

Abstract
Background
There is increasing evidence implicating atrial mitochondrial dysfunction in the pathogenesis of atrial fibrillation. In this study, we explored whether alogliptin, a dipeptidyl peptidase‐4 inhibitor, can prevent mitochondrial dysfunction and atrial remodeling in a diabetic rabbit model.
Methods and Results
A total of 90 rabbits were randomized into 3 groups as follows: control group (n=30), alloxan‐induced diabetes mellitus group (n=30), and alogliptin‐treated (12.5 mg/kg per day for 8 weeks) diabetes mellitus group (n=30). Echocardiographic and hemodynamic assessments were performed in vivo. The serum concentrations of glucagon‐like peptide‐1, insulin, and inflammatory and oxidative stress markers were measured. Electrophysiological properties of Langendorff‐perfused rabbit hearts were assessed. Mitochondrial morphology, respiratory function, membrane potential, and reactive oxygen species generation rate were assessed. The protein expression of transforming growth factor β1, nuclear factor κB p65, and mitochondrial biogenesis–related proteins were measured by Western blot analysis. Diabetic rabbits exhibited left ventricular hypertrophy and left atrial dilation without obvious hemodynamic abnormalities, and all of these changes were attenuated by alogliptin. Compared with the control group, higher atrial fibrillation inducibility in the diabetes mellitus group was observed, and markedly reduced by alogliptin. Alogliptin decreased mitochondrial reactive oxygen species production rate, prevented mitochondrial membrane depolarization, and alleviated mitochondrial swelling in diabetic rabbits. It also improved mitochondrial biogenesis by peroxisome proliferator–activated receptor‐γ coactivator 1α/nuclear respiratory factor‐1/mitochondrial transcription factor A signaling regulated by adiponectin/AMP‐activated protein kinase.
Conclusions
Dipeptidyl peptidase‐4 inhibitors can prevent atrial fibrillation by reversing electrophysiological abnormalities, improving mitochondrial function, and promoting mitochondrial biogenesis.
Keywords: atrial fibrillation, dipeptidyl peptidase‐4 inhibitors, mitochondrial biogenesis, mitochondrial function
Subject Categories: Arrhythmias, Electrophysiology
Clinical Perspective
What is New?
Impaired mitochondrial function and biogenesis was demonstrated in left atrial tissue from a diabetic rabbit model with preserved cardiac function.
Dipeptidyl peptidase‐4 inhibitors can potentially improve mitochondrial function and biogenesis and ameliorate atrial arrhythmic substrate in diabetes mellitus.
What are the Clinical Implications?
Mitochondria are attractive upstream targets for prevention of atrial fibrillation in patients with diabetes mellitus.
Dipeptidyl peptidase‐4 inhibitors can act upstream to prevent mitochondrial abnormalities and, in turn, induce reverse electrophysiological and structural remodeling of the atrium.
Introduction
Atrial fibrillation (AF) is the most frequent sustained arrhythmia in clinical practice and presents a high risk of thromboembolism.1 Its incidence is increasing worldwide owing to an aging population and higher prevalence of cardiovascular risk factors. Of the latter, diabetes mellitus (DM) is one of the most common chronic metabolic diseases. Findings from experimental and clinical studies suggest a strong relationship between DM and AF.2, 3, 4 Our group has previously demonstrated that oxidative stress and inflammation play an important role in the pathogenesis of AF in diabetic rabbits, and antioxidant therapies have proved beneficial in preventing the atrial structural and electrical remodeling.4, 5 However, these processes may only partially explain the high risk of AF in DM patients. Other potential mechanisms include alterations in energy metabolism, cellular calcium handling, ion channel function, and aberrant conduction.6, 7 All of these cellular processes are energy dependent and therefore require normal mitochondrial function.
The mitochondria are the cellular powerhouses responsible for ATP generation, forming a network surrounding the sarcoplasmic reticulum, myofilaments, and transverse tubules. They have additional functions such as signaling transduction, redox state control, and cellular apoptosis.8 In addition to oxidative stress, abnormalities in calcium handling, mitochondria DNA damage, and mitochondrial dysfunction readily disrupt cardiac rhythms through depleting energy supply to the ion channels and transporters. An emerging body of evidence implicates disturbances of energy metabolism in the pathogenesis of AF.9, 10, 11 Recently, Yan et al12 found that adiponectin, a metabolism‐related protein secreted by adipose tissue that possesses potent cardioprotective effects, attenuated myocardial infarction injury and improved mitochondrial biogenesis by AMP‐activated protein kinase (AMPK)/peroxisome proliferator–activated receptor‐γ coactivator 1α (PGC‐1α) signaling in diabetic hearts. Moreover, impaired mitochondrial biogenesis and energy metabolism were observed during rapid atrial pacing‐induced AF in rabbits.13 However, limited data exist as to whether adiponectin/AMPK/PGC‐1α signaling contributes to mitochondrial dysfunction and initiation or maintenance of AF in diabetic hearts.
Dipeptidyl peptidase‐4 (DPP‐4) inhibitors are glucose‐lowering agents for the treatment of type 2 DM mainly by inhibiting the degradation of glucagon‐like peptide‐1 (GLP‐1). Besides glycemic control, DPP‐4 inhibitors have been shown to have cardiovascular protective effects in the ischemic heart model.14, 15 The DDP‐4 inhibitor alogliptin was found to shorten the duration of AF in a heart failure model of rabbit induced by ventricular tachycardia pacing.16 Keller et al17 reported that saxagliptin, a DPP‐4 inhibitor, can restore vascular mitochondrial adaptation to exercise in a diabetic rodent model and increase the expression of mitochondrial complexes, cytochrome c, endothelial nitric oxide synthase, and PGC‐1α. Furthermore, vildagliptin and sitagliptin attenuated cardiac dysfunction and prevented cardiac mitochondrial dysfunction in obese insulin‐resistant rats.18 However, the effects of DPP‐4 inhibitors on the atrial metabolic disturbance associated with AF are not completely understood. Therefore, we aimed to investigate the effects of alogliptin, a DPP‐4 inhibitor, on atrial mitochondrial remodeling and AF associated with metabolic stress.
Methods
Experimental Animals and Protocol
This study was approved by the Experimental Animal Administration Committee of Tianjin Medical University and Tianjin Municipal Commission for Experimental Animal Control.
Japanese white rabbits (1.8–2.2 kg) were purchased from Beijing Medical Animals Research Institute. A computer was used to generate 90 different random numbers corresponding to the rabbits. The first 30 rabbits were taken as the control group, the middle 30 rabbits as the DM group, and the remaining 30 rabbits as the alogliptin‐treated DM (DM‐A) group. In each group, the first 10 rabbits were used for the first part of the experiments (including echocardiographic, hemodynamic, histological, and serum biochemical and oxidative stress–related markers examination, as well as Western blot and real‐time polymerase chain reaction [PCR] analysis). The middle 10 rabbits were used for the electrophysiological studies, and the remaining 10 rabbits were used for examinations of mitochondrial function.
Diabetic Rabbit Model
In the DM and DM‐A groups, 120 mg/kg of 5% alloxan (alloxan monohydrate, Sigma Aldrich Chemical) dissolved in sterile normal saline were injected intravenously into the marginal ear vein of rabbits for induction of DM. Fasting blood glucose was measured 48 hours later, and rabbits with glucose levels ≥14 mmol/L were used for subsequent experimentation. If the fasting blood glucose level did not reach diagnostic criteria after 48 hours, then the same dose of alloxan was injected again. If the fasting blood glucose level still did not meet the criteria, the rabbit was excluded from the study. This process was repeated until a sufficient number of diabetic rabbits were induced. After successful induction of DM, fasting blood glucose concentration was monitored weekly using the glucometer Optium Xceed (Abbott Laboratories MediSense Products). After the establishment of DM, animals in the DM‐A group were administrated alogliptin (12.5 mg/kg per day; Tianjin Takeda Pharmaceuticals Co, Ltd) for 8 weeks.
Echocardiographic Assessment
After 8 weeks, transthoracic echocardiography was performed by blinded operators. The rabbits were anesthetized with 3% pelltobarbitalum natricum (30 mg/kg) and placed on the table in the left lateral decubitus position. Echocardiographic parameters were obtained in the parasternal long‐axis view using a GE Vingmed machine (Vivid 7/Vingmed General Electric) equipped with a 7.5‐MHz standard pediatric probe. Left atrial (LA) anteroposterior diameter, left ventricular posterior wall thickness, interventricular septal thickness, left ventricular end‐diastolic dimension, and left ventricular end‐systolic dimension were measured using both 2‐dimensional and M‐mode imaging during 5 consecutive cardiac cycles. Left ventricular ejection fraction was calculated according to our previous study.5 The average of 3 measurements was calculated for subsequent analysis.
Hemodynamic Study and Sample Collection
At the end of echocardiographic examination, each rabbit underwent right carotid artery cannulation for measuring hemodynamic parameters during ECG monitoring. Heart rate, aortic systolic blood pressure, diastolic blood pressure, and mean blood pressure were recorded carefully after a stabilization period using a BL‐ICF biological function detection system (Chengdu Taimeng Science and Technology Co, Ltd). A cannula was then inserted through the aortic valve to the left ventricle to measure the ventricular end‐diastolic pressure and maximal and minimal rates of the rise in left ventricular pressure.
