

Abstract
Discovery of genetic mechanisms for resistance to obesity and diabetes may illuminate new therapeutic strategies for the treatment of this global health challenge. We used the polygenic Lean mouse model, selected for low adiposity over 60 generations, to identify thiosulfate sulfurtransferase (Tst, Rhodanese) as a candidate obesity-resistance gene with selectively increased adipocyte expression. Elevated adipose Tst expression correlated with indices of metabolic health across diverse mouse strains. Transgenic overexpression of Tst in adipocytes protected mice from diet-induced obesity and insulin-resistant diabetes. Tst gene deficiency markedly exacerbated diabetes whereas pharmacological TST activation ameliorated diabetes in mice in vivo. Mechanistically, TST selectively augmented mitochondrial function combined with degradation of reactive oxygen species and sulfide. In humans, adipose TST mRNA correlated positively with adipose insulin sensitivity and negatively with fat mass. Genetic identification of Tst as a beneficial regulator of adipocyte mitochondrial function may have therapeutic significance for type 2 diabetes.
Keywords: obesity, type 2 diabetes, obesity-resistance, leanness, insulin resistance, insulin sensitivity genetics, quantitative trait loci, thiosulfate sulfurtransferase, Rhodanese
Obesity prevalence appears to have plateaued at ~40%1,2 suggesting that environmental (hypercaloric diet, sedentary lifestyle) penetrance is limited to a sub-population that is genetically susceptible to obesity. Indeed, a substantial and stable proportion of the population has remained non-obese despite the modern obesogenic onslaught3. Determining the genetic basis for this resilience may illuminate novel intervention strategies for obesity and its associated cardiometabolic diseases. To discover novel genes contributing to healthy low adiposity we employed a unique polygenic Lean mouse line that was generated by selection over 60 generations for extreme low adiposity. Lean mice (4% of body weight is fat mass) exhibit metabolically healthy leanness4, not lipodystrophy. The major quantitative trait loci (QTL) underlying divergent adiposity between Lean mice and their counter-selected Fat mouse line (23% of body weight is fat mass) were described5, 6, facilitating our search for positional candidate lean genes. Direct experimental evidence7 and QTL mapping also ruled out known brain-regulated appetite and energy expenditure (Lep, Lepr, Npy, Mc4r) systems as driving adiposity divergence. Lean mice thus allow insight into the genetic landscape of healthy leanness manifested though peripheral mechanisms. We reasoned that selection on adiposity divergence, independent of lean mass, would enrich for genes with direct actions in the adipose tissue. To identify adipose tissue-specific candidate lean genes, strict inclusion criteria were applied to transcriptomic profiles from three distinct white adipose tissue (WAT) depots (subcutaneous, epididymal and mesenteric) versus liver, muscle and kidney of the Lean and Fat mice. Gene mRNAs exhibiting coordinate elevation only in the Lean WAT depots were then prioritized using adiposity QTL information. Genetic and functional validation supported nuclear-encoded mitochondrial TST as a novel Lean line adiposity-reducing gene, with a dominant and broader influence on metabolic disease-resistance across species.
RESULTS
Elevated adipose Tst expression links leanness and metabolic health
To prioritize positional candidate lean genes from within major adiposity QTLs5, 6 we selected for increased mRNA levels specifically in WAT of Lean mice8. Candidate gene mRNA levels had to be >2-fold higher in all three Lean versus Fat WAT depots but comparable in the liver, muscle and kidney. Higher Tst mRNA was validated in an independent microarray comparison of Lean versus Fat subcutaneous WAT. 11 genes fulfilled tissue-specific expression criteria (Est AI427515, Csprs, Cyp2e1, Fv1, Ms4a4c, Ms4a6c, Pik3cg, Pon1, Rassf6, Sah and Tst). 2 genes fulfilled the QTL inclusion criteria (Pik3cg; Chr12, Tst; Chr15). Pik3cg failed validation in the independent microarray. Only the nuclear-encoded mitochondrial thiosulfate sulfur-transferase (Tst, synonym Rhodanese), positioned on chromosome 15 (78,399,556–78,405,859) within the F-line obesity QTL3 (Fob3)5, 6, fulfilled all our inclusion criteria. Tst mRNA was ~7-fold higher in the Lean compared to the Fat line across the 3 WAT depots (Fig. 1a).
