

ABSTRACT
Monoclonal antibodies (mAbs) are a rapidly growing drug class for which great efforts have been made to optimize certain molecular features to achieve the desired pharmacokinetic (PK) properties. One approach is to engineer the interactions of the mAb with the neonatal Fc receptor (FcRn) by introducing specific amino acid sequence mutations, and to assess their effect on the PK profile with in vivo studies. Indeed, FcRn protects mAbs from intracellular degradation, thereby prolongs antibody circulation time in plasma and modulates its systemic clearance. To allow more efficient and focused mAb optimization, in vitro input that helps to identify and quantitatively predict the contribution of different processes driving non-target mediated mAb clearance in vivo and supporting translational PK modeling activities is essential. With this aim, we evaluated the applicability and in vivo-relevance of an in vitro cellular FcRn-mediated transcytosis assay to explain the PK behavior of 25 mAbs in rat or monkey. The assay was able to capture species-specific differences in IgG-FcRn interactions and overall correctly ranked Fc mutants according to their in vivo clearance. However, it could not explain the PK behavior of all tested IgGs, indicating that mAb disposition in vivo is a complex interplay of additional processes besides the FcRn interaction. Overall, the transcytosis assay was considered suitable to rank mAb candidates for their FcRn-mediated clearance component before extensive in vivo testing, and represents a first step toward a multi-factorial in vivo clearance prediction approach based on in vitro data.
KEYWORDS: Neonatal Fc receptor (FcRn), monoclonal antibody, clearance, in vitro-to-in vivo correlation, transcytosis, cellular assays, pharmacokinetics
Introduction
Monoclonal antibodies (mAbs) have developed into one of the fastest growing drug classes. Considering the competitive environment within the pharmaceutical sector and the demand for mAbs against new targets, novel discovery platforms and antibody engineering are rapidly evolving, allowing the generation of molecules with engineered variable mAb domains to decrease immunogenicity,1 bispecific mAbs targeting more than one antigen with optimized affinity,2 fragment antigen-binding (Fab) domains3 and fragment crystallizable (Fc)-fusion proteins.4,5 In addition, great efforts have been made to optimize mAb clearance in vivo by e.g., modulating the Fc interaction with the neonatal Fc receptor (FcRn)6,7 or by adapting mAb physicochemical properties to prevent increased unspecific cellular uptake via pinocytosis.8,9 To develop effective and safe biologic drugs with reduced efficacious dose and more convenient administration route/frequency, mAbs must have suitable pharmacokinetic (PK) properties. Thus, preclinical optimization and characterization of mAb PK/pharmacodynamics (PD) profile and its translation to human is an essential step in biologics drug discovery.10
The Fc region of immunoglobulin G (IgG)-based antibodies plays an important role in determining their in vivo PK profile. One major process is their pH-dependent binding to FcRn, for which the Fc region has strong affinity at pH 6.0, but weak affinity at pH 7.4. FcRn is located within endosomes in endothelial cells lining blood vessels and haematopoietic cells. In addition to IgG molecules, it interacts with serum albumin using a different binding site.11 FcRn ligands (i.e., IgG and albumin) in circulation are taken up by cells via non-specific fluid-phase pinocytosis, followed by binding to FcRn in the acidic environment (pH ∼6.0) of the endosome.12,13 This complexation protects the ligand from lysosomal degradation, enabling the ligand-FcRn complex to be recycled back to the cell surface. At pH 7.4, the “protected” ligand bound to FcRn is then released from the complex to the extracellular space. Thereby, FcRn maintains IgG and albumin homeostasis in human and animal serum, and transfers maternal IgGs from the mother to the fetus over the placental barrier.13
The effect of mAb-FcRn interactions on plasma systemic clearance of mAbs has been extensively studied in preclinical species and in man, using various mutant mAbs with abolished14,15 or enhanced16–18 FcRn affinity. These studies confirmed that the interaction with FcRn strongly affects the PK properties of therapeutic mAbs. Therefore, its accurate in vitro assessment, with a readout suitable for the quantitative prediction of FcRn-mediated PK properties in vivo, would be of great help to support drug candidate optimization/selection and to ensure an appropriate PK profile according to the desired drug product profile, as well as to minimize animal testing early in discovery.19
Preclinical PK properties of biologic drug candidates are currently assessed in vivo mostly in mice and in non-human primates. The preclinical PK parameters are typically translated to human using allometric scaling,20 with or without the inclusion of “target-mediated drug disposition” (TMDD), depending on the specific case and the availability of kinetic information on the target. For mAbs exhibiting TMDD, the PK profile is often described by a 2-compartmental model with linear and nonlinear elimination. Both the linear clearance (CL) and nonlinear CL (Vmax) of mAbs are typically scaled allometrically, with the Michaelis-Menten constant (Km) assumed to be equal in monkeys and humans based on similar in vitro target binding characteristics or identical target protein sequences.21 However, in vitro tools that help to quantitatively scale all the different processes contributing to mAb disposition in vivo from animals to human are currently not available.
A routinely used in vitro approach for the qualitative study of mAb-FcRn interaction is the surface plasmon resonance (SPR) technique, which allows affinity measurement of a mAb for the FcRn at different pH conditions.22 However, in certain cases, affinity-based physicochemical FcRn methodologies failed to correlate with the observed clearance in vivo.23 These limitations are likely due to additional biochemical and biophysical properties of the mAbs, along with their FcRn affinity, that influence the in vivo PK, such as “unspecific”/charge-dependent binding characteristics of the mAbs during the cellular uptake process via pinocytosis, the pH-dependent dissociation of the FcRn-IgG complexes and the effect of TMDD at non-target saturating dose levels.24 More recently, new methodology using an FcRn-coated affinity column was introduced by Schlothauer et al.,14 where mAb-receptor binding and release was measured in a pH-dependent fashion and was correlated with the in vivo half-life of 3 mAbs.
Due to the complexity of the FcRn-mediated mAb recycling phenomenon in vivo, comprising interactions of the antibody with the cell membrane, mAb uptake via fluid-phase pinocytosis, FcRn-mediated intracellular sorting/trafficking/salvage from lysosomal degradation and finally mAb release at the cell membrane, we aimed to evaluate an in vitro tool mimicking the entire IgG-FcRn-interaction cycle in living cells, which is a more physiologically-like system.
Previously, Tesar et al. and Prator et al. described the use of FcRn-transfected Madin-Darby Canine kidney (MDCK) cells to assess FcRn-mediated transcytosis of mAbs. They monitored the flux of IgGs and other Fc-containing molecules across a tight cell monolayer plated in a Transwell® system, using acidic pH in the donor compartment and neutral pH in the acceptor compartment. This pH gradient strongly reduced IgG recycling back into the donor chamber and at the same time enabled FcRn-mediated endocytotic cellular uptake, which altogether maximized unidirectional FcRn-mediated trafficking.25,26
Here, we describe the application of this cellular assay for understanding the in vivo FcRn-mediated protection from lysosomal catabolism of chimeric, human and humanized mAbs. Even though IgG recycling in vivo, which takes place, for example, in vascular endothelial cells, and the transcytotic IgG movement are not identical processes, the transcytotic IgG trafficking in vitro under optimized conditions may be used as a surrogate for IgG recycling in vivo. Using this in vitro transcellular flux as primary assay readout, a selected set of mAbs was tested in rat or human FcRn-expressing MDCK cells to differentiate their properties and to quantitatively correlate the data with their in vivo clearance values in rats and cynomolgus monkeys. Such a systematic evaluation of the correlation between in vitro parameters (FcRn-mediated mAb flux) and in vivo clearance, referred to here as in vitro-to-in vivo correlation (IVIVC), has not been available until now, but is considered essential to understand the multi-factorial mAb clearance mechanism in vivo by applying in vitro methodologies.
Results
Assessment of FcRn-mediated transcytosis of antibodies in rat or human FcRn receptor over-expressing cells
According to the described previously methods by Tesear et al.25 and Praetor et al.,26 MDCK cells transfected with either rat or human FcRn were cultured as tight monolayers and the antibody transport assay was conducted in the presence of a pH gradient (donor/acceptor compartment buffer at pH 6.0/8.0) to maximize uni-directional IgG transcytotic transport. The incubation without a pH gradient (donor/acceptor compartment buffer at pH 8.0/8.0) should allow determination of “unspecific,” receptor-independent antibody uptake. We conducted initial confirmatory studies with a 3H-labeled humanized mAb bearing no mutations in the FcRn binding region (herein named as wildtype “Wt” IgG) and targeting an antigen absent in the cells used for this assay. FITC-labeled dextran (MW 150 kDa), which is restricted to paracellular permeability, was included as a marker of monolayer tightness under the applied assay conditions.