Subsequently, blood samples were obtained from the carotid artery for measurements of serum biochemical, inflammatory, and oxidative stress markers. The animals were then immediately euthanized and the LA tissues were collected, frozen immediately in liquid nitrogen, and stored at −80°C. Small pieces of LA tissue were immersed in 10% formaldehyde and 2.5% glutaraldehyde for histological and ultrastructural studies, respectively.
Serum Biochemical, Inflammatory, and Oxidative Stress Measurements
Serum biochemical examination including fasting glucose at the 8th week, total cholesterol, triglycerides, low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, and creatinine level were detected using a full automatic biochemical analyzer. Fasting insulin and GLP‐1 level were assessed using a rabbit insulin ELISA kKit (Wuhan Huamei Biological Engineering Co, Ltd) and rabbit GLP‐1 ELISA kit (Shanghai Huding Biological Technology Co) according to the manufacturers' instructions, respectively. A lipid peroxidation malondialdehyde (MDA) assay kit (Nanjing Jianchen Bioengineering Institute) was used to detect the serum MDA level, and another oxidative stress–related marker serum 8‐hydroxy‐2′‐deoxyguanosine (8‐OHdG) was assessed by a rabbit 8‐OHdG ELISA kit (Shanghai Huding Biological Technology Co). Serum antioxidant enzyme superoxide dismutase activity was evaluated by an antioxidant enzyme activities kit (Nanjing Jianchen Bioengineering Institute). Finally, the inflammation marker high‐sensitivity C‐reactive protein level was also detected using a rabbit high‐sensitivity C‐reactive protein ELISA kit (Wuhan Huamei Biological Engineering Co, Ltd).
Histological and Ultrastructural Analyses
The LA myocardium was cut at 4‐μm intervals and stained with hematoxylin and eosin and Masson's trichrome stains to evaluate the cardiomyocyte diameter and the extent of interstitial fibrosis, respectively. The cell borders were measured in the short‐axis view with a visible mononucleus from 10 random fields, and an average of 40 cardiomyocytes per animal were analyzed from each animal in the group. Micrographs were digitized using Photoshop 7.0 (Adobe). To quantify the areas of interstitial fibrosis in the LA myocardium, the blue pixel content of the digitized images, excluding the perivascular fibrotic areas, were measured relative to the total tissue area using Image‐Pro Plus 6.0 Scion image software (Scion Corporation).
Atrial tissues that had been fixated in 2.5% glutaraldehyde for 2 hours were used for ultrastructural analysis. After being further fixated in 1% osmium tetroxide, dehydrated in ethanol, and embedded in Epon, ultrathin sections were cut from each sample. Finally, each ultrathin section was counterstained with uranium acetate and lead citrate and evaluated under H‐7650 transmission electron microscope (Hitachi).
Surface ECGs and Electrophysiological Studies
Surface ECGs were performed before the electrophysiological study. To evaluate the electrophysiological properties of rabbit hearts, median sternotomy was performed under anesthetization with 3% pelltobarbitalum natricum (30 mg/kg), and the hearts were quickly removed and retrogradely perfused through the ascending aorta with 37°C Tyrode's solution equilibrated with 5% CO2 and 95% O2. The perfusion pressure was maintained at 80 to 95 mm Hg. The Tyrode's solution (pH 7.3–7.4) consisted of the following (mmol/L): NaCl 130, KCl 5.6, NaHCO3 24.2, CaCl2 2.2, MgCl2 0.6, NaH2PO4 1.2, and glucose 12.
Four silver bipolar electrodes were attached to the high right atrium, high left atrium, low left atrium, and right ventricular apex, respectively. A custom‐made computer software program (Electrophysiological Recording System, TOP‐2001, HTONG Company) was used to deliver 2‐fold diastolic pacing threshold currents with a 2‐ms pulse duration. The atrial effective refractory period (AERP) at high right atrium, high left atrium, and low left atrium were measured at basic cycle lengths of 150, 200, and 250 ms using programmed extra‐stimuli, which was defined as the longest S1S2 interval that failed to capture the atrium. The S2 extra‐stimulus was delivered after a train of 8 basic S1S1 stimuli, and the S1S2 interval was decreased by an interval of 2 ms until atrial refractoriness was reached. AERP dispersion was defined as the difference between the longest AERP and the shortest AERP at 4 different sites. The interatrial conduction time (IACT), defined as the duration from the high right atrium pacing stimulus to the beginning of the high left atrium stimulus, was measured at basic cycle lengths of 150, 200, and 250 ms during high right atrium pacing. Atrioventricular Wenckebach cycle length was measured by right atrial incremental pacing. AF induction was tested by burst pacing (cycle length of 50 ms) for 1 second, which was performed 5 times with 30‐second intervals at amplitude of 5 V. AF was defined as rapid, irregular atrial response longer than 1000 ms.
Isolation of Mitochondria From Atrial Tissues
Rabbits were euthanized with 3% pelltobarbitalum natricum and median sternotomy was immediately performed. A portion of ≈100 mg of the atrial tissue was quickly dissected and minced in an ice‐cold isolation medium containing mannitol 220 mmol/L, sucrose 70 mmol/L, HEPES 5 mmol/L, PMSF 1 mmol/L, BSA 0.2% (w/v), and pH 7.4. The minced blood‐free tissue was homogenized using a manual glass homogenizer with 6 passes (0–4°C). Subsequently, the homogenate was centrifuged at 1000g for 10 minutes and the liquid supernatant was collected, which was then centrifuged at 10 000g for 10 minutes. The major constituent of the deposit was mitochondrial pellet, which was suspended in 0.5 mL of the conversational medium containing mannitol 220 mmol/L, sucrose 70 mmol/L, HEPES 5 mmol/L, and PH 7.4.
The mitochondrial isolation procedures were completed within 1 hour after the rabbits were euthanized. Mitochondrial protein content was assayed using a BSA protein assay reagent kit (Thermo Scientific).
Mitochondrial Respiration
Mitochondrial respiratory function was measured polarographically at 25°C using a Clark‐type oxygen electrode (Oroboros Instruments). In a 3‐mL closed thermostatic and magnetically stirred glass chamber, respiration medium (mannitol 225 mmol/L, sucrose 70 mmol/L, EDTANa2 1 mmol/L, KH2PO4 20 mmol/L, K2HPO4 20 mmol/L, and BSA 1 mg/mL, PH 7.4) was saturated with ambient oxygen to reach a concentration of 258 μmol/L. After an equilibration period, 300 μg of mitochondrial protein was added to the reaction system. Upon stabilization of the mitochondrial oxygen consumption, a 15‐μL mixture of 0.8 mol/L malic acid and 1 mol/L glutamic acid was added to initiate the state 2 respiration. After stable state 2 respiration was established, state 3 respiration was initiated by the addition of 200 nmol/L ADP. When all of the ADP had been phosphorylated to ATP, the respiratory rate returned to state 4. The respiratory control ratio was calculated as the ratio of the respiratory rate in state 3 to that in state 4.
Mitochondrial Membrane Potential
Mitochondrial membrane potential (Δψ) was assessed with tetraethyl benzimidazolyl carbocyanine iodide cationic dye, which exhibited potential‐dependent accumulation in mitochondria, resulting in a fluorescence emission shift from 525 nm (green) to 590 nm (red). Therefore, loss of Δψ was detectable by the decrease in the red to green fluorescence emission ratio.19 The experiments were conducted at 25°C in 2 mL of respiration medium with 300 μg of mitochondrial protein, and tetraethyl benzimidazolyl carbocyanine iodide dye equilibration was allowed for 10 minutes. Mitochondrial respiratory function was initiated by a 15‐μL mixture of 0.8 mol/L malic acid and 1 mol/L glutamic acid, and the alteration of the fluorescence emission was detected.
Mitochondrial Reactive Oxygen Species Production
According to a study by Bo et al,20 mitochondrial reactive oxygen species (ROS) generation was assessed using fresh mitochondrial suspensions with the dichlorodihydrofluorescein diacetate probe. Mitochondrial protein 300 μg was added to a quartz cuvette containing 3 mL of phosphate buffer (KCl 130 mmol/L, MgCl2 43 mmol/L, NaH2PO4 20 mmol/L, glucose 30 mmol/L, malate 2 mmol/L, and PH 7.4) and 2 μL of 2.5 mmol/L dichlorodihydrofluorescein diacetate, which was dissolved in 1.25 mmol/L methanol and kept in the dark at 0°C. The mixture was incubated at 37°C for 15 minutes, and dichlorodihydrofluorescein diacetate formation was determined fluorometrically at the excitation wavelength of 499 nm and emission wavelength of 521 nm at 37°C for 2 minutes using a Cary Eclipse Fluorescence spectrophotometer (Varian). The dichlorodihydrofluorescein diacetate fluorescence was normalized to the fold of the control group.21
Western Blot Analysis
Western blotting was performed to assess the expression of proteins on mitochondrial biogenesis in 3 groups. Atrial tissue protein (n=10 for each group) was extracted by lysis buffer containing 150 mmol/L sodium chloride, 10 mmol/L Tris, 0.01 mol/L EDTA 4 Na, 1% NP40, 10 μg/mL Aprotein, 10 μg/mL leupeptin, 1 mmol/L PMSF, 1 mmol/L Na3VO4, and 10 mmol/L NaF. The lysates were centrifuged at 15 000g for 15 minutes, and the supernatants were collected. The protein concentration was determined using the Bradford colorimetric method. Total protein was fractionated by electrophoresis and transferred onto PVDF sheets (Millipore) and separately incubated with a specific antibody targeting adiponectin (1:2000; Abcom), AMPK (1:2000; Abcom), phosphorylated AMPK (1:2000; Abcom), PGC‐1α (1:1000; Abcom), transcription of nuclear respiratory factor‐1 (NRF‐1) (1:1000; Abcom), and mitochondrial transcription factor A (Tfam) (1:1000; Abcom), followed by incubation with appropriate peroxidase‐conjugated secondary antibodies. The reactions were visualized with Western Lightning Chemiluminescence Reagent (Millipore). The blots were exposed to autoradiographic film (Fujifilm Holdings Corp) according to the manufacturer's instructions.
