed heart failure–related proteins are shown (A). Differentially expressed transforming growth factor β (TGFβ) and Wnt‐related proteins are shown (B).
Figure S7. Overexpression of general control nonderepressible 2 (GCN2) activates amino acid response (AAR) and reduces collagen deposition. Normal human cardiac fibroblasts were transfected with BacMam virus encoding a constitively active form of GCN2 or treated with halofuginone in the presence of ficoll medium. The expression of total GCN2, phosphorylated GCN2 (p‐GCN2), total eukaryotic translation initiation factor 2α (eIF2α), phosphorylated eIF2α (peIF2α), and tubulin were detected by Western blot (A). Deposition of collagen was determined by immunohistochemistry (B) and quantitated by image analysis (C, n=3). A BacMam virus that does not carry any protein was used as negative control and had no effect on AAR activation or collagen deposition at similar multiplicity of transfection (MOT, data not shown).
Figure S8. Overexpression of general control nonderepressible 2 (GCN2) activates amino acid response (AAR) and enhance autophagy in cardiomyocytes. Human induced pluripotent stem cell (iPSC)‐derived cardiomyocytes were transfected with a BacMam virus encoding a constitutively active form of GCN2. The expression of total GCN2, phosphorylated GCN2 (p‐GCN2), total eukaryotic translation initiation factor 2α (eIF2α), phosphorylated‐eIF2α (p‐eIF2α), microtubule‐associated proteins 1A/1B light chain (LC3BI and LC3BII) and tubulin were detected by Western blot (A). Band intensity values normalized by the housekeeper tubulin were used to compare relative expression levels. LC3B ratio is shown as the normalized value of LC3BII over LC3BI (B).