Fig. 1 displays the basolateral to apical transport or flux of this 3H-labeled Wt IgG across human and rat FcRn over-expressing MDCK cells compared with “empty vector” (EV) transfected MDCK cells without FcRn as the functional negative control. The IgG flux at pH 6.0/8.0 across FcRn-transfected cells, displayed as apparent permeability “Papp” in nanometers per second (nm/s), ranged from 13 to 17 nm/s depending on the species, and was about 10-fold higher than across EV cells (1.1 nm/s), indicating a pronounced FcRn-mediated trafficking of the antibody. As expected, the antibody flux at pH 8.0/8.0 was significantly reduced due to the known weak mAb affinity for the FcRn receptor at pH 8.0, resulting in a flux of 5–6 nm/s, likely reflecting “unspecific ” antibody uptake processes, such as pinocytosis, followed by FcRn-mediated transcellular trafficking. The control FITC-dextran showed negligible paracellular fluxes of ∼1.0 nm/s, confirming the integrity of the cell layer under the applied assay conditions.
Figure 1.
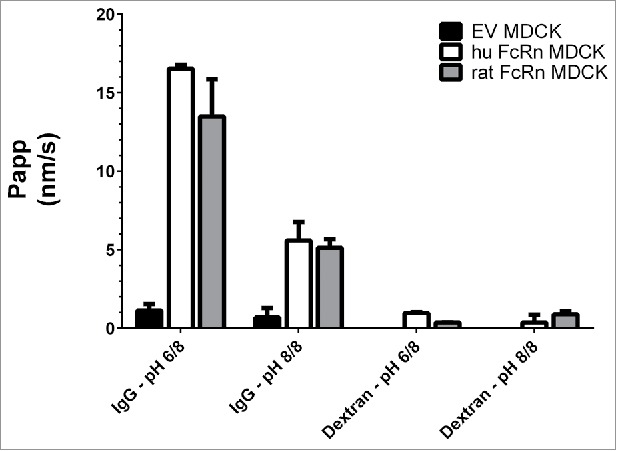
Basolateral to apical flux (Papp) of a 3H-IgG and FITC-dextran across rat and human FcRn over-expressing MDCK cells and EV cells. The highest Papp was observed in the cell line over-expressing FcRn and under pH 6.0/8.0, indicating a specific FcRn-mediated antibody transport. Data is shown as mean ± SD; n = 3 (separate wells).
Species selectivity of rat and human FcRn-mediated IgG transcytosis
Rat, mouse, or chicken IgGs do not bind to the human FcRn, whereas IgGs from all mentioned species, apart from chicken, bind to rat FcRn.27 To assess whether these species-selective IgG-FcRn interactions were captured in the studied assay, the flux of FITC-labeled mouse, rat, human and chicken IgGs was measured in both rat and human FcRn cell systems at pH 6.0/8.0 and pH 8.0/8.0. The chicken IgG served here as non-binding control. As depicted in Fig. 2, human FcRn over-expressing cells enabled a high flux of the human IgG at pH 6.0/8.0, and low fluxes of immunoglobulins from animal species. Rat FcRn over-expressing cells transported rat, mouse, and human IgG with a respectively increasing flux, which was in line with the higher affinity of human IgG for rat FcRn compared with rat or mouse IgGs.15
Figure 2.
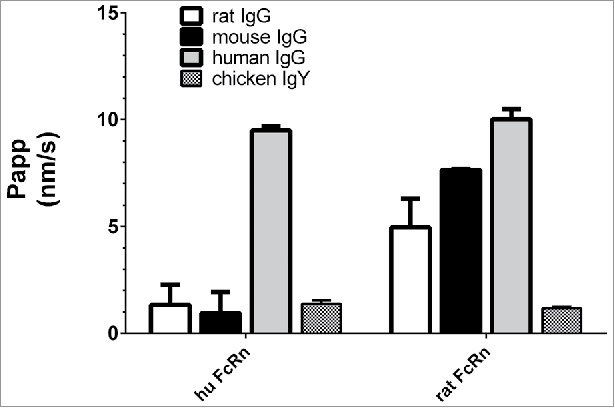
Basolateral to apical flux (Papp) of FITC-labeled rat, mouse, human IgGs and chicken IgY across human FcRn and rat FcRn MDCK cells with a pH gradient (6.0/8.0). The inability of rodent IgG to bind to human FcRn was confirmed. Data is shown as mean ± SD; n = 3 (separate wells).
These data confirm the expected species-specific FcRn-trafficking of IgGs in the rat and human FcRn cell systems. Further investigations addressing FcRn saturation by endogenous plasma IgGs, and their effect on therapeutic mAb flux in vitro are described in the supplemental material.
Trancytosis assay validation with 3H-labeled Fc-mutated IgGs
To validate the predictability of human and rat FcRn transcytosis assay for FcRn-driven in vivo CL of mAbs, the cellular flux of 3 IgG1 molecules was evaluated. These mAbs contain an identical Fab part (anti-digoxigenin = <DIG>) but distinct Fc mutations known to modulate the FcRn affinity. IgG Wildtype (Wt) has unmodified FcRn binding ability (without mutation in the FcRn-binding region of the Fc part). In the IgG AAA (Triple A) mutant M252A/H310A/H435A, 3 of the main amino acids associated with the Fc-FcRn interactions were replaced by alanine, abolishing the FcRn binding completely.14 This mutant was reported to have a very high in vivo CL (in rodents and monkey) and a short-terminal elimination half-life.28 In the IgG YTE mutant M252Y/S254T/T256E, 3 amino acids were replaced, enabling the IgG to form additional hydrogen bonds with FcRn that enhance binding at pH 6.0 which in monkey and human leads to an increased terminal half-life16 In addition, we included a 3H-labeled chicken IgY as a non-binding control.
Human FcRn Trancytosis assay
As depicted in Fig. 3, the paracellular marker showed a negligible flux (Papp of 0–2 nm/s) across monolayers of both cell lines, demonstrating the tightness of the cell layer under the applied assay conditions. Chicken IgY and AAA IgG mutant, which do not bind to human FcRn, showed equally low transcytosis values in human FcRn cells. The Wt IgG reached a 2-fold higher flux than the 2 non-binders and the FcRn-binding-enhanced YTE IgG mutant displayed a transcytosis rate of ∼20 nm/s. The flux differences of the 3 Fc mutants (AAA vs. Wt vs. YTE) were statistically significant from each other (P value ≤ 0.04 in paired t-test).
Figure 3.
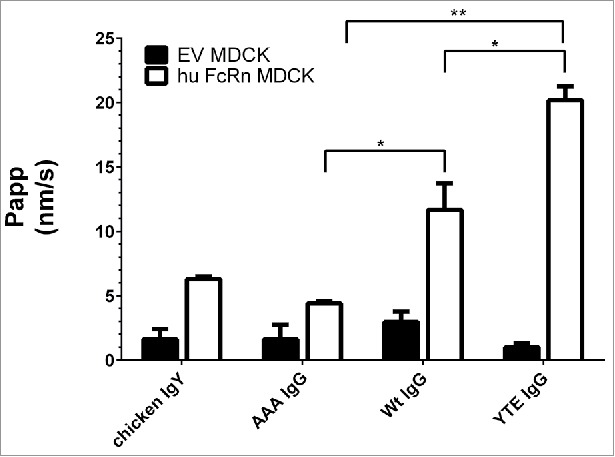
Transcytosis of different 3H-labeled IgG Fc mutants and non-binding control (chicken IgY) at pH 6.0/8.0 in human FcRn MDCK cells and EV MDCK cells as negative control. AAA, Wt, and YTE IgG clearly differed by their flux (Papp). Paired t-test P value (AAA vs. Wt IgG) = 0.024; P value (YTE vs. Wt IgG) = 0.041; P value (AAA vs. YTE IgG) = 0.002; Data is shown as mean ± SD; n = 3 (separate wells).
The transcytosis of all compounds across EV MDCK cells was consistently in the background range and similar to the dextran flux as expected due to the lack of human FcRn expression. Overall, the measured transcytotic flux increased with increasing mAb affinity to the FcRn receptor.