Real‐Time PCR
Total RNA was isolated with TRIzol reagent (Invitrogen) and the RNA sample was quantified using fluorescence spectrophotometry and then reverse transcribed at 42°C for 60 minutes with the Oligod (T) 18 Primers (TakaRa Biotechnology) and SuperScript RT (Invitrogen). A 20‐μL reaction system including 10 μL of FastStart Universal SYBR Green Master (ROX; Roche), 0.5 μL of forward primers (10 mmol/L), 0.5 μL of reverse primers (10 mmol/L), and 2 μL of complementary DNA was applied to the ABI 7500 Real‐Time PCR System (Applied Biosystems). The relative quantification was calculated as the 2−ΔΔCT, and GAPDH was used as an internal control. The primers used are shown in Table 1.
Table 1.
Primers Used for Real‐Time PCR
| Fragments | Primers | |
|---|---|---|
| PGC‐1α | Sence | 5′‐AGCCTCTTTGCCCAGATCTT‐3′ |
| Anti‐sence | 5′‐GGCAATCCGTCTTCATCCAC‐3′ | |
| NRF‐1 | Sence | 5′‐GAACAGAATTGGGCCACGTT‐3′ |
| Anti‐sence | 5′‐GTTGAGTGCCATGGTGACTG‐3′ | |
| Tfam | Sence | 5′‐TGCTTACAGGGTAGAGTGGG‐3′ |
| Anti‐sence | 5′‐GCTTTGCCTGTGATGAACCA‐3′ | |
NRF‐1 indicates nuclear respiratory factor‐1; PCR, polymerase chain reaction; PGC‐1α, peroxisome proliferator–activated receptor‐γ coactivator 1α; Tfam, mitochondrial transcription factor A.
Statistical Analysis
Data were presented as mean±SEM. Comparisons among the 3 groups were analyzed for statistical significance using 1‐way ANOVA followed by Bonferroni correction for comparisons between 2 groups. Repeated measures ANOVA test was performed to compare the electrophysiological parameters, which were measured at different basic cycle lengths. A P<0.05 was considered statistically significant.
Results
Echocardiographic and Hemodynamic Studies
Representative echocardiographic images of the atria from the 3 groups are shown in Figure 1. Compared with the control group, the LA diameter, interventricular septal thickness, and left ventricular posterior wall thickness were significantly increased in both the DM and DM‐A groups (P<0.01). However, in the DM‐A group, interventricular septal thickness was decreased compared with the DM group (P<0.01) and LA diameter and left ventricular posterior wall thickness showed a nonstatistically significant decrease, suggesting that alogliptin can partially inhibit structural remodeling. No difference in left ventricular ejection fraction was observed. Although there was a trend that the maximal decreasing rate of left intraventricular pressure in the DM group was lower than in both the control and DM‐A groups, all the hemodynamic parameters were not significantly different. Table 2 summarizes the baseline characteristics of echocardiographic and hemodynamic studies.
Figure 1.
Representative echocardiographic imaging of the atria for the 3 different groups. A, control group; (B) diabetes mellitus (DM) group; (C) alogliptin‐treated DM group.
Table 2.
Hemodynamic and Echocardiographic Studies
| Control Group (n=10) | DM Group (n=10) | DM‐A Group (n=10) | P Value | |
|---|---|---|---|---|
| HR, beats per min | 283.0±5.6 | 278.3±3.4 | 279.5±5.3 | 0.777 |
| SBP, mm Hg | 125.88±2.22 | 128.79±2.38 | 125.61±1.70 | 0.512 |
| DBP, mm Hg | 82.94±2.38 | 85.06±3.22 | 85.59±2.15 | 0.754 |
| MBP, mm Hg | 98.24±2.48 | 100.38±2.97 | 101.43±1.76 | 0.651 |
| LVEDP, mm Hg | 0.73±0.20 | 1.02±0.45 | 0.74±0.50 | 0.844 |
| +dp/dtmax, mm Hg/m | 3807.04±248.13 | 3565.89±260.21 | 3758.58±247.53 | 0.776 |
| −dp/dtmax, mm Hg/m | 2355.20±239.61 | 1536.55±124.89 | 2240.09±154.59 | 0.080 |
| LAD, mm | 6.09±0.32 | 8.59±0.26a | 7.71±0.29a | <0.001 |
| IVS, mm | 1.78±0.13 | 2.75±0.11a | 2.29±0.06a, b | <0.001 |
| PWLV, mm | 1.66±0.12 | 2.48±0.12a | 2.28±0.07a | <0.001 |
| LVEDD, mm | 11.96±0.20 | 12.20±0.46 | 12.10±0.28 | 0.876 |
| LVESD, mm | 7.81±0.19 | 7.79±0.21 | 8.15±0.19 | 0.381 |
| LVEF, % | 56.67±1.24 | 55.65±1.63 | 56.94±1.70 | 0.824 |
| RVEDD, mm | 4.81±0.32 | 5.03±0.18 | 5.21±0.21 | 0.525 |
| Heart weight ratio (1/1000) | 2.60±0.07 | 2.72±0.07 | 2.68±0.08 | 0.724 |
Values are expressed as mean±SEM. DBP indicates diastolic blood pressure; DM‐A, alogliptin‐treated diabetes mellitus (DM); +dp/dtmax, maximal increasing rate of left intraventricular pressure; −dp/dtmax, maximal decreasing rate of left intraventricular pressure; HR, heart rate; IVS, interventricular septum; LAD, left atrial diameter; LVEDD, left ventricular end‐diastolic dimension; LVEDP, left ventricular end diastolic pressure; LVEF, left ventricular ejection fraction; LVESD, left ventricular end‐systolic dimension; MBP, mean blood pressure; PWLV, left ventricular posterior wall; RVEDD, right ventricular end‐diastolic dimension; SBP, systolic blood pressure.
Compared with the control group, P<0.01.
Compared with the DM group, P<0.05.
Serum Biochemical and Oxidative Stress–Related Examination
As shown in Table 3, fasting glucose at the 8th week was higher (P<0.01) while the levels of insulin and GLP‐1 were lower (P<0.01) in the DM group compared with the control group. Following alogliptin treatment for 8 weeks, serum concentration of GLP‐1 was higher in the DM‐A group when compared with the DM group (P<0.05). The levels of insulin and GLP‐1 were still lower than those in the control group (P<0.01). The levels of creatinine, triglycerides, total cholesterol, and low‐ and high‐density lipoprotein cholesterol were not significantly different among the 3 groups. There were increased MDA, 8‐OHdG, and high‐sensitivity C‐reactive protein concentrations (P<0.01) and decreased superoxide dismutase activity (P<0.05) in the DM group compared with the control group (P<0.01). Of note, these inflammatory and oxidative stress markers could be attenuated by alogliptin treatment (P<0.01 or 0.05), except superoxide dismutase activity.
Table 3.
Serum Biochemical, Oxidative Stress, and Inflammation Examination
| Control Group (n=10) | DM Group (n=10) | DM‐A Group (n=10) | P Value | |
|---|---|---|---|---|
| Glucose, mmol/L | 5.35±0.35 | 15.98±0.96a | 11.57±0.52a, b | <0.001 |
| Insulin, mmol/L | 17.36±1.19 | 8.01±0.59a | 10.19±0.87a | <0.001 |
| GLP‐1, pmol/L | 1.21±0.14 | 0.35±0.02a | 0.80±0.07a, b | <0.001 |
| Creatinine, μmol/L | 81.47±5.70 | 90.56±6.23 | 91.21±3.58 | 0.365 |
| TC, mmol/L | 1.43±0.12 | 1.57±0.11 | 1.61±0.14 | 0.551 |
| Triglycerides, mmol/L | 1.06±0.13 | 1.16±0.14 | 1.09±0.12 | 0.774 |
| LDL‐C, mmol/L | 0.28±0.06 | 0.35±0.05 | 0.38±0.05 | 0.445 |
| HDL‐C, mmol/L | 0.41±0.05 | 0.45±0.03 | 0.49±0.04 | 0.403 |
| SOD, U/mL | 472.10±23.49 | 398.45±19.43c | 425.72±12.17 | 0.037 |
| hs‐CRP, mg/L | 1.78±0.36 | 5.89±0.62a | 3.89±0.42b, c | <0.001 |
| MDA, nmol/mL | 11.00±0.75 | 16.9±1.07a | 13.77±0.91b | 0.001 |
| 8‐OHdG, ng/mL | 1.62±0.18 | 3.67±0.24a | 2.04±0.19d | <0.001 |
Values are expressed as mean±SEM. 8‐OHdG indicates 8‐hydroxy‐2′‐deoxyguanosine; DM‐A, alogliptin‐treated diabetes mellitus (DM); GLP‐1, glucagon‐like peptide‐1; HDL‐C, high‐density lipoprotein cholesterol; hs‐CRP, high‐sensitivity C‐reactive protein; LDL‐C, low‐density lipoprotein cholesterol; MDA, malondialdehyde; SOD, superoxide dismutase; TC, total cholesterol.