Figure 1.

TST is elevated in adipose tissues from lean mice. (a) Ratio of Tst mRNA levels (n = 3) in Lean (L) versus Fat (F) mice in tissue from subcutaneous (SC), epididymal (Epi) and visceral (Vis) fat or liver (Liv), muscle (Mus) and kidney (Kid). (b) Tst mRNA levels (top) in SC fat of F (white bar) or L (n = 16) and derived congenic lines (black bars) U12, M and M2 (n = 8). Chromosome 15 composition (bottom) derived from F (white infill), L (black infill) and origin-undetermined regions (grey infill). Arrow, Tst gene position. (c) TST (yellow balloons) modulates 1. Cyanide and H2S, 2. Reactive oxygen species (O2−) with thioredoxin (purple cylinder) and 3. respiratory complex enzymes (e.g. cII, succinate dehydrogenase). “Fe-S”, iron-sulfur cluster, red “S, S-S”, sulfur, sulfane-sulfur. (d) Representative western blot image (n = 6) of TST protein (top) and quantification (bottom) of the ratio of TST and β-actin levels in SC fat of L mice on control diet (black bars) or high fat (HF) diet (dark grey bars) and F mice control diet (white bars) or HF diet (light grey bars). (e–g) Adipose Tst mRNA correlations (Pearsons’) with fat mass (e) blood glucose (f) and adiponectin mRNA levels (g) across 23 mouse strains. (h) Tst expression from segregating Tst locus in the diversity outbred (DO) strains (top) and LOD score (bottom). Blue bar, Tst position. n = 277 DO mice. (i) SC fat RNA-Seq Tst mRNA level correlated with female adiposity in founder DO strains. n = 6 per strain, combined control and HFD. (j) Representative western blot (top) and quantification (bottom) of TST protein levels in SC fat of C57BL/6J fed control (black bars, n = 6) or HFD (n = 6) for 16 weeks (dark grey bars) or 10 week-old C57BL/6J (black bar, n = 6) and leptin-deficient (Lepob) mice (white bar, n = 5). In a,b,d,j data are mean ± SEM. In (b) †† = P <0.01 versus F, §§ = P <0.01 versus L, by 1-way ANOVA. In d and j ** = P <0.01 effect of diet and †† = P < 0.01, ††† = P < 0.001 effect of line or genotype by 2-way ANOVA.
To support causality over simple association with leanness, we sought evidence that increased Tst mRNA originated from a cis-acting effect within the QTL. To this end we employed congenic lines of mice with increasingly refined Lean line Fob3 QTL chromosome 15 segments around the Tst locus introgressed into an otherwise Fat line genetic background (lines U12, M and M2). The U12 line carries a ~35 Mbp QTL (Fob3b) that accounted for a 20% reduction in Fat line adiposity5. The M line carries a ~6.8 Mbp sub-QTL (Fob3b2)6 that accounted for an 8.1% reduction in Fat line adiposity (Supplementary Table 1a). The M2 line was developed specifically for this study and carries a ~2.8 Mbp sub-sub-QTL (Fob3b2-M2; Supplementary Fig. 1) that accounted for an 8.4% reduction in Fat line adiposity (Supplementary Table 1a). Comparable fat-reduction in M2 versus M suggested the leanness effect was fully captured by this smallest sub-sub genetic interval (Supplementary Table 1b, Supplementary Fig. 1b–e). Adipose Tst mRNA levels were higher in all three congenic lines than in the parental Fat line, supporting Tst as a positional candidate lean gene (Fig. 1b). Allele-dosage studies in Fob3b2-M2 heterozygotes supported elevated Lean-line Tst allele mRNA as cis-mediated (Supplementary Fig. 1f,g). Co-segregating metabolic protection of the locus was confirmed in Fob3b2-M2-line mice (Supplementary Fig. 1h,i). We used haplotype analysis of the M2 interval to restrict for causal genomic regions (non-identical-by-descent). 20 of 90 genes remained positional candidates, of which only Tst exhibited co-ordinate up-regulation in all 3 WAT depots of Lean mice (Fig. 1a) and in WAT from Fob3b2-M2 Lean allele homozygotes generated from an F2 cross with the Fat line (Supplementary Fig. 2). Tst also scored as the top functional and positional candidate gene by bioinformatics approaches (Supplementary Fig. 2).