To maximize the resolution of the assay, the effect of different incubation times (3 vs. 5 hrs) and pH gradients (pH 6.0/8.0 vs. pH 6.0/7.4) were assessed with the same Fc mutants and the chicken IgY as a non-binding control (Fig. 4). The pH 6.0/7.4 gradient is more physiologically relevant because the pH of the donor compartment matches that of early endosomes (pH 6.0), while the acceptor compartment pH resembles the blood (pH 7.4). These adjustments led to a better differentiation between the test molecules and the controls. Also, to allow easier comparison among the mAb transcytosis levels within one experiment, trancytosis data in Fig. 4 were reported as “normalized flux” (NF), where the Papp values are divided by the Papp of the chicken IgY, which represent no FcRn-mediated flux and were arbitrarily set to a flux of 1.0.
Figure 4.
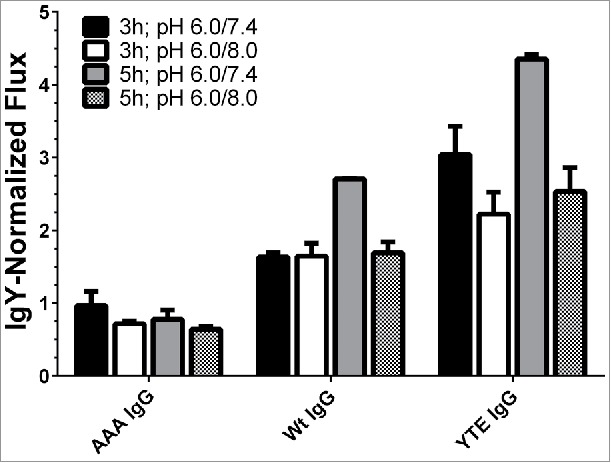
Effect of incubation time and donor/acceptor chamber pH on transcytosis of IgG mutants by human FcRn-over-expressing cells. The application of 5 h incubation time and a pH gradient of 6.0/7.4 differentiated the various molecules best. The fluxes were normalized to the flux of chicken IgY. Data is shown as mean ± SD; n = 3 (separate wells).
Rat FcRn Trancytosis assay
The same Fc mutants and control molecules were additionally tested in the same assay set up with rat FcRn-over-expressing MDCK cells. Molecules are often screened in rodents for their PK properties, making the comparison of rat and human FcRn receptors of interest. Table 1 lists the flux values of the different mutants obtained in the rat FcRn system against the results from the human FcRn system (see also Fig. 3). For easier comparison, both data sets were normalized to the respective transcytosis rates of IgY. AAA IgG and IgY had similarly low fluxes across rat and human FcRn MDCK cells, whereas the Wt IgG was transported approximately twice as efficiently. The YTE IgG mutant, however, reached the same transcytosis rate as the Wt IgG in the rat FcRn system, even though in the human FcRn assay the YTE IgG flux markedly exceeded the Wt IgG. Thus, apparently the YTE mutation interacts differently with the FcRn of these 2 species.
Table 1.
In vitro flux of IgG mutants across human and rat FcRn MDCK cells at pH 6.0/7.4.
| Tested antibody | Flux across rat FcRn cells | Flux across human FcRn cells |
|---|---|---|
| IgY | 1.0 ± 0.11 | 1.0 ± 0.03 |
| AAA IgG | 1.34 ± 0.26 | 0.70 ± 0.02 |
| Wt IgG | 2.72 ± 0.31 | 1.95 ± 0.12 |
| YTE IgG | 2.41 ± 0.38 | 3.11 ± 0.06 |
Only the YTE mutant showed a species difference in FcRn interaction leading to enhanced flux across human FcRn, but Wt-IgG-like flux across rat FcRn cells. The fluxes were normalized to the IgY flux obtained in the same experiment. Data is shown as mean ± SD; n = 3 to 6 (separate wells).
In vivo PK evaluation of 3H-labeled Fc mutants in Wistar rats
To evaluate the relationship between in vitro Papp and in vivo clearance, the 5 previously introduced 3H-labeled molecules with different FcRn binding affinities (IgY, anti-Digoxigenin-hu AAA IgG, anti-Digoxigenin-hu IgG Wt IgG and anti-Digoxigenin-hu YTE IgG) were administered as single intravenous dose (i.v.) to male Wistar rats. The plasma concentrations of these molecules were monitored up to 336 or 540 h post dose (Fig. S1 in supplemental material) and their respective PK parameters were determined via non-compartmental PK analysis (Table S1 in supplemental material). The plasma exposure of IgY and AAA IgG dropped rapidly after administration, leading to a very high systemic total CL of 36.4 mL/day/kg for the AAA mutant and 52.5 mL/day/kg for the IgY. As expected, the Wt IgG and the YTE IgG showed 6.5-fold and 7.9-fold lower CL than the AAA-IgG, respectively, as a result of the FcRn-mediated protection from mAb degradation in vivo. Interestingly, the YTE and Wt IgG showed a very similar PK profile. This confirmed that the YTE mutation does not lead to a half-life-extending effect in rodents, in contrast to the effect seen in monkey or human,16 and revealed one limitation of the use of rodents as a suitable model to evaluate PK of particular Fc-engineered biologics.
Correlation of antibody FcRn-mediated transcytosis in vitro to in vivo systemic clearance in rat
To evaluate the in vivo relevance of the FcRn transcytosis assay, the flux obtained in rat FcRn-over-expressing cells for the IgY, AAA IgG, wt IgG and YTE IgG was compared with their corresponding systemic clearance derived from single dose PK studies in rats (Table S1 in supplemental material).
As FcRn-mediated Ab binding is in vivo a salvage mechanism reducing antibody clearance, higher transcytosis-mediated flux is expected to result in a lower in vivo clearance. Indeed, the IVIVC followed an inverse relationship to the in vitro flux (Fig. 5). Although the data set is limited, the rat in vitro FcRn assay was able to correctly rank mAbs with respect to their FcRn-mediated salvage from degradation in vivo.
Figure 5.
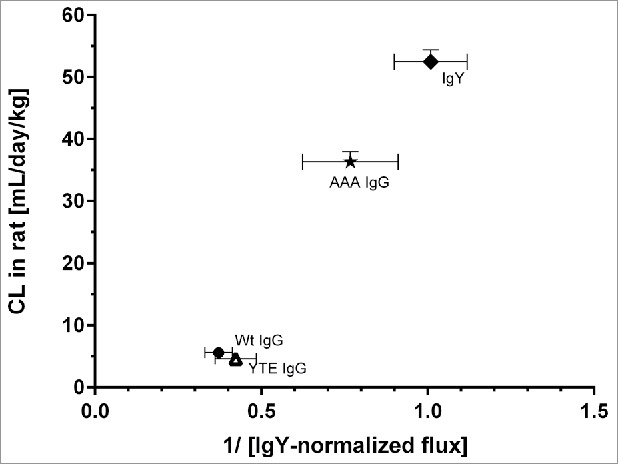
Mean systemic clearance observed in rat after single mAb intravenous administration plotted against the reciprocal normalized flux obtained from the rat FcRn cellular transcytosis assay. IgY-normalized in vitro flux of 5 mAbs were inversely proportional with the corresponding in vivo CL values in rat. Data is shown as mean ± SD.
Correlation of antibody FcRn-mediated transcytosis in vitro to in vivo systemic clearance in cynomolgus monkeys
Cynomolgus monkey (henceforth referred to as “monkey”) is the most predictive preclinical species for the assessment of mAb PK in human, since monkey FcRn shares a 96% sequence identity (98% homology) with human FcRn.20 Additionally, the effect of Fc engineering on mAb PK in human was described to be well reflected in this species.17 Therefore, in vivo monkey data were used to establish a correlation to in vitro human FcRn transcytosis data. The in vitro transcytosis data from the human FcRn system was compared with systemic clearance data observed in monkey PK studies performed for 24 mAbs. To avoid contribution of non-FcRn-mediated CL in the IVIVC, such as target-mediated antibody disposition and immunogenicity, only PK studies performed at target-saturating mAb doses were included and we excluded the data from animals displaying circulating anti-drug antibodies.29
The in vitro flux from the human FcRn cellular system and the in vivo PK parameters derived in monkey are summarized in Table 2. Flux values were normalized to the Papp value of the wildtype (Wt) <DIG> IgG1 “mAb 2,” the control mAb used in assay validation experiments (Fig. 3 to 5). This enabled a quantitative comparison of Papp values across different experiments reducing potential interference by fluctuation of cellular FcRn expression and functionality.