Compared with the control group, P<0.01.
Compared with the DM group, P<0.05.
Compared with the control group, P<0.05.
Compared with the DM group, P<0.01.
Surface ECGs and Electrophysiological Studies
The findings from surface ECGs showed no statistical differences among the 3 groups (Table 4). Electrophysiological studies demonstrated slight prolongation in AERPs in the DM group compared with the control group, with alogliptin treatment reversing the prolonged AERPs. No significant difference was observed among the 3 groups except high right atrium ERP at the basic cycle length of 250 ms (Figure 2A through 2C). Notably, AERP dispersion and interatrial conduction time in the DM group were significantly increased compared with the control group, which was attenuated by alogliptin treatment (Figure 2D and 2E). A representative AF episode induced by the burst pacing is shown in Figure 2G. In the control group, AF was induced in 20% of rabbits, and a smaller increase was observed in the DM‐A model by 10%, compared with control, than the DM, which had an absolute increase of 60%. The specific values of the electrophysiological studies are shown in Table 4.
Table 4.
Surface ECGs and Electrophysiological Parameters
| Control Group (n=10) | DM Group (n=10) | DM‐A Group (n=10) | P Value | |
|---|---|---|---|---|
| Surface ECG parameters, ms | ||||
| P‐wave duration | 35.87±1.80 | 41.13±1.29 | 38.25±1.84 | 0.106 |
| PR interval | 64.63±1.58 | 70.88±1.88 | 67.50±2.56 | 0.121 |
| QRS duration | 33.13±1.04 | 35.50±1.48 | 33.63±1.60 | 0.460 |
| QT interval | 138.38±4.45 | 139.00±3.45 | 136.88±2.98 | 0.916 |
| Electrophysiological parameters | ||||
| SCL | 355.30±9.77 | 353.90±12.74 | 349.30±9.50 | 0.919 |
| IACT | ||||
| 150 ms | 22.70±1.32 | 35.60±1.91a | 23.90±1.54b | <0.001 |
| 200 ms | 21.10±1.31 | 34.30±2.06a | 24.00±1.43b | <0.001 |
| 250 ms | 23.00±1.25 | 35.80±1.01a | 25.70±1.19b | <0.001 |
| P Value | 0.543 | 0.801 | 0.597 | |
| AVWCL | 162.00±5.44 | 167.50±5.01 | 172.50±3.82 | 0.318 |
| HRAERP | ||||
| 150 ms | 72.20±2.12 | 73.60±2.65 | 77.40±2.25 | 0.286 |
| 200 ms | 77.20±1.77 | 83.00±3.98 | 80.80±2.25 | 0.357 |
| 250 ms | 73.80±1.59 | 86.00±2.29a, c | 78.60±2.97 | 0.004 |
| P Value | 0.165 | 0.021 | 0.630 | |
| LLAERP | ||||
| 150 ms | 80.20±1.50 | 88.80±4.70 | 85.00±3.62 | 0.243 |
| 200 ms | 84.80±2.48 | 91.60±5.34 | 86.40±4.48 | 0.522 |
| 250 ms | 84.60±2.15 | 95.60±6.17 | 88.80±2.92 | 0.184 |
| P Value | 0.229 | 0.683 | 0.769 | |
| HLAERP | ||||
| 150 ms | 73.20±1.82 | 82.20±3.76 | 77.40±2.05 | 0.078 |
| 200 ms | 80.20±3.13 | 85.60±4.36 | 83.80±3.13 | 0.563 |
| 250 ms | 79.00±2.75 | 87.00±6.01 | 83.80±2.59 | 0.394 |
| P Value | 0.151 | 0.770 | 0.158 | |
| AERPD | 17.60±1.71 | 32.00±3.14a | 23.80±2.16d | 0.001 |
Values are expressed as mean±SEM. AERPD indicates atrial effective refractory period dispersion; AVWCL, atrioventricular Wenckebach cycle length; DM‐A, alogliptin‐treated diabetes mellitus (DM); HLAERP, high left atrium effective refractory period; HRAERP, high right atrium effective refractory period; IACT, interatrial conduction time; LLAERP, low left atrium effective refractory period; SCL, sinus cardiac length.
Compared with the control group, P<0.01.
Compared with the DM group, P<0.01.
Compared with the basic cycle length of 150 ms, P<0.05.
Compared with the DM group, P<0.05.
Figure 2.
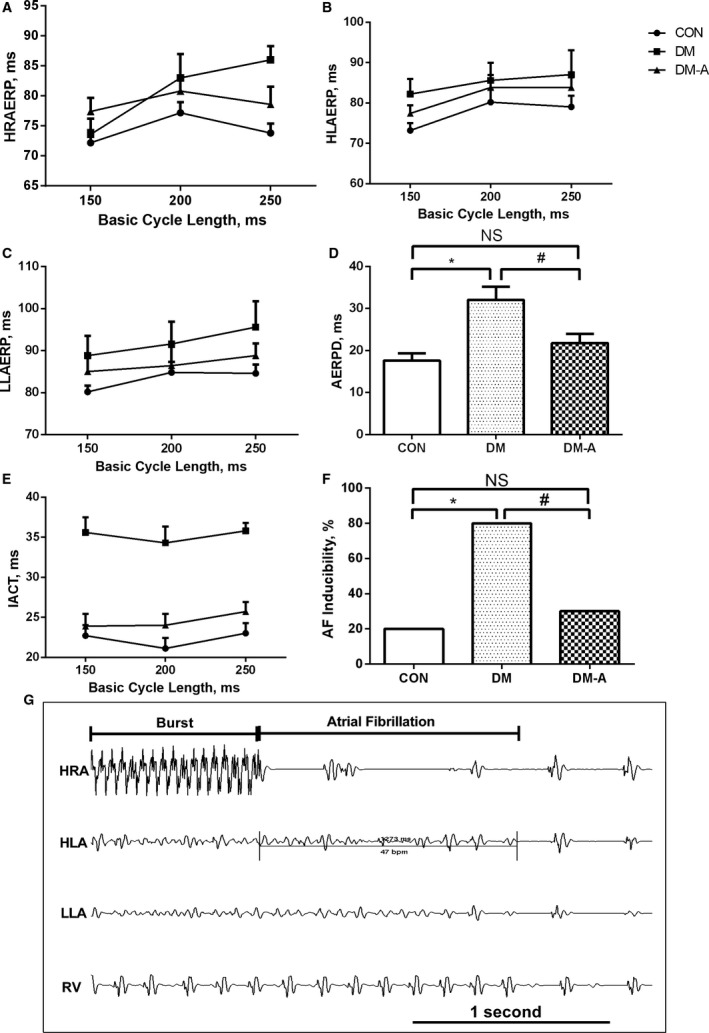
Electrophysiological measurements and atrial fibrillation (AF) induction rate. A through C, High right atrium effective refractory period (HRAERP), high left atrium effective refractory period (HLAERP), and low left atrium effective refractory period (LLAERP) at basic cycle lengths of 150, 200, and 250 ms. D, Atrial effective refractory period dispersion (AERPD) of the 3 groups. E, Interatrial conduction time (IACT) at basic cycle lengths of 150, 200, and 250 ms in the 3 groups. F, the inducibility of AF in the 3 groups. G, Representative AF episodes induced by burst pacing. *Compared with the control (CON) group, P<0.05; #Compared with the diabetes mellitus (DM) group, P<0.05. n=10 per group. DM‐A indicates alogliptin‐treated DM group; HLA, high left atrium; HRA, high right atrium; LLA, low left atrium; NS, not significant; RV, right ventricular.
LA Interstitial Fibrosis and Atrial Myocyte Ultrastructural Changes
Figures 3 and 4 showed the representative histological sections from the left atrium in the 3 groups. Compared with the control group, extensive interstitial fibrosis and increased cross‐sectional areas of atrial cardiomyocytes were observed in the DM group and these were attenuated by the treatment of alogliptin. The ultrastructure of LA cardiomyocytes is shown in Figure 5. In the control group, regular sarcomere organization and uniformly sized mitochondria between sarcomeres were observed. However, the LA tissues from the DM group showed severe disintegrated myofilaments and swelling mitochondria that were accompanied by fractured cristae. After treatment with alogliptin, partial disintegration of myofilaments and slight swelling of mitochondria were observed.
Figure 3.