Given the strong genetic evidence for Tst as a causal lean gene, we considered the mechanistic rationale for its effects. TST was identified more than 80 years ago as a rhodanese enzyme activity involved in cyanide detoxification9, with the clinical antidote thiosulfate (S2O3) acting as a TST substrate for sulfane-sulfur transfer9,10. This did not immediately suggest a purpose for TST in fat cell function or adiposity. However, TST was linked to modulation of iron-sulfur clusters and regulatory cysteine residues of succinate dehydrogenase11 and modulation of NADH dehydrogenase12, two key mitochondrial respiratory complexes. In addition, TST degraded reactive oxygen species (ROS) with thioredoxin in cell-free systems13 and participated in sulfide (H2S) metabolism14 through its interaction with the sulfur dioxygenase ETHE115. Unfolded TST may also chaperone cytosolic 5S rRNA to the mitochondria16. Since mitochondrial ROS17–19 and recently H2S20,21 were implicated in adipocyte function, we hypothesized that elevated TST activity beneficially alters these pathways (Fig 1c).
During candidate lean gene validation we found that adipose TST was unexpectedly up-regulated when Lean mice were challenged with HFD (Fig. 1d). Moreover, Lean mice remarkably lost fat mass (Supplementary Table 2a–b) with HFD. Our previous study reported fat loss with HFD in Lean mice before4 that could not be fully explained by divergent eating or running behaviour7,22, supporting alternative leanness mechanisms. In contrast, adipose TST was reduced in Fat line mice (Fig. 1d) that gained fat mass with the HFD (Supplementary Table 2). We then investigated whether TST was broadly related to adiposity in mice. Adipose Tst mRNA correlated negatively with fat mass and plasma glucose (Fig. 1e,f) but positively with adiponectin mRNA across 23 commonly used mouse strains (Fig. 1g). RNA-Seq data from the Diversity Outbred (DO) panel of mice23 revealed an expression QTL (eQTL) at the Tst locus, whereby Tst intervals derived from lean founder mouse strains had higher liver Tst mRNA levels (Fig. 1h). Higher adipose Tst mRNA was also found in representative lean DO founder strains compared to obesity-susceptible strains (Fig. 1i and Supplementary Fig. 3a). Adipose Tst mRNA (Supplementary Fig. 3b,c) and protein (Fig. 1j) was markedly lower in HFD-fed obesity-prone male C57BL/6J mice and in leptin-deficient, C57BL/6JLepob/ob mice.
Overexpression of Tst in adipocytes preserves metabolic health
We next tested the hypothesis that elevated adipose Tst was causal for leanness. It was first established that Tst was expressed predominantly in mature 3T3-L1 (Fig. 2a) and primary adipocytes (Fig 2b). We then generated mice overexpressing Tst ~2-fold in the mature adipocytes (Fig. 2c) using the adiponectin promoter24 (Ad-Tst mice). Tst mRNA was unchanged in brown adipose tissue (BAT) or bone of Ad-Tst mice (Supplementary Fig. 4a,b). Ad-Tst mice were of initially comparable weight to their non-transgenic littermates (Fig. 2d) but resisted HFD-induced obesity (Fig. 2d,e, Supplementary Fig. 4c,d) and had smaller fat cell size (Fig. 2f) despite similar food intake (Supplementary Fig. 4e). Ad-Tst mice exhibited higher energy expenditure on chow and HFD (Supplementary Fig. 4f,g) with a lower respiratory exchange ratio on HFD indicative of preferential fat oxidation (Fig. 2g and Supplementary Fig. 4g), particularly mid-way through the diurnal cycle (Fig. 2g) in comparison to the C57BL/6N controls. Physical activity was comparable between genotypes (Supplementary Fig. 4h). Overexpression of Tst did not increase BAT or WAT Ucp1 mRNA level (Supplementary Fig. 4i,j) but was associated with elevated basal hepatic Cpt1a mRNA levels (Fig. 2h) and increased fat oxidation in liver (Fig. 2i).