Table 2.
Human in vitro and cynomolgus monkey in vivo data used for the in vitro-in vivo correlation
|
In vivo data |
In vitro data | |||||
|---|---|---|---|---|---|---|
| Tested antibody | Description | Dose (mg/kg) | Terminal half life (h) | Total CL (mL/day/kg) | Number of animals | Wt IgG-normalizedin vitro fluxpH 6.0/7.4 |
| mAb 1 | hu IgG1, AAA mutant; FcRn non-binder | 0.3 | 53 | 16.1 | 2 | 0.31 ± 0.01 |
| mAb 2 | hu IgG1, FcRn Wt binder | 0.3 | 79 | 6.49 | 2 | 1.00 ± 0.09 |
| mAb 3 | hz IgG1, FcRn Wt binder | 10 | 310 ± 57 | 3.12 ± 0.55 | 4 | 1.64 ± 0.23 |
| mAb 4 | hu IgG1, FcRn Wt binder | 75 | 269 | 5.23 | 2 | 0.97 ± 0.07 |
| mAb 5 | hu IgG1, FcRn Wt binder | 0.3 | 108 | 3.84 | 2 | 0.63 ± 0.09 |
| mAb 6 | hu IgG1, AAA; FcRn non-binder | 0.3 | 32 | 15.4 | 2 | 0.25 ± 0.01 |
| mAb 7 | hu IgG1, FcRn Wt binder with FcRn-unrelated CL component | 1 | 178 | 24.7* | 3 | 0.84 ± 0.1 |
| mAb 8 | chimeric IgG (hu Fc), FcRn Wt binder | 5 | NC* | 3.60* | 2 | 0.91 ± 0.1 |
| mAb 9 | chimeric IgG (hu Fc), FcRn Wt binder | 0.8 | 380 ± 89 | 2.46 ± 0.35 | 3 | 0.95 ± 0.18 |
| mAb 10 | hu IgG1, enhanced FcRn-binding; positive charge patches | 20 | 288 | 2.86 | 2 | 4.14 ± 0.82 |
| mAb 11 | hz IgG1, FcRn Wt binder (bevacizumab, Avastin) | 5 | Deng et al20 | 3.97 | Deng et al20 | 0.83 ± 0.07 |
| mAb 12 | hu IgG1, FcRn Wt binder | 20 | 235 ± 156 | 4.52 ± 1.07 | 3 | 1.27 ± 0.1 |
| mAb 13 | hu IgG1, enhanced FcRn-binding | 20 | 137 ± 73 | 3.51 ± 0.8 | 3 | 3.42 ± 0.15 |
| mAb 14 | hz IgG1, FcRn Wt binder (trastuzumab, Herceptin) | 5 | Deng et al20 | 5.52 | Deng et al20 | 1.77 ± 0.03 |
| mAb 15 | hu IgG4, FcRn Wt binder | 2.5 | 355 ± 34 | 2.56 ± 0.33 | 3 | 1.10 ± 0.2 |
| mAb 16 | hu IgG1, FcRn Wt binder | 10 | 59 | 9.81 | 2 | 1.03 ± 0.09 |
| mAb 17 | hz IgG1, FcRn Wt binder | 10 | 194 | 5.88 | 2 | 1.06 ± 0.05 |
| mAb 18 | hz IgG1, FcRn Wt binder | 0.15, 1.5, 15, 150 | 150* | 5.86* | 8 | 1.41 ± 0.06 |
| mAb 19 | hu IgG1, FcRn Wt binder | 100 | 282 | 4.49 | 2 | 1.49 ± 0.26 |
| mAb 20 | hz IgG1, FcRn Wt binder | 50 | NC* | 3.34* | 3 | 2.13 ± 0.35 |
| mAb 21 | hu IgG1, FcRn Wt binder | 1 | 218 ± 74.6 | 3.74 ± 0.32 | 4 | 1.07 ± 0.34 |
| mAb 22 | hz IgG1, FcRn Wt binder | 30 | 240 | 4.61 | 2 | 0.95 ± 0.07 |
| mAb 23 | hz IgG1, FcRn Wt binder | 30 | 94.2 | 3.57 | 2 | 1.11 ± 0.16 |
| mAb 24 | hu IgG1, FcRn Wt binder | 150 | 242 | 8.16* | 6 | 2.75 ± 0.19 |
PK parameters estimated by population approach, no SD applicable
mAb systemic clearance from non- compartmental (NCA) PK analysis and terminal half-life after intra-venous dose to monkeys is reported together with the respective mAb wt-normalized Papp from human cellular FcRn assay (pH 6.0/7.4). Data are given as mean values ± standard deviation in case of > 2 subjects or measurements.
hu: human; hz: humanized; Wt: wildtype mAb without mutations of the FcRn-binding sites; TMDD: target-mediated drug disposition.
Fig. 6A depicts the Wt-normalized in vitro flux (Wt-NF) of these 24 mAbs across human FcRn-over-expressing MDCK cells plotted against their respective in vivo CL in cynomolgus monkeys.
Figure 6.

(A) Correlation of reciprocal Wt IgG-normalized in vitro flux (pH gradient 6.0/7.4) of 7 mAbs across human FcRn MDCK cells with in vivo clearance in cynomolgus monkey. Data is plotted as mean values ± SD. Corresponding pairs of Fc mutant and Wt IgG are labeled with the same symbol (closed triangle, star, cross). The greyish area roughly indicates mAbs for which in vivo CL is hypothesized to be affected by “decelerating” processes, for mAbs in the white area “CL-accelerating” processes are thought to be involved. (B) Correlation of reciprocal Wt IgG-normalized in vitro flux (pH gradient 6.0/7.4) of 20 mAbs with wildtype FcRn binding ability across human FcRn MDCK cells with in vivo clearance in cynomolgus monkey. Data is plotted as mean values ± SD for CL and as reciprocal mean values of the Wt IgG-NF. The greyish area roughly indicates mAbs for which in vivo CL is hypothesized to be affected by “decelerating” processes, for mAbs in the white area “CL-accelerating” processes are thought to be involved.
For easier visualization, we plotted the reciprocal Wt-normalized in vitro flux (Wt-NF) values across human FcRn-over-expressing MDCK cells against a mAb's respective in vivo CL in cynomolgus monkeys. Fig. 6A only shows the data from mAbs mutated in their FcRn binding region and their corresponding Wt mAbs: AAA IgG No. 1 vs. Wt IgG No. 2 (star), AAA IgG No. 6 vs. Wt IgG No. 5 (cross), FcRn-enhanced IgG No. 13 vs. Wt IgG No. 12 (closed triangle) and the FcRn-enhanced IgG No. 10 (corresponding Wt IgG n.a.). Similar to the rat IVIVC (Fig. 5), the plot revealed an inverse relationship between these 2 parameters.
The 2 AAA-mutated mAbs (mAb 1 and 6) showed a very high systemic CL in monkeys of about 15 mL/day/kg, due to the lack of FcRn-mediated recycling. This also led to low in vitro flux values in FcRn transcytosis assay compared with their respective Wt IgGs.
On the other hand, 2 mAbs with enhanced FcRn affinity (mAbs 10 and 13) both showed low in vivo CL of 2.9 (mAb 10) and 3.5 mL/day/kg (mAb 13), as their FcRn-mediated recycling and thus plasma residence time was increased. This consistently translated into enhanced in vitro Wt-normalized flux ratios (NF) of 4.1 (mAb 10) and 3.5 (mAb 13) in our cell system.
Most of the tested molecules, however, consisted of Wt-FcRn binders that had different complementarity-determining regions, and thus different molecular properties. Their respective in vivo CL values in monkey were very diverse, ranging from 2 to 25 mL/day/kg, which exceeds the range of 2–10 mL/day/kg reported for other FcRn binding competent IgG1s.20 The respective range of in vitro Wt-NF flux at pH 6.0/7.4 was 0.6 to 2.8 (Table 2). When plotting these in vitro and in vivo data (Fig. 6 B), however, no linear correlation could be derived.