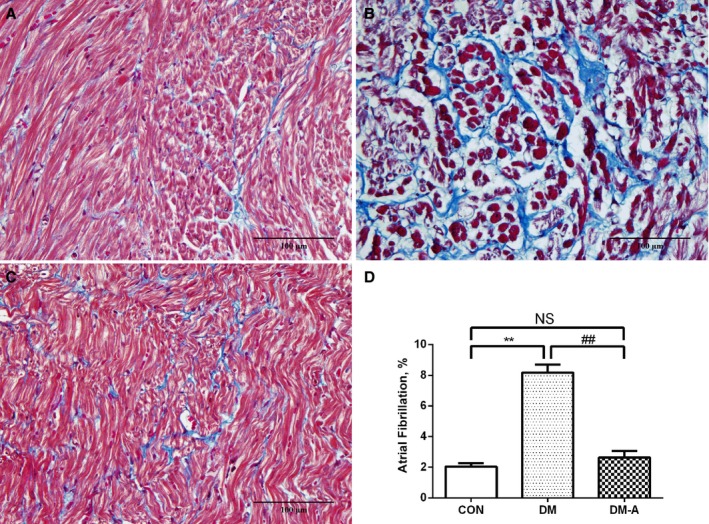
Left atrial interstitial fibrosis in the 3 groups. A, Control group; (B) diabetes mellitus (DM) group; (C) alogliptin‐treated DM (DM‐A) group. D, Illustrates the quantitative ratio of the area of fibrosis to the area of the reference tissue. **Compared with the control (CON) group, P<0.01; ##Compared with the DM group, P<0.01. NS indicates not significant. n=6 per group.
Figure 4.
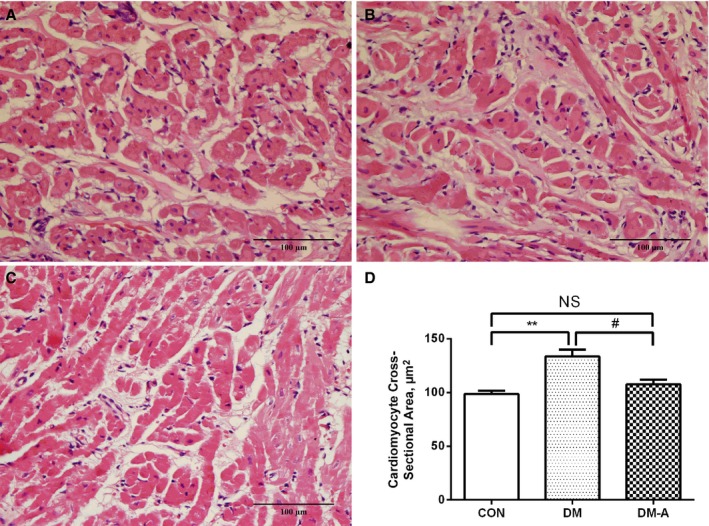
Left atrial cardiomyocyte mean cross‐sectional area in the 3 groups. A, Control (CON) group; (B) diabetes mellitus (DM) group; (C) alogliptin‐treated DM (DM‐A) group, D, Illustrates cardiomyocyte cross‐sectional area. **Compared with the control group, P<0.01; #Compared with the DM group, P<0.05. NS indicates not significant. n=6 per group.
Figure 5.

Comparison of left atrial ultrastructure among the 3 groups. A, Control group; (B) diabetes mellitus (DM) group; (C) alogliptin‐treated DM (DM‐A) group.
Mitochondrial Respiratory Function, Membrane Potential, and ROS Generation
In the DM group, state 3 respiration rate was significantly decreased compared with the control group; however, alogliptin treatment increased state 3 respiration rate compared with the DM group (P<0.05; Figure 6A). No significant difference of state 4 respiration rate was observed among the 3 groups (Figure 6B). As a result, the DM group showed a lower respiratory control ratio than the other 2 groups (P<0.05; Figure 6C). Mitochondrial Δψ in the DM group was reduced compared with the control group, and this was reversed by alogliptin treatment (P<0.05 or 0.01; Figure 6D). On the contrary, the DM group showed markedly higher mitochondrial ROS generation rate than the control group, and alogliptin treatment significantly suppressed the mitochondrial ROS generation rate (P<0.01; Figure 6E).
Figure 6.
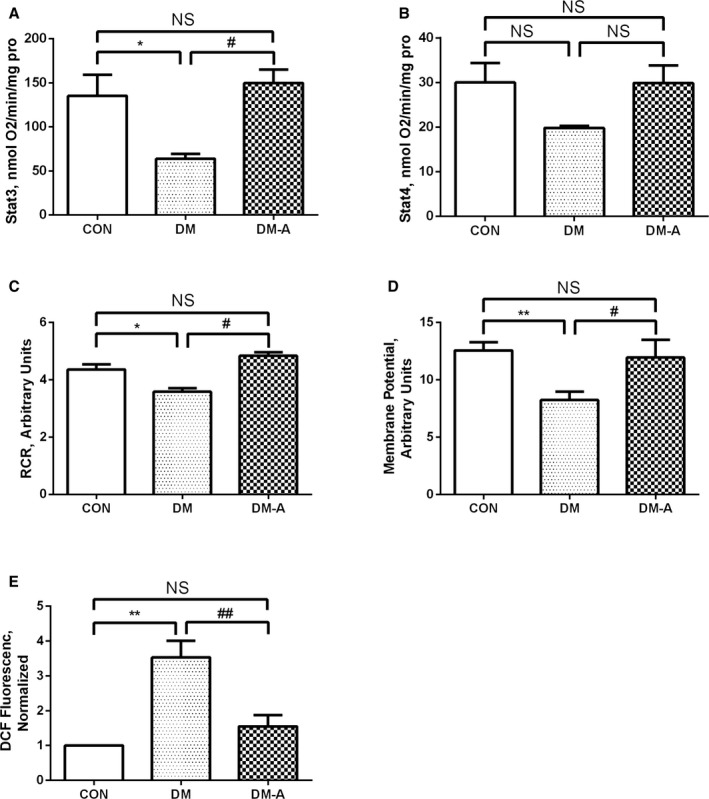
Effects of alogliptin on mitochondrial state 3 respiration rate (A), state 4 respiration rate (B), respiratory control ratio (RCR) (C), mitochondrial membrane potential (D), and reactive oxygen species levels (E). *P<0.05 vs the control (CON) group; **P<0.01 vs the CON group; # P<0.05 vs the diabetes mellitus (DM) group; ## P<0.01 vs the DM group. n=7 to 10. DCF indicates dichlorodihydrofluorescein diacetate; DM‐A, alogliptin‐treated DM group; NS, not significant.
PGC‐1α, NRF‐1, and Tfam mRNA Expression
As shown in Figure 7, decreases in the mRNA expression of PGC‐1α and Tfam (P<0.01) were observed in the DM group compared with the control group, which were reversed by alogliptin treatment (P<0.01). The mRNA expression of NRF‐1 was not significantly different among the 3 groups.
Figure 7.
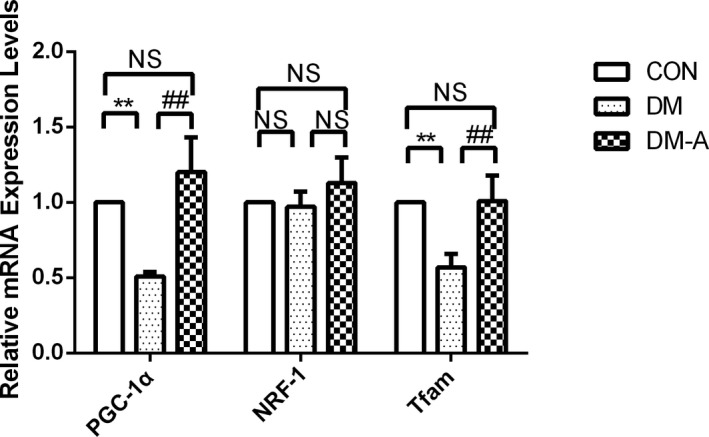
Peroxisome proliferator–activated receptor‐γ coactivator 1α (PGC‐1α), nuclear respiratory factor‐1 (NRF‐1), and mitochondrial transcription factor A (Tfam) mRNA expression estimated by real‐time polymerase chain reaction. **P<0.01 vs the control (CON) group; ## P<0.01 vs the diabetes mellitus (DM) group. n=3 to 6. DM‐A indicates alogliptin‐treated DM group; NS, not significant.
Transforming Growth Factor β1 and Nuclear Factor κB p65 Protein Expression in LA Tissue
Compared with that in the control group, the expression of transforming growth factor β1 (TGF‐β1) and nuclear factor κB (NF‐κB) p65 increased in the DM group (Figure 8). These changes were reversed by alogliptin treatment (P<0.01). No statistical differences in TGF‐β1 and NF‐κB p65 expression were observed between the control and DM‐A groups.
Figure 8.
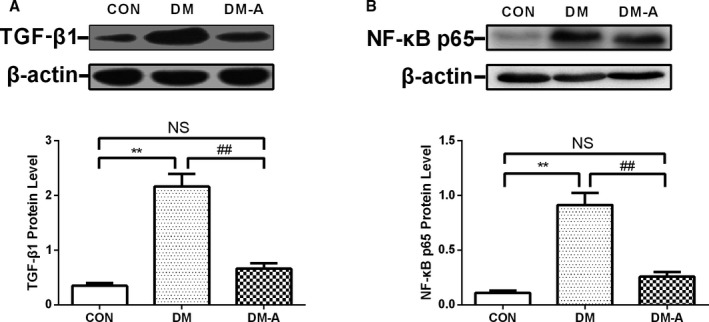
Transforming growth factor β1 (TGF‐β1) and nuclear factor κB (NF‐κB) p65 protein expression in left atrial tissue estimated by Western blot. A, Representative Western blot analysis of the expression of TGF‐β1. B, Protein expression of NF‐κB p65. **P<0.01 vs the control (CON) group; ## P<0.01 vs the diabetes mellitus (DM) group. n=3 to 6. DM‐A indicates alogliptin‐treated DM group; NS, not significant.