Figure 2.
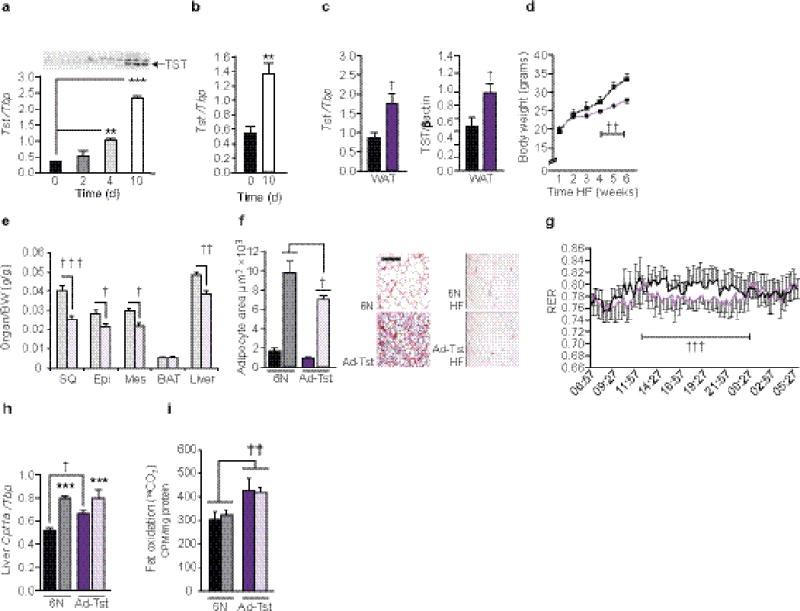
Adipocyte Tst overexpression drives obesity-resistance. (a) Western blot (top) of TST protein (arrow) and the Tst mRNA levels (bottom) in differentiating 3T3-L1 preadipocytes. n = 3, **, *** = P < 0.01, P < 0.001, by 1-way ANOVA. (b) Tst mRNA levels in mouse SVF. n = 3, ** = P <0.01 by t-test. (c) Quantification of Tst mRNA (left) and TST protein levels (right) in C57BL/6N littermates (black bars) or Ad-Tst mice (purple bars), n = 5. † = P <0.05 by t-test. (d) Bodyweight of C57BL/6N (black broken line) or Ad-Tst mice (purple broken line) fed high fat diet (HFD) for 6 weeks. n = 5, †† = P <0.01 by RM-ANOVA. (e) Organ-to-body weight ratio after 6 weeks HFD in C57BL/6N (6N, grey bars, n = 4) or Ad-Tst mice (pink bars, n = 5). †, ††, ††† = P <0.05, 0.01, 0.001, by 2-way ANOVA. (f) Fat cell size quantification (left) and representative images of adipose sections (right) in C57BL/6N mice fed control (black bar, n = 5) or HFD (grey bar, n = 6) for 6 weeks and Ad-Tst mice fed control (purple, n = 6) or HFD (pink bar, n = 6) for 6 weeks. Scale bar, 100 µm. † = P <0.01 effect of genotype by 2-way ANOVA. (g) Respiratory exchange ratio (RER) in C57BL/6N (black lines) versus Ad-Tst mice (purple lines) after 6 weeks HFD (n = 4). ††† = P <0.001, effect of genotype (mean RER,13:00 to 01:00) by t-test. (h,i) Hepatic Cpt1a mRNA levels (h) and 1-14C-palmitate oxidation (i) in groups as described in f. n = 6. *** = P < 0.001, effect of diet, † = P < 0.05 and †† = P < 0.01, effect of genotype by 2-way ANOVA. All data are mean ± SEM.