Based on these results, the CL of a mAb whose elimination mechanism is mainly FcRn-modulated is likely to follow this inverse relationship with its in vitro flux. If CL is dominated by additional “CL-accelerating” factors, mAbs may digress into the upper left part of the 2 IVIVC plots (roughly indicated by the white background area in Fig. 6A and B); in the case of “CL-decelerating” influences, such as low pinocytotic uptake, such mAbs might appear into the lower right part (greyish background area).
Discussion
The PK of mAbs is influenced by many factors, such as the biologic target(s) (including turnover, expression level and tissue distribution), affinity to FcRn receptor, immunogenicity, the presence of off-target binding, catabolism, and mAb physicochemical properties (e.g., isoelectric point and charge patch distribution).8,9,30 Several engineered antibodies bearing specific mutations in the FcRn-binding sites have been extensively studied with regards to their ability to prolong residence time in circulation.7
Preclinical in vivo PK studies are in general of great help for understanding mAb clearance mechanisms, and thus for predicting human PK profiles and identifying possible PK-related liabilities early in drug discovery.20 However, depending on antibody properties/format and intended human dose/administration route, factors like species-specific FcRn receptor and antigen(s) expression/turnover/affinity can remarkably affect mAb PK behavior, and consequently increase challenges and uncertainties in translating the PK profile from animal into human.
To characterize the interplay between various mAb clearance pathways and estimate which of them predominantly drive drug candidate disposition in vivo, the availability of in vitro tools to study and address in a quantitative manner specific clearance mechanisms may be of great help. Such in vitro readouts should support antibody optimization and improve the accuracy of human translations in early in drug discovery. As already established in small molecules drug discovery, in vitro screening of PK properties of mAbs can reduce optimization cycles, expand the number of candidates that can be evaluated, and can be beneficial to “refine,” “reduce” and “replace” animal studies (3Rs).31
Here, we characterized and applied a cellular FcRn transcytosis assay using cells expressing either rat or human FcRn receptor (basic assay principle previously introduced by Praetor et al.26 and Tesar et al.25), to assess mAb in vitro transcytosis rate and to use it as an indicator for the FcRn-mediated salvage from intracellular degradation of IgGs, also called “recycling,” known to occur in vivo.13
The derived in vitro transcytotic flux for 4 mAbs in the rat FcRn assay and 24 mAbs in the human FcRn assay was then compared with their in vivo clearance (CL) in rat or cynomolgus monkey, respectively, to evaluate whether this in vitro readout shows in vivo relevance, and consequently whether it could be reliably used in drug discovery projects as a screening and mechanistic tool for mAb candidate optimization and selection.
The cellular FcRn assay was clearly able to differentiate unspecific antibody uptake from FcRn-mediated uptake and transcytosis, as we only detected low background fluxes in “empty vector” MDCK cells lacking human/rat FcRn expression. In addition, mAb flux across human/rat FcRn-overexpressing MDCK cells at pH gradient conditions (pH 6.0/8.0) far exceeded the mAb flux obtained when pH was kept at 8.0 at both sides of the cell layer.
The use of FcRn-overexpressing cells and the application of a pH gradient to induce detectable FcRn-mediated endocytotic uptake followed by transcytosis markedly differs from the process occurring in vivo, where mAb endocytotic uptake occurs instead via fluid-phase pinocytosis only.
Additionally, both endogenous IgGs and therapeutic mAbs bind to FcRn, and inhibition of FcRn-IgG interactions by administration of very high dose of intravenous immunoglobulins is known to lead to accelerated pathogenic antibody CL in vivo and disease amelioration in auto-immune patients.18 In our FcRn in vitro assay, endogenous IgGs were intentionally not included in the donor compartment to ensure maximal dynamic range for the Papp values and allow mechanistic investigations and ranking of FcRn-mediated transcytosis of the studied mAb.
The FcRn transcytosis assay could capture species-specific differences in IgG transcytosis rate, as well as correctly rank wild type vs. Fc-mutated mAbs (AAA and YTE) according to their respective observed PK behavior in vivo. The different transcytosis rates of the diverse Fc mutants support the requirement of the FcRn receptor for transcellular trafficking of an IgG in the studied cell system, since low / no binding to FcRn receptor led to less transcytosis.
It is well known that human and mouse FcRn have different binding affinities for IgGs from different species. As reported by other authors,32,33 human FcRn only binds human, guinea pig and rabbit IgG, whereas mouse FcRn binds IgGs from many different species with high affinity. Human IgG1 binds cynomolgus monkey FcRn with a 2-fold higher affinity than human FcRn, and binds both, mouse and rat FcRn with a 10-fold higher affinity than human FcRn.15
In this work, for the YTE mutant, species-selective differences in rat and human FcRn IgG interactions were observed. This is probably related to differences in mAb pH-dependent binding properties for the 2 FcRn receptor homologues, and thus different binding/release behavior at endosomal and blood pH, respectively. Theoretically, an increased FcRn receptor binding affinity of a mAb at pH 6.0, as reported for the YTE mutant, should help the recycling of the mAb and thus reduce its clearance in vivo.16 However, as also descibed by Vaccaro et al. for a different Fc mutation,34 the YTE mutant might also possess enhanced binding affinity to murine FcRn at pH 7.4, resulting in a reduced release of the IgG to the blood stream and thus increased cellular degradation. Conversely, the binding of the YTE mAb to the human FcRn at pH 7.4 was low, resulting in an increased in vitro flux in the human FcRn cellular system, exceeding that of the wt-IgG. This indicates that the studied assay not only identifies differences in FcRn affinity at pH 6.0 across animal species, but can help to reveal the overall effect of the mutations on the various processes, including receptor binding, FcRn-IgG-complex trafficking and IgG release.
Given the additive nature of mAb systemic clearance process,9 expected to occur also for large molecules, and the lack of target antigen expression in our cellular system, a direct correlation between in vitro transcytotic flux and in vivo CL can be expected only for biologics whose clearance mechanism at the studied dose levels is modulated predominantly by its interactions with the FcRn receptor, and not in cases where CL is affected by other processes, such as the formation of anti-drug antibodies or other immunogenic reactions, by TMDD when non-saturating drug doses are studied, by specific sequence instability and catabolism, or by an enhanced non-specific cellular pinocytotic uptake.35
The rat FcRn in vitro flux to in vivo rat CL correlation shown here indicated an inverse relationship of the 2 parameters. When comparing the human FcRn in vitro flux to the observed in vivo CL in cynomolgus monkeys, again the AAA mutants with high CL displayed a low in vitro flux, whereas mAbs with enhanced FcRn affinity and consequently low CL showed a stronger transcytosis in the cellular assay. The Wt-FcRn binders with CL values from 2 to 25 mL/day/kg showed an in vitro “Wt IgG-normalized flux” (NF) range from 0.6 to 2.8. Therefore, mAbs within this range could be considered to have “normal” FcRn interaction properties. For this mAb cluster, no obvious quantitative correlation between the in vivo CL and the FcRn-mediated flux in vitro was observed, despite the absence of TMDD at the tested dose levels and immunogenicity in vivo.
The fact that mAbs with very similar in vitro flux, still showed considerable differences in in vivo CL could be explained by additional CL processes, apart from FcRn recycling, not present in the cellular assay. One example is mAb 7, which showed an extremely high systemic CL in monkey (25 mL/day/kg) even though it had a non-mutated Fc scaffold and its in vitro flux was similar to a mAb with normal in vivo CL < 10 mL/day/kg.20 The more mAbs digress from a linear relationship between in vitro FcRn-mediated transcytosis and in vivo CL, the more their PK is expected to be affected by additional “CL-accelerating” processes, such as enhanced pinocytosis or off-target binding, or “decelerating” processes, such as low unspecific uptake.
It should be noted that, in contrast to the PK data available for rat where most mAbs shared the same Fab structure and epitope (anti-DIG) and were evaluated in the same in vivo experiment, the clearance data set used for the human FcRn flux/monkey CL-IVIVC were obtained at different dose levels, with different study designs and bioanalytical assays, and using different monkey colonies (thus animals of various ages/origins), which can increase data variability and reduce the accuracy of the IVIV correlation. In addition, the mAbs tested in monkey PK studies have different scaffolds and are not directed against the same target epitope, resulting in large differences in physicochemical properties and 3D structures, whose effect on FcRn-mediated recycling and cellular trafficking occurring in vivo might not be captured in this cellular system. Additionally, cynomolgus monkey and human FcRn receptor sequence/expression are very similar but not entirely identical.20 To further evaluate this aspect and to calibrate the IVIVC with clinical data, we are currently selecting more mAbs for in vitro testing for which systemic clearance in man is available from Roche clinical trials or reported in literature. For mAbs with monkey PK that does not translate well into man due to species-dependent FcRn differences, the IVIVC is expected to improve. However, the correlation could also get worse if additional FcRn-unrelated clearance processes take place in man which are absent in monkey, such as a human-specific off-target binding.