Mitochondrial Biogenesis–Related Protein Expression in LA Tissue
As shown in Figure 9A, the expression of adiponectin was significantly decreased in the DM group compared with the control group (P<0.01), changes that were reversed by alogliptin treatment (P<0.01). The expression of AMPK was not significantly different among the 3 groups (Figure 9B). DM resulted in a decrease in phosphorylated AMPK protein expression (Figure 9C; P<0.01), which was ameliorated by alogliptin treatment (P<0.01). In comparison with the control group, the protein levels of PGC‐1α, NRF‐1, and Tfam were significantly reduced in the DM group (Figure 9D through 9F; P<0.05 or 0.01). Treatment with alogliptin increased the expression of PGC‐1α, NRF‐1, and Tfam, suggesting that alogliptin can improve the mitochondrial biogenesis of atrium myocytes, which was depressed by DM.
Figure 9.
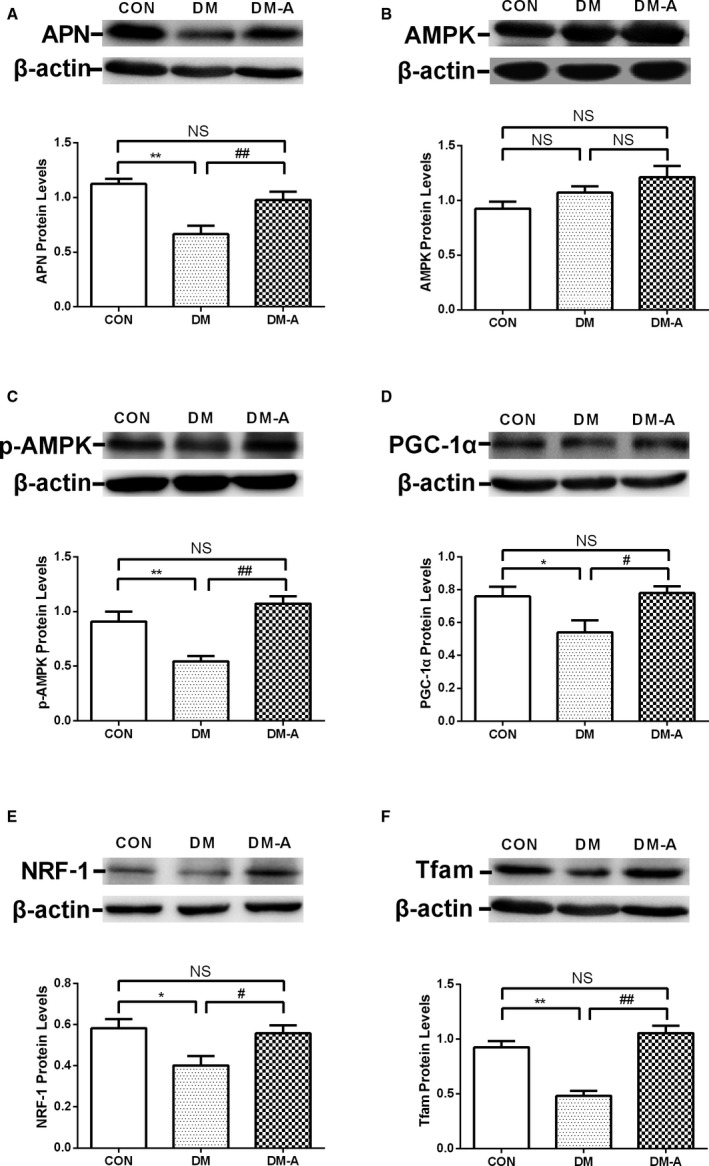
Protein levels of mitochondrial biogenesis‐related proteins in left atrial tissue estimated by Western blot. A through F, adiponectin, AMP‐activated protein kinase (AMPK), phosphorylated AMPK (p‐AMPK), peroxisome proliferator–activated receptor‐γ coactivator 1α (PGC‐1α), nuclear respiratory factor‐1 (NRF‐1), and mitochondrial transcription factor A (Tfam) protein levels in the 3 groups. *P<0.05 vs the control (CON) group; **P<0.01 vs the CON group; # P<0.05 vs the diabetes mellitus (DM) group; ## P<0.01 vs the DM group. n=3 to 6. DM‐A indicates alogliptin‐treated DM group; NS, not significant.
Discussion
In the present study, we identified the beneficial effects of the DPP‐4 inhibitor alogliptin on atrial structural and electrical remodeling in an alloxan‐induced diabetic rabbit model, providing further evidence that mitochondrial remodeling may be a possible mechanism in causing AF in the DM setting. The major findings of this study are as follows: (1) alogliptin attenuated DM‐induced atrial structural remodeling such as LA interstitial fibrosis and atrial myocyte hypertrophy and suppressed effectively serum MDA, 8‐OHdG, and high‐sensitivity C‐reactive protein levels in diabetic rabbits; (2) alogliptin ameliorated atrial mitochondrial swelling, prevented atrial mitochondrial respiratory dysfunction, preserved mitochondrial membrane potential, and reduced mitochondrial ROS production in diabetic rabbits; (3) AERP dispersion, interatrial conduction time, and AF inducibility increased in diabetic hearts and alogliptin effectively prevented this electrical remodeling; (4) PGC‐1α and Tfam mRNA expression in the LA tissue were decreased in the DM group and were elevated by alogliptin; and (5) LA myocardial protein expressions of TGF‐β1 and NF‐κB p65 increased in diabetic states and were attenuated by alogliptin. Mitochondrial biogenesis–related protein expressions of adiponectin, phosphorylated AMPK, PGC‐1α, NRF‐1, and Tfam in the DM group were decreased, while alogliptin promoted the expression of these proteins.
Although the precise pathophysiological mechanisms leading to AF in patients with DM has not been fully elucidated, structural and electrical remodeling are 2 major synergistic contributors to the AF substrate.22 Atrial interstitial fibrosis and replacement fibrosis are hallmarks of arrhythmogenic structural remodeling, producing electrical conduction heterogeneity and disturbance23 and eventually AF. The results from our previous studies have demonstrated that alloxan‐induced diabetic rabbit models exhibit atrial interstitial fibrosis and increased AF inducibility associated with prolonged AERP dispersion and interatrial conduction time.5, 24 The data from this study further support the notion that AF is associated with atrial interstitial fibrosis and electrical conduction heterogeneity. Extensive interstitial fibrosis as well as a high protein level of TGF‐β1 in LA tissue were observed in the DM group. TGF‐β is recognized as a major stimulator of fibrous tissue deposition and can induce a profibrotic phenotype. In the setting of DM, hyperglycemia, generation of advanced glycation end‐products, inflammation, and oxidative stress may directly activate resident cardiac fibroblasts and induce a matrix‐synthetic phenotype by the activation of fibrogenic growth factors, especially TGF‐β1.25
Moreover, accumulating evidence suggests that oxidative stress plays a pivotal role in the development and perpetuation of AF.4, 5, 21, 24 Increased ROS levels can damage proteins, lipids, and DNA (especially mitochondria DNA), and induce inflammation by augmenting cytokine production from activated inflammatory cells, which, in turn, precipitates further tissue damage.26 In addition, ROS are involved in cardiac structural and electrical remodeling, which increase the susceptibility to AF.27 In our study, the content of lipid oxidation products MDA and DNA oxidation products 8‐OHdG were sharply raised in the circulation of diabetic rabbits, providing indirect evidence that increased oxidative stress is involved. The redox imbalance may contribute to the altered gene regulation in the DM setting. One of the most important mediators of this change in transcriptional regulation is NF‐κB. In the LA tissues, we found that the protein level of NF‐κB was significantly increased in the DM group. NF‐κB is a redox‐sensitive transcription factor that regulates a multitude of genes involved in inflammation.28 It can upregulate the TGF‐β pathway29 and decrease the amplitude of the Na+ current,30 both of which predispose to reentry.31
Although there are several sources of ROS in cardiac tissue, including NADPH oxidase, xanthine oxidase, and uncoupled nitric oxide synthase, mitochondria have been proposed as the major ROS source for long‐term AF and age‐related functional decline.32 Electron leakage from complex I and III is associated with the generation of ROS that has been implicated in arrhythmogenesis.33 Inhibition of complex II can prevent hyperglycemia‐induced ROS production in bovine endothelial cells and the subsequent activation of protein kinase C, the formation of advanced glycation end‐products, sorbitol accumulation, and NF‐κB activation.34 Xie et al's study21 indicated that inhibition of mitochondrial ROS production can reduce AF susceptibility and improve mitochondrial function. Montaigne et al35 found that atrial tissue from patients in whom postoperative AF developed exhibited a significantly enriched cluster of downregulated genes involved in redox reactions and upregulation of mitochondrial manganese superoxide dismutase activity, both of which were regarded as indirect evidence of increased mitochondria‐targeted ROS. In our study, we provide direct evidence that mitochondrial ROS generation rate in the DM group was significantly increased, which contributed to the arrhythmic substrate.