We next investigated if adipocyte Tst overexpression improved metabolic status. Adiponectin concentrations were higher in Ad-Tst mice (Fig. 3a), consistent with elevated hepatic fat oxidation and an anti-diabetic phenotype25. On control diet, Ad-Tst mice showed mildly improved glucose homeostasis (Supplementary Fig. 5a,b) and were markedly protected from impaired glucose homeostasis after 6 weeks HFD compared to C57BL/6N mice (Supplementary Fig. 5c,d). Supportive of maintained metabolic health even before body weight divergence (Fig. 2d), euglycemic clamps revealed higher whole-body insulin-sensitivity (Fig. 3b and Supplementary Table 3) and glucose uptake in muscle and adipose tissue of Ad-Tst mice (Fig. 3c) compared to C57BL/6N mice after 2 weeks HFD. Adipose tissue from Ad-Tst mice exhibited higher Glut4 (official gene symbol Slc2a4) mRNA (Supplementary Fig. 5e) and GLUT4 protein levels (Fig. 3d), supporting primary adipose insulin-sensitization26. Supportive of this, 6 weeks HFD suppressed adipose phospho-AKT levels in C57BL/6N littermates but not Ad-Tst mice (Fig. 3e) and endogenous insulin suppressed free fatty acid (NEFA) release to a greater extent in Ad-Tst mice in vivo (Supplementary Fig. 5f). Indices of lipolytic capacity such as the beta-3 adrenergic receptor (Adrb3) mRNA levels were suppressed by HFD in C57BL/6N littermates but not Ad-Tst mice (Fig. 3f). Indeed, Ad-Tst mice showed basally lower phospho660-hormone-sensitive lipase (HSL) protein levels that were maintained with HFD, whereas C57BL/6N littermates showed a distinct suppression of phospho660-HSL levels with HFD (Fig. 3g). Adipose tissue triglyceride lipase (ATGL) showed a selective reduction in Ad-Tst adipose tissue with HFD (Fig. 3h). PERILIPIN 1 (PLIN1) protein levels responded similarly to HFD in C57BL/6N and Ad-Tst mice (Supplementary Fig. 5g). To directly test if lipolytic capacity was altered in vivo, we administered the β3 agonist CL316,243 (CL) and found that Ad-Tst mice exhibited enhanced NEFA release (Fig. 3i). Further, adipocytes from Ad-Tst mice (basal Ad-Tst: 16 ± 4, versus C57BL/6N: 27 ± 3 pmol/min/103 adipocytes, n = 6) showed enhanced responsiveness to CL (Ad-Tst: +171 ± 20, versus C57BL/6N: +141 ± 13 pmol/min/103 adipocytes, n = 6, P < 0.01) and pronounced suppression of CL-induced lipolysis in response to 0.5 nM insulin (CL + insulin Ad-Tst: −126 ± 16, versus C57BL/6N: −82 ± 8 pmol/min/103 adipocytes, n = 6, P < 0.01, data are mean ± SEM, compared by 2-way ANOVA) in vitro.
Figure 3.