After intracellular uptake in vivo, an antibody can either enter the “recycling pathway” occurring via FcRn-receptor interactions, or the “degradative (lysosomal) pathway.”36 This is dependent on the degree of interaction with the FcRn receptor and the level of occupancy of the FcRn receptors in the endosomes, which is a saturable process.
In vitro, due to the high expression level of FcRn in the transfected MDCK cells we used, which is much higher than under physiologic conditions, it is likely that most/all of the IgG molecules internalized under pH gradient conditions at pH 6.0/7.4 are further trafficked intracellularly by FcRn and no saturation occurs. At the same time, an unspecific cellular uptake of a mAb, which might be enhanced by certain physicochemical properties, e.g., large positive charge patches, can also occur under pH gradient conditions and contribute to the flux measured in this work at pH 6.0/7.4. It should be noted that an antibody molecule showing a high degree of unspecific cellular uptake (i.e., non-target- and non FcRn-mediated) occurring via fluid-phase pinocytosis is expected to be more prone to undergo intracellular degradation. Indeed, a higher fraction of the total antibody amount is internalized independently of its binding to FcRn and is thus not protected by lysosomal degradation.8 Therefore, increased unspecific cellular uptake of a mAb might counteract the prolongation of in vivo residence time expected by an increased FcRn binding affinity.37
These considerations are well in line with the observations for mAb 10, which was specifically designed to display enhanced FcRn affinity and a certain positive charge patch distribution to increase pinocytotic cellular uptake.8,38 This combination resulted in increased flux in the transcytosis assay, but a residence time in monkeys comparable to non-mutated IgG1, as pinocytosis and FcRn-mediated effects highly likely compensate each other in vivo.
This counter-activity of pinocytosis and FcRn-mediated salvage in vivo is also the reason why we chose the pH gradient conditions for the IVIVC. When applying pH 7.4 in both chambers, mAb uptake will solely happen via pinocytosis. Thus, we can assume that a mAb with high FcRn affinity, but low pinocytotic uptake, would reach a similar or even lower transcytotic flux than a mAb with average FcRn affinity and high pinocytotic uptake. As described above, a mAb with high pinocytotic uptake behavior undergoes a more rapid clearance as opposed to a mAb with low non-specific cellular uptake potential. Thus, the pinocytotic uptake component is considered to be a confounding element for our in vitro readout, where an enhanced transcytotic flux is in general interpreted as a hint for low in vivo CL due to proper FcRn salvage.
In summary, our cellular FcRn assay was able to detect and quantitatively measure the FcRn-mediated transcytosis occurring in vitro for various therapeutic IgGs. Beside describing further validation of the in vitro FcRn assays initially reported by Praetor26 and Tesar,25 this work evaluates for the first time the correlation between in vitro transcytosis and in vivo preclinical PK data for 25 antibody-based molecules. Despite the discussed limitations and need for further mechanistic investigations before quantitatively using the in vitro flux for, for example, physiologically based PK modeling purposes, this cellular assay looks promising for a first in vitro evaluation of potential liabilities in antibody FcRn-mediated clearance, with the aim to rank candidates and reduce the number of molecules tested in vivo. Although it does not cover all the biologic processes involved in mAb recycling and degradation in vivo, it helps to understand the contribution of the FcRn-mediated recycling to mAb PK and highlights that mAb disposition in vivo is a phenomenon likely more complex and more peculiar for each antibody than has been considered so far.
Material and methods
Antibodies
Fluorescein isothiocyanate (FITC)-labeled antibodies (rat IgG, mouse IgG and chicken IgY) were purchased from Jackson ImmunoResearch (Cat.No. 012-090-003, 015-090-003 and 003-090-003, respectively), FITC-human IgG from Sigma-Aldrich (Cat.No. F 9636). The paracellular marker FITC-dextran (150 kDa) was obtained from Sigma-Aldrich. Chimeric, human or humanized unlabeled antibodies with/without Fc mutations were generated at Hoffmann- La Roche as part of various discovery and development projects; due to company policy on chemical name disclosure, structures and targets for Roche mAbs are not reported here. Fc mutants were designed in a way to specifically alter the FcRn-binding affinity of the molecule. All other antibodies were considered as «wildtype» FcRn-binders. All tested unlabeled mAbs are listed in Table 2.
3H-labeling of antibodies
Five molecules (anti-Digoxigenin-YTE (M252Y/S254T/T256E) IgG; anti-Digoxigenin AAA (M252A/H310A/H435A) IgG; anti-Digoxigenin Wildtype IgG; chicken IgY; human Wt IgG Fc fragment) were 3H-labeled according to the following protocol in a representative example. Chicken IgY was purchased from Jackson ImmunoResearch, the anti-DIG IgGs had been generated internally.
Four mg (0.0274 µmol) of antibody in 377 µL formulation buffer (20 mM His, 140 mM NaCl, pH 6) was diluted with 100 µL labeling buffer (DPBS pH 8.5, pH was adjusted with 1 N aq. NaOH) and placed into a 3500 MWCO Midi D-Tube™ Dialyzer. The solution was dialyzed against labeling buffer; the buffer was changed 3 times each after 45 minutes and stored in the refrigerator overnight. 13.1 µg (6.6 mCi, 0.074 µmol) of [3H]-N-succinimidyl propionate (NSP) was transferred into a 1.5 mL Eppendorf LoBind tube and dissolved in 15 µL dimethylsulfoxide. Antibody solution was added to the Eppendorf vial and the solution was shaken for 15 minutes at room temperature. After this time, 1 µL of 1 M lysine, dissolved in labeling buffer, was added to stop the reaction. The reaction solution was transferred into a 3500 MWCO Midi D-Tube™ Dialyzer and dialyzed against the formulation buffer. The buffer was changed 3 times each after 45 minutes and stored in the fridge overnight. The protein concentration was determined by UV at 280 nm (Eppendorf BioSpectrometer). The radioactivity was determined by liquid scintillation counting (Hidex 300SL and ULTIMA GOLD™ cocktail). Specific activities were achieved in the range of 900 µCi/mg to 1200 µCi/mg. The radiochemical purity of > 95% was determined by size-exclusion chromatography.
Cell culture
MDCK cells transfected with rat FcRn and rat β2m were kindly provided by Prof. Pamela Bjorkman (CalTech, Los Angeles, US) and were maintained according to Tesar et al.25 in SMEM (Gibco, Life Technologies), supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich), 10′000 IU/µg/mL penicillin-streptomycin (Gibco, Life Technologies), 2mM L-Glutamine (Gibco, Life Technologies) and 0.2 mg/mL geneticin (Gibco, Life Technologies) at 37°C, 5% CO2. MDCK cells transfected with human FcRn and empty vector (EV) were kindly provided by Prof. Walter Hunziker, (Singapore University) and were maintained according to Praetor et al. in DMEM (Gibco, Life Technologies), supplemented with 10% FBS (Sigma-Aldrich), 10′000 IU/µg/mL penicillin-streptomycin, 20 mM HEPES (Gibco, Life Technologies) and 0.5 mg/mL geneticin at 37°C, 5% CO2. All the cells were passaged twice per week.
For experiments requiring polarized cell monolayers, cells were seeded at superconfluent density (0.25 × 106 cells/mL) onto 6.5 mm diameter 24-well Transwell® polycarbonate filters, 0.4 µm pore size (Corning Costar), with 0.3 and 1.0 mL of media in the apical and basolateral reservoirs, respectively. In the Transwell® system, rat FcRn MDCK cells were maintained in MEM (Sigma-Aldrich) supplemented with 10% FBS, 10′000 IU/µg/mL penicillin-streptomycin and 0.2 mg/mL geneticin at 37°C, 5% CO2, were fed on day 3 after initial seeding and used for experiments on the fourth day post plating. Human FcRn and EV MDCK cells were maintained in the same medium and conditions as in the flask were fed the second or third day after initial seeding and used for experiments on the third or fourth day post plating, respectively.