In addition to mitochondrial respiration, complex activity, and calcium retention capacity, mitochondrial oxidative stress has been regarded as a main cause of mitochondrial dysfunction. Emerging evidence implicates mitochondrial dysfunction in arrhythmogenesis.26 Except the adverse effect of ROS, mitochondrial dysfunction can directly lead to reduced peak sodium current33 and downregulation of connexin 43,36 result in cytosolic calcium overload,37 and cause the depolarization of Δψ and the opening of the sarcolemmal ATP–sensitive potassium channel,38 all of which contribute to an increased propensity for reentrant‐type arrhythmias. Evidence suggests that preoperative mitochondrial dysfunction of the atrial myocardium may be an important determinant of AF risk after coronary artery bypass grafting surgery in patients with metabolic syndrome.35 Anderson et al39 reported mitochondrial dysfunction in the atrial myocardium of 11 patients with DM. In this study we found that mitochondria from the LA tissue of diabetic rabbits showed severe disintegrated myofilaments and swelling mitochondria that was accompanied by fractured cristae, as well as decreased Δψ and mitochondrial respiration. Optimal mitochondrial Δψ is important for ATP generation and for maintaining the complete function of mitochondria. The mitochondrial permeability transition pore plays a critical role in preserving the normal Δψ. Accumulating ROS levels and calcium overload can trigger the opening of mitochondrial permeability transition pores to molecules <1500 Da in molecular weight, causing mitochondria to become further depolarized, leading to Δψ collapse, cytochrome c release, mitochondrial swelling, and cellular apoptosis.40 Another structure associated with normal Δψ is uncoupling proteins. Activation of uncoupling proteins has beneficial effects on mitochondrial functions, including balancing the Δψ and reducing ROS formation and cellular oxidative damage.41 Both oxidative stress and cellular apoptosis are related to atrial remodeling.
Mitochondrial biogenesis, a dynamically regulated process, is essential for its maintenance of quantity and function, which plays an important role for cells and tissues in surviving and recovering from mitochondrial damage. Recently, research suggested that impaired mitochondrial function and biogenesis in the myocardium may represent an important pathological feature of DM.42 PGC‐1α is a crucial promoter of mitochondrial biogenesis, regulated by AMPK and induced by NRF‐1 and Tfam.12, 43 Adiponectin concentration is known to be decreased in metabolic syndrome, which can be present in patients with type 1 DM, and higher adiponectin concentration is associated with a lower prevalence of metabolic syndrome.44 Adiponectin can improve mitochondrial biogenesis and reduce oxidative stress by activating AMPK or other pathways.12, 45, 46 In our study, we found that the mRNA level of PGC‐1α and Tfam, as well as the protein level of PGC‐1α, NRF‐1, and Tfam in the LA tissue were decreased in the DM group, keeping with the notion that mitochondrial biogenesis was impaired. Furthermore, the protein level of adiponectin and phosphorylated AMPK in the DM group was decreased, suggesting that adiponectin/AMPK/PGC‐1α signaling may contribute to the mitochondrial biogenesis in alloxan‐induced type 1 diabetic hearts.
Alogliptin is a novel antidiabetic agent that is part of the DPP‐4 inhibitor class. Consistent with a previous report,16 our study demonstrated that DPP‐4 inhibitors have favorable effects in ameliorating arrhythmic substrate, improving electrophysiological abnormalities, and reducing AF inducibility. Previously, it has been shown that DPP‐4 inhibitors decrease plasma and brain oxidative stress levels in high‐fat diet–induced insulin‐resistant rats, and restored impaired brain mitochondrial function.47 Another study showed that DPP‐4 inhibitors ameliorated cardiac dysfunction in hearts subjected to ischemia‐reperfusion injury by attenuating cardiac mitochondrial dysfunction and cardiomyocyte apoptosis.48 In the present study, we found that DPP‐4 inhibitors decreased mitochondrial ROS production rate, prevented mitochondrial membrane depolarization, alleviated mitochondrial swelling, and increased mitochondrial respiration function. Of note, DPP‐4 inhibitors increased the expression of adiponectin, AMPK, PGC‐1α, NRF‐1, and Tfam, indicating that alogliptin can improve the mitochondrial biogenesis by PGC‐1α/NRF‐1/Tfam signaling regulated by adiponectin/AMPK. By contrast, DPP‐4 inhibitors can improve the mitochondrial biogenesis by GLP‐1 receptor signaling. Recently, Takada et al49 found that DPP‐4 inhibitors improved exercise capacity and mitochondrial biogenesis in mice with heart failure via activation of GLP‐1. Similarly, GLP‐1 and GLP‐1 receptor agonist exendin‐4 treatment triggered an increase in mitochondrial mass, mitochondrial density, mitochondrial membrane potential, and oxygen consumption, which was accompanied by upregulation of PGC‐1α, the key regulator of mitochondrial biogenesis.50
Study Limitations
There were several limitations to the present study that should be acknowledged. First, we used only one antidiabetic medication in this study without a comparison with other glucose‐lowering agents that could also affect AF. Second, the changes in the ionic currents, such as INa, ICa,L, IKr, Ito, and especially the IKATP channel and the related functional change that would play a main role in this setting, as well as connexin in atria, were not investigated. Also, serum angiotensin‐II level, which could aggravate atrial fibrosis, was not measured. Third, the mitochondrial ROS generation rate was measured, but ROS levels in mitochondria and myocardial cells could be quantified in future studies. Fourth, a causal relationship between mitochondrial dysfunction (especially mitochondrial oxidative stress) and AF was not established. Finally, the roles of distinct components of signal transduction pathways in mitochondrial biogenesis need further clarification.
Conclusions
DPP‐4 inhibitors have the potential to ameliorate arrhythmic substrate in a rabbit model, and our study supports further research in the role of DDP‐4 inhibitors in patients with DM.
Sources of Funding
This work was supported by grants (81570298, 30900618, and 81270245 to Liu) from the National Natural Science Foundation of China, Tianjin Natural Science Foundation (16JCZDJC34900 to Liu). Tse is supported by the Croucher Foundation of Hong Kong.
Disclosures
None.
(J Am Heart Assoc. 2017;6:e005945 DOI: 10.1161/JAHA.117.005945.)28507060
Contributor Information
Guangping Li, Email: tjcardiol@126.com.
Tong Liu, Email: liutongdoc@126.com.
References
- 1. Lip GY, Lane DA. Stroke prevention in atrial fibrillation: a systematic review. JAMA. 2015;313:1950–1962. [DOI] [PubMed] [Google Scholar]
- 2. Benjamin EJ, Levy D, Vaziri SM, D'Agostino RB, Belanger AJ, Wolf PA. Independent risk factors for atrial fibrillation in a population‐based cohort. The Framingham Heart Study. JAMA. 1994;271:840–844. [PubMed] [Google Scholar]
- 3. Lip GY, Varughese GI. Diabetes mellitus and atrial fibrillation: perspectives on epidemiological and pathophysiological links. Int J Cardiol. 2005;105:319–321. [DOI] [PubMed] [Google Scholar]
- 4. Liu T, Zhao H, Li J, Korantzopoulos P, Li G. Rosiglitazone attenuates atrial structural remodeling and atrial fibrillation promotion in alloxan‐induced diabetic rabbits. Cardiovasc Ther. 2014;32:178–183. [DOI] [PubMed] [Google Scholar]
- 5. Fu H, Li G, Liu C, Li J, Wang X, Cheng L, Liu T. Probucol prevents atrial remodeling by inhibiting oxidative stress and TNF‐alpha/NF‐kappaB/TGF‐beta signal transduction pathway in alloxan‐induced diabetic rabbits. J Cardiovasc Electrophysiol. 2015;26:211–222. [DOI] [PubMed] [Google Scholar]
- 6. Tadic M, Cuspidi C. Type 2 diabetes mellitus and atrial fibrillation: from mechanisms to clinical practice. Arch Cardiovasc Dis. 2015;108:269–276. [DOI] [PubMed] [Google Scholar]
- 7. Tse G, Lai ET, Tse V, Yeo JM. Molecular and electrophysiological mechanisms underlying cardiac arrhythmogenesis in diabetes mellitus. J Diabetes Res. 2016;2016:2848759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang KC, Bonini MG, Dudley SC Jr. Mitochondria and arrhythmias. Free Radic Biol Med. 2014;71:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tu T, Zhou S, Liu Z, Li X, Liu Q. Quantitative proteomics of changes in energy metabolism‐related proteins in atrial tissue from valvular disease patients with permanent atrial fibrillation. Circ J. 2014;78:993–1001. [DOI] [PubMed] [Google Scholar]
- 10. Opacic D, van Bragt KA, Nasrallah HM, Schotten U, Verheule S. Atrial metabolism and tissue perfusion as determinants of electrical and structural remodelling in atrial fibrillation. Cardiovasc Res. 2016;109:527–541. [DOI] [PubMed] [Google Scholar]