Adipocyte Tst overexpression drives insulin sensitisation and maintained lipolytic capacity. (a) Adiponectin in C57BL/6N fed control (black bar) or HFD for 6 weeks (grey bar) Ad-Tst mice fed control (purple bar) or HFD (pink bar). n = 6, *** = P < 0.01, †† = P < 0.01 by 2 way ANOVA (b) Blood glucose (left) and glucose infusion rate (right) during euglycemic clamps (n = 6) in C57BL/6N (black broken lines) and Ad-Tst mice (purple broken lines) after 2 weeks of HF diet. †† = P < 0.01 (c) Metabolic index (Rg, n = 6) in quadriceps muscle (Quad) and epididymal fat (Epi) of C57BL/6N (grey bar) or Ad-Tst mice (pink bar). †† = P < 0.01. (d) GLUT4 protein, (n = 5, 6, 5, 5), representative western blot image of (e) phospho-AKT/total AKT protein (n = 3, 3, 4, 3), (f) β3-adrenergic receptor (Adrb3) mRNA, (n = 5, 5, 5, 6) and representative western blot image of (g) phospho-660-HSL/total HSL protein (n = 3, 3, 4, 4) and (h) ATGL protein (all n = 3), groups as in (a). (i) Plasma NEFA after CL316,243 administration (1 µg/g BW) to C57BL/6N (black line, n = 5) or Ad-Tst mice (purple line, n = 8). Data are mean ± SEM. * = P < 0.05, *** = P<0.001, † = P < 0.05, †† = P < 0.01, by 2 way ANOVA (c – h) or RM ANOVA (i).
Tst deficiency and TST activation have reciprocal metabolic effects
We then determined if loss of Tst gene function, shown by complete loss of TST enzyme activity (Supplementary Fig. 5h), was causal for obesity or diabetes. Tst−/− mice had comparable gross body weight, energy expenditure and fat mass to C57BL/6N littermates after 6 weeks HFD (Fig 4a,b and Supplementary Fig. 5i). Nevertheless, histologically, HFD fed Tst−/− mice exhibited mild adipocyte hypertrophy in vivo (Fig. 4c). To explore this further, we differentiated adipose stromal vascular cells (SVF) in vitro and found that SVF from Tst−/− mice exhibited greater late-stage lipid accumulation than C57BL/6N SVF (day 9; Fig. 4d), coincident with Tst expression in late adipogenesis (Fig. 2a,b). Transcriptional regulators of early preadipocyte differentiation were comparable between genotypes in the SVF (Supplementary Fig. 5j). Despite comparable adiposity, glucose intolerance was observed in Tst−/− mice and this was exacerbated with HFD compared to C57BL/6N mice (Fig. 4e). The GTT also revealed impaired NEFA suppression despite comparable endogenous insulin output in Tst−/− compared to C57BL/6N mice on HFD (Fig. 4e), an indicator of adipose insulin resistance that was confirmed in primary adipocytes from Tst−/− mice in vitro (Fig. 4f).
Figure 4.

The effects of Tst gene knockout and TST activation on diabetes in mice. (a) Body weight gain in C57BL/6N (black broken lines, n = 6) and Tst−/− mice (blue broken lines, n = 5) with 6 weeks HFD. (b,c) Day and night RER (n = 6, 5), (b) and (c) adipocyte area (n = 4) in C57BL/6N mice (6N, grey bars) or Tst−/− mice (KO, light blue bars, n = 5) after 6 weeks HFD. † = P < 0.05 by t-test. (d) Representative images for lipid staining (top) of SVF from C57BL/6N (6N, black bar, n = 6) or Tst−/− mice (KO, blue bar, n = 5) at day 6 and day 9 of differentiation. Scale bar, 25 mm. Insets, day 9 at higher magnification. Scale bar, 500 µm. Quantification (bottom) of day 9 lipid. † = P < 0.05 by t-test. (e) Glucose tolerance (left), insulin excursion (middle) and NEFA excursion (right) in C57BL/6N mice (black lines, n = 4) or Tst−/− mice (blue lines, n = 5) fed control diet (solid lines) or HFD (broken lines, n = 5 per genotype) for 6 weeks. † = P < 0.05, †† = P < 0.01 by RM ANOVA (HFD). (f) 3HGlucose incorporation into lipid in C57BL/6N (black bars) or Tst−/− mice (blue bars) adipocytes. † = P < 0.05 by 2-way ANOVA. n = 11. (g) Glucose tolerance in HFD-fed C57BL/6N (black lines) or Tst−/− mice (blue lines) after 6 weeks of water (solid lines) or thiosulfate administration (broken lines). n = 6. †† = P < 0.01, * = P < 0.05 by 2-way ANOVA. (h) Quantification of water intake (left) and an illustrative image of the urine output in the cage (right). Scale bar, 5 cm. (i,j) insulin tolerance (i) and glycosylated hemoglobin (j) in C57BL/KsJ-Leprdb/db mice treated with water (white bars, in-fills, n = 5) or thiosulfate (STS, orange bars, in-fills, n = 6). C: reference C57BL/6J HbA1c values (n = 2). Data are mean ± SEM. * = P < 0.05, ** = P < 0.01, *** = P < 0.001 by RM-ANOVA (g, i) or t-test (h). †† = P < 0.01 (g, j).