Prior to incubations with the test compounds, the designated receiver plate was blocked for 24 h with 1% bovine serum albumin (BSA) at 4°C to prevent mAb adsorption to the plate surface. Cells were serum-starved for 2 to 2.5 h in their respective medium without FBS, washed once with HEPES (Sigma-Aldrich)-buffered Hanks' balanced salt solution with Ca2+ and Mg2+ (HBSS++; Gibco, Life Technologies) pH 7.4 and finally pre-incubated for 30 minutes with MES (Sigma-Aldrich)-buffered HBSS++ pH 6.0 (donor compartment) or HEPES-HBSS++ pH 7.4 (acceptor compartment) or with HEPES-HBSS++ pH 7.4 in both compartments as a control.
Quantitative transcytosis assay
Test antibody (3H-labeled, fluorescently labeled or unlabeled) was added at the indicated final concentration in HBSS++ pH 6.0, pH 7.4 or 8.0 (the 2 latter for control experiments) and 0.1% BSA in 0.6 mL in the basolateral chamber, which served as donor compartment. The apical chamber was filled with 0.2 mL of HBSS++ pH 7.4 or pH 8.0 containing as well 0.1% BSA. FITC-dextran (150 kDa) at 100 nM was incubated as paracellular marker in separate wells for each experiment. Plates were then incubated at 37°C or 4°C (for control measurements), 5% CO2, for the indicated durations. At the end of the experiment, samples were taken from the acceptor and donor compartment and for 3H-labeled compounds, concentration was measured via liquid-scintillation counting (LSC) in LumaPlate-96, White Opaque Microplate with Scintillate Coated on the Bottom (Perkin Elmer) in a Top Count Plate reader NXT (HTS) on the following day. Fluorescently labeled molecules were measured in 384-wells black plate (Greiner) in a Multiplate reader system for fluorescence (Perkin Elmer) and concentration calculation was based on calibration curves for each compound in the given buffer system. Unlabeled human IgGs were analyzed by quantitative ELISA.
ELISA (enzyme-linked immunosorbent assay)
Concentrations of unlabeled human(ized) IgGs were determined by appropriate enzyme-linked immunoassays. Anti-human-IgG, Fcγ-specific and Anti-human F(ab)2-F(AB2)-Biotin (Jackson Immunoresearch), were used for capturing and detection, respectively, in combination with Poly-HRP40-Streptavidin (Fitzgerald) and Supersignal chemiluminescent (Thermo scientific). PBS +0.1% Tween 20® was used as wash buffer and PBS +0.5%BSA +0.05% Tween 20® was used as blocking and assay buffer. All washing steps were performed in the Biotek ELx405 washer: 4 times after blocking and incubation with the samples, and 6 times after incubation with the detection Ab and the HRP-conjugate. All incubation steps with exception of the coating period (4°C overnight, no shaking) were performed at room temperature (RT) with shaking at 450 RPM.
ELISA plates (Nunc ImmunoMaxiSorp) were coated with 0.1 µg/mL of capturing antibody in PBS followed by at least 1 hour of blocking. Then, serially diluted assay samples (1:8, 1:32 and 1:128) and a calibration curve (range detection: 34 pg/mL −25 ng/mL) for each test compound prepared in assay buffer were added and incubated for 1.5 to 2 hours. Subsequently, bound test IgG was detected by incubating with 50 ng/mL of detection antibody for 1.5 to 2 hours, followed by incubation with 1 ng/mL of HRP-conjugate for 20 to maximally 30 minutes. Finally, the chemiluminescence substrate was added and the signal was measured in the Multiplate reader system for luminescence (Perkin Elmer). The data was processed with SoftMax Pro Data Acquisition and Analysis Software (Molecular Devices).
Single-dose Pharmacokinetic Studies with 3H-antibodies in rats
Adult male Wistar rats weighing ∼250 g were obtained from Harlan Laboratories (Horst, Netherlands) and housed at F. Hoffmann La-Roche, Basel, in a controlled environment (temperature, humidity, and 12 h light/dark cycle) with ad libitum access to food and water. All rodent studies were conducted with the approval of the local veterinary authority in strict adherence to the Swiss federal regulations on animal protection and to the rules of the Association for Assessment and Accreditation of Laboratory Animal Care International. The tested antibodies were the following: chicken IgY, anti-Digoxigenin-hu IgG-AAA mutation (AAA IgG), anti-Digoxigenin-hu Wt IgG (Wt IgG); anti-Digoxigenin-hu IgG-YTE mutation (YTE IgG).16 All molecules were administered in their 3H-labeled form.
Four rats per compound received 30 µCi 3H-antibody in 500 µL histidine buffer (20 mM histidine, 140 mM NaCl, pH 6) by i.v. tail vein injection.
Blood samples were collected from the tail vein under mild isoflurane anesthesia into K2EDTA coated polypropylene tubes at 0.83, 1, 3, 7, 24, 48, 72, 96, 168, 240, 336, 408, 504, 576, 672, 744, and 840 hours after administration. Plasma was prepared within 30 min by centrifugation at 3000 g for 5 min at 4°C and frozen immediately. Then 10 µL of plasma and 3.5 mL of Ultima Gold® (Perkin-Elmer) were then mixed in 6 mL polyethylene tubes (Perkin Elmer) and the radioactivity counts were measured by a liquid scintillation analyzer (Tri-Carb 3100 TR, Packard Instruments).
Single-dose Pharmacokinetic Studies with cold antibodies in monkeys
Adult cynomolgus monkeys were used for the PK studies. All monkey studies were approved by the Roche Ethical Committee on Animal Welfare and conducted with the approval of the local veterinary authorities and conducted an Roche Innovation Center Basel or at audited Contract Research Organizations. Most of the tested antibodies originated from various internal Roche R&D projects; due to company policy on chemical name disclosure, structures and targets for 22 Roche mAbs are not reported here. The tested compounds were administered as single i.v. injection and the antibody concentrations in plasma or serum were determined by ELISA methodologies. Details of the individual studies are reported in Table 2.
Data analysis
In vitro data was evaluated with Microsoft Excel 2010 (Microsoft). Plotting was performed with Microsoft Excel 2010 and GraphPad Prism 6. Analysis of in vivo PK data was performed with Phoenix WinNonlin by NCA (Certara).
Supplementary Material
Disclosure of potential conflicts of interest
All authors are current or former employees of Roche, Pharmaceutical Research and Early Development, and hold a financial interest in Roche.
Acknowledgments
We thank Maria Stella Gruyer, Peter Schrag, and Claudia Senn for conducting part of the rat and cynomolgus monkey PK studies and Eginhard Schick for his support with the ELISA method establishment (all from Pharmaceutical Sciences, Pharma Research and Early Development, Roche Innovation Center Basel). Moreover, we would like to thank Hubert Kettenberger, Alexander Haas, Mathias Mueller, Dieter Muri, Thomas Hartung, Eike Hoffmann, Erwin van Prujenbrook, Tilman Schlothauer, Jens Fischer, Silke Metz, Marcus Schmid, and Jens Niewoehner from the TMo Department, Roche Innovation Center Basel, Zurich, and Munich for their advices, for the QC of 3H-labeled mAbs, and for the supply of the mAbs for the IVIVC. We are also grateful to Kevin Brady, Matthias Fueth, Martin Lechmann, Niels Janssen, Wolfgang Richter, Hans-Peter Grimm, Antje-Christine Walz, Erich Koller, and Cristina Bertinetti-Lapatki from Pharmaceutical Sciences, Roche Innovation Center Basel, for the mAb supply, cell characterization, monkey PK data and valuable scientific discussions. Finally, we acknowledge CalTech, Singapore University, for providing the cells and Prof. Dr. Oliver Muehlemann from the University of Bern.