- 11. Liu GZ, Hou TT, Yuan Y, Hang PZ, Zhao JJ, Sun L, Zhao GQ, Zhao J, Dong JM, Wang XB, Shi H, Liu YW, Zhou JH, Dong ZX, Liu Y, Zhan CC, Li Y, Li WM. Fenofibrate inhibits atrial metabolic remodelling in atrial fibrillation through PPAR‐alpha/sirtuin 1/PGC‐1alpha pathway. Br J Pharmacol. 2016;173:1095–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yan W, Zhang H, Liu P, Wang H, Liu J, Gao C, Liu Y, Lian K, Yang L, Sun L, Guo Y, Zhang L, Dong L, Lau WB, Gao E, Gao F, Xiong L, Wang H, Qu Y, Tao L. Impaired mitochondrial biogenesis due to dysfunctional adiponectin‐AMPK‐PGC‐1alpha signaling contributing to increased vulnerability in diabetic heart. Basic Res Cardiol. 2013;108:329. [DOI] [PubMed] [Google Scholar]
- 13. Dong J, Zhao J, Zhang M, Liu G, Wang X, Liu Y, Yang N, Liu Y, Zhao G, Sun J, Tian J, Cheng C, Wei L, Li Y, Li W. beta3‐Adrenoceptor impairs mitochondrial biogenesis and energy metabolism during rapid atrial pacing‐induced atrial fibrillation. J Cardiovasc Pharmacol Ther. 2016;21:114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hocher B, Sharkovska Y, Mark M, Klein T, Pfab T. The novel DPP‐4 inhibitors linagliptin and BI 14361 reduce infarct size after myocardial ischemia/reperfusion in rats. Int J Cardiol. 2013;167:87–93. [DOI] [PubMed] [Google Scholar]
- 15. Chinda K, Palee S, Surinkaew S, Phornphutkul M, Chattipakorn S, Chattipakorn N. Cardioprotective effect of dipeptidyl peptidase‐4 inhibitor during ischemia‐reperfusion injury. Int J Cardiol. 2013;167:451–457. [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto T, Shimano M, Inden Y, Takefuji M, Yanagisawa S, Yoshida N, Tsuji Y, Hirai M, Murohara T. Alogliptin, a dipeptidyl peptidase‐4 inhibitor, regulates the atrial arrhythmogenic substrate in rabbits. Heart Rhythm. 2015;12:1362–1369. [DOI] [PubMed] [Google Scholar]
- 17. Keller AC, Knaub LA, Miller MW, Birdsey N, Klemm DJ, Reusch JE. Saxagliptin restores vascular mitochondrial exercise response in the Goto‐Kakizaki rat. J Cardiovasc Pharmacol. 2015;65:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Apaijai N, Pintana H, Chattipakorn SC, Chattipakorn N. Effects of vildagliptin versus sitagliptin, on cardiac function, heart rate variability and mitochondrial function in obese insulin‐resistant rats. Br J Pharmacol. 2013;169:1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reers M, Smiley ST, Mottola‐Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC‐1 dye. Methods Enzymol. 1995;260:406–417. [DOI] [PubMed] [Google Scholar]
- 20. Bo H, Jiang N, Ma G, Qu J, Zhang G, Cao D, Wen L, Liu S, Ji LL, Zhang Y. Regulation of mitochondrial uncoupling respiration during exercise in rat heart: role of reactive oxygen species (ROS) and uncoupling protein 2. Free Radic Biol Med. 2008;44:1373–1381. [DOI] [PubMed] [Google Scholar]
- 21. Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX, Marks AR. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5:11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lip GY, Fauchier L, Freedman SB, Van Gelder I, Natale A, Gianni C, Nattel S, Potpara T, Rienstra M, Tse HF, Lane DA. Atrial fibrillation. Nat Rev Dis Primers. 2016;2:16016. [DOI] [PubMed] [Google Scholar]
- 23. Tse G, Yeo JM. Conduction abnormalities and ventricular arrhythmogenesis: the roles of sodium channels and gap junctions. Int J Cardiol Heart Vasc. 2015;9:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qiu J, Zhao J, Li J, Liang X, Yang Y, Zhang Z, Zhang X, Fu H, Korantzopoulos P, Liu T, Li G. NADPH oxidase inhibitor apocynin prevents atrial remodeling in alloxan‐induced diabetic rabbits. Int J Cardiol. 2016;221:812–819. [DOI] [PubMed] [Google Scholar]
- 25. Lin Y, Li H, Lan X, Chen X, Zhang A, Li Z. Mechanism of and therapeutic strategy for atrial fibrillation associated with diabetes mellitus. ScientificWorldJournal. 2013;2013:209428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tse G, Yan BP, Chan YW, Tian XY, Huang Y. Reactive oxygen species, endoplasmic reticulum stress and mitochondrial dysfunction: the link with cardiac arrhythmogenesis. Front Physiol. 2016;7:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yeh YH, Hsu LA, Chen YH, Kuo CT, Chang GJ, Chen WJ. Protective role of heme oxygenase‐1 in atrial remodeling. Basic Res Cardiol. 2016;111:58. [DOI] [PubMed] [Google Scholar]
- 28. Jiang Y, Jiang LL, Maimaitirexiati XM, Zhang Y, Wu L. Irbesartan attenuates TNF‐alpha‐induced ICAM‐1, VCAM‐1, and E‐selectin expression through suppression of NF‐kappaB pathway in HUVECs. Eur Rev Med Pharmacol Sci. 2015;19:3295–3302. [PubMed] [Google Scholar]
- 29. Fei AH, Wang FC, Wu ZB, Pan SM. Phosphocreatine attenuates angiotensin II‐induced cardiac fibrosis in rat cardiomyocytes through modulation of MAPK and NF‐kappaB pathway. Eur Rev Med Pharmacol Sci. 2016;20:2726–2733. [PubMed] [Google Scholar]
- 30. Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, Dudley SC Jr. NF‐kappaB‐dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am J Physiol Cell Physiol. 2008;294:C372–C379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tse G, Lai ET, Lee AP, Yan BP, Wong SH. Electrophysiological mechanisms of gastrointestinal arrhythmogenesis: lessons from the heart. Front Physiol. 2016;7:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu M, Liu H, Dudley SC Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010;107:967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. [DOI] [PubMed] [Google Scholar]
- 35. Montaigne D, Marechal X, Lefebvre P, Modine T, Fayad G, Dehondt H, Hurt C, Coisne A, Koussa M, Remy‐Jouet I, Zerimech F, Boulanger E, Lacroix D, Staels B, Neviere R. Mitochondrial dysfunction as an arrhythmogenic substrate: a translational proof‐of‐concept study in patients with metabolic syndrome in whom post‐operative atrial fibrillation develops. J Am Coll Cardiol. 2013;62:1466–1473. [DOI] [PubMed] [Google Scholar]
- 36. Sovari AA, Rutledge CA, Jeong EM, Dolmatova E, Arasu D, Liu H, Vahdani N, Gu L, Zandieh S, Xiao L, Bonini MG, Duffy HS, Dudley SC Jr. Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circ Arrhythm Electrophysiol. 2013;6:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation‐contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Faivre JF, Findlay I. Action potential duration and activation of ATP‐sensitive potassium current in isolated guinea‐pig ventricular myocytes. Biochim Biophys Acta. 1990;1029:167–172. [DOI] [PubMed] [Google Scholar]
- 39. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate‐specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54:1891–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res. 2012;111:1237–1247. [DOI] [PubMed] [Google Scholar]
- 41. Akhmedov AT, Rybin V, Marin‐Garcia J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail Rev. 2015;20:227–249. [DOI] [PubMed] [Google Scholar]
- 42. Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy‐Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, Staels B. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130:554–564. [DOI] [PubMed] [Google Scholar]
- 43. Wang Y, Zhao X, Lotz M, Terkeltaub R, Liu‐Bryan R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator‐activated receptor gamma coactivator 1alpha. Arthritis Rheumatol. 2015;67:2141–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Blaslov K, Bulum T, Zibar K, Duvnjak L. Relationship between adiponectin level, insulin sensitivity, and metabolic syndrome in type 1 diabetic patients. Int J Endocrinol. 2013;2013:535906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gan L, Yan J, Liu Z, Feng M, Sun C. Adiponectin prevents reduction of lipid‐induced mitochondrial biogenesis via AMPK/ACC2 pathway in chicken adipocyte. J Cell Biochem. 2015;116:1090–1100. [DOI] [PubMed] [Google Scholar]
- 46. Qiao L, Kinney B, Yoo HS, Lee B, Schaack J, Shao J. Adiponectin increases skeletal muscle mitochondrial biogenesis by suppressing mitogen‐activated protein kinase phosphatase‐1. Diabetes. 2012;61:1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pintana H, Apaijai N, Chattipakorn N, Chattipakorn SC. DPP‐4 inhibitors improve cognition and brain mitochondrial function of insulin‐resistant rats. J Endocrinol. 2013;218:1–11. [DOI] [PubMed] [Google Scholar]
- 48. Chinda K, Sanit J, Chattipakorn S, Chattipakorn N. Dipeptidyl peptidase‐4 inhibitor reduces infarct size and preserves cardiac function via mitochondrial protection in ischaemia‐reperfusion rat heart. Diab Vasc Dis Res. 2014;11:75–83. [DOI] [PubMed] [Google Scholar]
- 49. Takada S, Masaki Y, Kinugawa S, Matsumoto J, Furihata T, Mizushima W, Kadoguchi T, Fukushima A, Homma T, Takahashi M, Harashima S, Matsushima S, Yokota T, Tanaka S, Okita K, Tsutsui H. Dipeptidyl peptidase‐4 inhibitor improved exercise capacity and mitochondrial biogenesis in mice with heart failure via activation of glucagon‐like peptide‐1 receptor signalling. Cardiovasc Res. 2016;111:338–347. [DOI] [PubMed] [Google Scholar]
- 50. Kang MY, Oh TJ, Cho YM. Glucagon‐like peptide‐1 increases mitochondrial biogenesis and function in INS‐1 rat insulinoma cells. Endocrinol Metab (Seoul). 2015;30:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]