Therapeutic potential was assessed with the TST substrate thiosulfate10, 27 which ameliorated HFD-induced glucose intolerance in C57BL/6N but not Tst−/− mice (Fig. 4g) and modestly reduced weight gain without altered fecal fat excretion (Supplementary Fig. 6a,d). We then showed that thiosulfate markedly ameliorated the existing polydipsia, polyuria (Fig. 4h), insulin-resistance (Fig. 4i; glucose decrement at 60 minute as % of 30 minute value after 2 mU/g BW humulinS: water-treated: −21 ± 4%, thiosulfate-treated: −35 ± 4%, P < 0.05 by t-test) and elevated glycosylated haemoglobin (%HbA1c) of diabetic C57BL/KsJ-Lepdb/db mice (Fig. 4j) but did not alter body weight or fat mass (Supplementary Fig. 6e,f).
TST impacts multiple mitochondrial pathways
We next sought evidence for mitochondrial processes that could support a beneficial metabolic effect of elevated TST (Fig. 1c). Adipose tissue from Ad-Tst mice exhibited higher protein levels for the Fe-S-containing succinate dehydrogenase (SDH) B subunit (Supplementary Fig. 7a). Higher SDHB protein was maintained with HFD in adipose tissue of Ad-Tst but not C57BL/6N mice (Fig. 5a). Adipose Complex-V F1 alpha subunit, Fe-S containing mitochondrial aconitase (ACO2) and cytosolic ACO1 (Supplementary Fig. 7a–c) levels were comparable between Ad-Tst and C57BL/6N littermates. Thiosulfate was not a mitochondrial substrate per se (Supplementary Fig. 7d) and tended to suppress pyruvate/malate (complex I, NADH dehydrogenase) metabolism, but augmented succinate metabolism (Fig. 5b). In contrast, the TST inhibitor 2-propenyl thiosulfate (2-PTS)28 abolished succinate metabolism (Fig. 5b).
Figure 5.