References
- 1.Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K. Engineering the variable region of therapeutic IgG antibodies. Mabs-Austin 2011; 3:243-52; PMID:21406966; https://doi.org/ 10.4161/mabs.3.3.15234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klein C, Schaefer W, Regula JT. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. Mabs-Austin 2016; 8:1010-20; PMID:27285945; https://doi.org/ 10.1080/19420862.2016.1197457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson AL. Antibody fragments Hope and hype. Mabs-Austin 2010; 2:77-83; PMID:20093855; https://doi.org/ 10.4161/mabs.2.1.10786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kontermann RE. Half-life extended biotherapeutics. Expert Opin Biol Th 2016; 16:903-15; PMID:26967759; https://doi.org/ 10.1517/14712598.2016.1165661 [DOI] [PubMed] [Google Scholar]
- 5.Ayyar BV, Arora S, O'Kennedy R. Coming-of-Age of Antibodies in Cancer Therapeutics. Trends Pharmacol Sci 2016; 37:1009-28; PMID:27745709; https://doi.org/ 10.1016/j.tips.2016.09.005 [DOI] [PubMed] [Google Scholar]
- 6.Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010; 10:345-52; PMID:20414207; https://doi.org/ 10.1038/nri2747 [DOI] [PubMed] [Google Scholar]
- 7.Mimoto F, Kuramochi T, Katada H, Igawa T, Hattori K. Fc engineering to improve the function of therapeutic antibodies. Curr Pharm Biotechnol 2016; 17:1298-1314; PMID:27552846; https://doi.org/0.2174/1389201017666160824161854 [DOI] [PubMed] [Google Scholar]
- 8.Li B, Tesar D, Boswell CA, Cahaya HS, Wong A, Zhang JH, Meng YG, Eigenbrot C, Pantua H, Diao JY, et al.. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. Mabs-Austin 2014; 6:1255-64; PMID:25517310; https://doi.org/ 10.4161/mabs.29809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Datta-Mannan A, Lu JR, Witcher DR, Leung D, Tang Y, Wroblewski VJ. The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. Mabs-Austin 2015; 7:1084-93; PMID:26337808; https://doi.org/ 10.1080/19420862.2015.1075109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leavy O. Therapeutic antibodies: past, present and future. Nat Rev Immunol 2010; 10:297; PMID:20422787; https://doi.org/ 10.1038/nri2763 [DOI] [PubMed] [Google Scholar]
- 11.Sand KMK, Bern M, Nilsen J, Noordzij HT, Sandlie I, Andersen JT. Unraveling the interaction between FcRn and albumin: opportunities for design of albumin-based therapeutics. Front Immunol 2015; 5:1-21; PMID:25674083; https://doi.org/ 10.3389/fimmu.2014.00682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martins JP, Kennedy PJ, Santos HA, Barrias C, Sarmento B. A comprehensive review of the neonatal Fc receptor and its application in drug delivery. Pharmacol Therapeut 2016; 161:22-39; PMID:27016466; https://doi.org/ 10.1016/j.pharmthera.2016.03.007 [DOI] [PubMed] [Google Scholar]
- 13.Giragossian C, Clark T, Piche-Nicholas N, Bowman CJ. Neonatal Fc receptor and its role in the absorption, distribution, metabolism and excretion of immunoglobulin G-Based biotherapeutics. Curr Drug Metab 2013; 14:764-90; PMID:23952252; https://doi.org/ 10.2174/13892002113149990099 [DOI] [PubMed] [Google Scholar]
- 14.Schlothauer T, Rueger P, Stracke JO, Hertenberger H, Fingas F, Kling L, Emrich T, Drabner G, Seeber S, Auer J, et al.. Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. Mabs-Austin 2013; 5:576-86; PMID:23765230; https://doi.org/ 10.4161/mabs.24981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdiche YN, Yeung YA, Chaparro-Riggers J, Barman I, Strop P, Chin SM, Pham A, Bolton G, McDonough D, Lindquist K, et al.. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. Mabs-Austin 2015; 7:331-43; PMID:25658443; https://doi.org/ 10.1080/19420862.2015.1008353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robbie GJ, Criste R, Dall'Acqua WF, Jensen K, Patel NK, Losonsky GA, Griffin MP. A novel investigational Fc-Modified humanized monoclonal antibody, motavizumab-YTE, has an extended Half-Life in healthy adults. Antimicrob Agents Ch 2013; 57:6147-53; PMID:24080653; https://doi.org/ 10.1128/AAC.01285-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dall'Acqua WF, Kiener PA, Wu HR. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281:23514-24; PMID:16793771; https://doi.org/ 10.1074/jbc.M604292200 [DOI] [PubMed] [Google Scholar]
- 18.Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. Mabs-Austin 2011; 3:422-30; PMID:22048693; https://doi.org/ 10.4161/mabs.3.5.16983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Souders CA, Nelson SC, Wang Y, Crowley AR, Klempner MS, Thomas W Jr.. A novel in vitro assay to predict neonatal Fc receptor-mediated human IgG half-life. mAbs 2015; 7:912-21; PMID:26018774; https://doi.org/ 10.1080/19420862.2015.1054585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data What have we learned? Mabs-Austin 2011; 3:61-6; PMID:20962582; https://doi.org/ 10.4161/mabs.3.1.13799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong JQ, Salinger DH, Endres CJ, Gibbs JP, Hsu CP, Stouch BJ, Hurh E, Gibbs MA. Quantitative prediction of human pharmacokinetics for monoclonal antibodies retrospective analysis of monkey as a single species for First-in-Human prediction. Clin Pharmacokinet 2011; 50:131-42; PMID:21241072; https://doi.org/ 10.2165/11537430-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 22.Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu JR, Witcher DR, Wroblewski VJ. FcRn Affinity-Pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab Dispos 2012; 40:1545-55; PMID:22584253; https://doi.org/ 10.1124/dmd.112.045864 [DOI] [PubMed] [Google Scholar]
- 23.Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol Immunol 2006; 43:1462-73; PMID:16139891; https://doi.org/ 10.1016/j.molimm.2005.07.032 [DOI] [PubMed] [Google Scholar]
- 24.Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu J, Witcher DR, Wroblewski VJ. FcRn affinity-pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab Dispos 2012; 40:1545-55; PMID:22584253; https://doi.org/ 10.1124/dmd.112.045864 [DOI] [PubMed] [Google Scholar]
- 25.Tesar DB, Tiangco NE, Bjorkman PJ. Ligand valency affects transcytosis, recycling and intracellular trafficking mediated by the neonatal Fc receptor. Traffic 2006; 7:1127-42; PMID:17004319; https://doi.org/ 10.1111/j.1600-0854.2006.00457.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Praetor A, Ellinger I, Hunziker W. Intracellular traffic of the MHC class I-like IgG Fc receptor, FcRn, expressed in epithelial MDCK cells. J Cell Sci 1999; 112:2291-9; PMID:10381385. [DOI] [PubMed] [Google Scholar]
- 27.Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol 2001; 13:1551-9; PMID:11717196; https://doi.org/ 10.1093/intimm/13.12.1551 [DOI] [PubMed] [Google Scholar]
- 28.Martin WL, West AP Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell 2001; 7:867-77; PMID:11336709; https://doi.org/ 10.1016/S1097-2765(01)00230-1 [DOI] [PubMed] [Google Scholar]
- 29.Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: Impact on PK/PD and efficacy. Aaps J 2012; 14:296-302; PMID:22407289; https://doi.org/ 10.1208/s12248-012-9340-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al.. A strategy for risk mitigation of antibodies with fast clearance. Mabs-Austin 2012; 4:753-60; PMID:23778268; https://doi.org/ 10.4161/mabs.22189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russell WM. The development of the three Rs concept. Altern Lab Anim 1995; 23:298-304; PMID:11656565. [PubMed] [Google Scholar]
- 32.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; https://doi.org/ 10.1038/nri2155 [DOI] [PubMed] [Google Scholar]
- 33.Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: Implications for therapeutic antibodies. Int Immunol 2001; 13:1551-9; PMID:11717196; https://doi.org/ 10.1093/intimm/13.12.1551 [DOI] [PubMed] [Google Scholar]
- 34.Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. P Natl Acad Sci USA 2006; 103:18709-14; PMID:17116867; https://doi.org/ 10.1073/pnas.0606304103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Datta-Mannan A, Lu J, Witcher DR, Leung D, Tang Y, Wroblewski VJ. The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies. mAbs 2015; 7:1084-93; PMID:26337808; https://doi.org/ 10.1080/19420862.2015.1075109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grevys A, Bern M, Foss S, Brodie DB, Moen A, Gunnarsen KS, Aase A, Michaelsen TE, Sandlie I, Andersen JT. Fc Engineering of Human IgG1 for Altered Binding to the Neonatal Fc Receptor Affects Fc Effector Functions. J Immunol 2015; 194:5497-508; PMID:25904551; https://doi.org/ 10.4049/jimmunol.1401218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs 2015; 7:483-93; PMID:25695748; https://doi.org/ 10.1080/19420862.2015.1016696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al.. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 2010; 23:385-92; PMID:20159773; https://doi.org/ 10.1093/protein/gzq009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.