TST beneficially modulates mitochondrial functions. (a) succinate dehydrogenase protein (SDHB) levels in C57BL/6N mice on control (black bar, n = 5) or with 6 weeks of HFD (HF, n = 6) diet (grey bar) and in Ad-Tst mice on control (purple bar, n = 5) or with 6 weeks HFD diet (pink bar, n = 6). † = P <0.05 effect of genotype by 2 way ANOVA. (b) Respiration of mitochondria (m) plus succinate (s), ADP (A), thiosulfate (STS), oligomycin (o) and FCCP (F) (left). Broken line; oxygen concentration; black line, Rate of change of oxygen (Slope). Average Slope of respiration (right) of succinate (black bars) or pyruvate/malate (grey bars) ± ADP, STS or 2-PTS. n = 3. ** = P < 0.01, † = P < 0.05 by 1-way ANOVA. (c) Superoxide dismutase (SOD2) protein level (n = 5, 5, 6, 6) and (d) peroxiredoxin-3 (Prdx3) mRNA level (n = 5, 5, 3, 5) in groups as described in a. *** = P < 0.001, †, †† = P < 0.05, < 0.01 by 2-way ANOVA. (e) Mitochondrial ROS in control (C, black bar, n = 22), shRNAmir-treated (grey bar, n = 14) or shRNATstmir-treated (white bar, n = 8) 3T3-L1 adipocytes. ** = P < 0.01, by 1 way ANOVA. (f) Adiponectin secretion from 3T3-L1 adipocytes treated with thiosulfate (0 (b) –10 mM). n = 4. * = P < 0.05, *** = P < 0.001 by 1-way ANOVA. (g) Sulfide effects on mitochondrial respiration from C57BL/6N (black lines) or Tst−/− mice (blue lines), axes as in b with (p), rotenone, pyruvate and malate. (h) Blood sulfide levels in C57BL/6N (black lines) or Tst−/− mice (blue lines). (i) Sulfide (0–500 µM) effects on NEFA release from 3T3-L1 adipocytes exposed to CL316,243 (0.1–10 nM). * = P < 0.05, ** = P < 0.01, effect of sulfide. CL effects omitted for clarity. (j) NEFA release from C57BL/6N (black bars) or Tst−/− mice (blue bars) adipocytes. n = 3. †† = P < 0.01 by t-test. Quantitative data are mean ± SEM.
Indicative of TST interactions with ROS, mitochondrial superoxide dismutase (SOD)-2 levels were higher in Ad-Tst adipose tissue (Fig. 5c) with no difference in the cytosolic SOD1 (Supplementary Fig. 7e) and adipose Prdx3 mRNA levels were higher in HFD-fed Ad-Tst mice compared to C57BL/6N littermates (Fig. 5d). Supporting a functional impact of TST on ROS, lentiviral shRNA-mediated Tst knockdown (Supplementary Fig. 7f) engendered higher mitochondrial ROS when cells were exposed to oxidant stress (Fig. 5e). Moreover, inhibition of TST with 2-PTS (Supplementary Fig. 7f) reduced (basal: 40 ± 1 ng/ml versus 10 µM 2-PTS: 25 ± 1 ng/ml, n = 3, P < 0.01), whereas thiosulfate increased, ROS-sensitive17, 19, 29 adiponectin release from 3T3-L1 adipocytes (Fig. 5f). Finally, a functional role for TST in H2S metabolism was established as isolated mitochondria from Tst−/− mice showed greater sensitivity to sulfide-mediated suppression of succinate respiration (Fig. 5g) and blood sulfide levels were markedly higher in Tst−/− compared to C57BL/6N mice in vivo (Fig. 5h). Dose-dependent sulfide suppression of 3T3-L1 adipocyte lipolysis (Fig. 5i) and reduced basal lipolysis in adipocytes from Tst−/− mice in vitro (Fig. 5j) supported TST modulation of H2S effects on adipocyte function.
Parallels for TST biology in humans
Supporting conservation of function in humans, we found that adipose tissue TST mRNA levels were higher in lean than obese (Fig. 6a) individuals from an Austrian population and correlated negatively with body mass index (BMI; r = −0.249, P = 1.7×10−10) in a large sample (n = 673) of human subcutaneous adipose tissue from Iceland30. Moreover, adipose TST mRNA was lower in obese subjects with type 2 diabetes and correlated negatively with BMI (Fig. 6b–d and Supplementary Table 4) in a Spanish cohort31. Adipose TST mRNA correlated positively with GLUT4, IRS1 and PPARG mRNA (Fig. 6e–g and Supplementary Table 4) independently of BMI after adjusting with multivariate linear regression (GLUT4, P < 0.001, IRS1, P < 0.006, PPARG, P < 0.001). TST mRNA also correlated with markers of lipolytic capacity and lipid droplet formation (Supplementary Fig. 8).
Figure 6.

Human adipose TST mRNA levels correlate with insulin sensitivity. (a) TST mRNA levels in human subcutaneous (SC) and visceral (Visc) adipose tissue from lean (black bars) and obese (white bars) women from a Viennese cohort. n = 8, *P < 0.05 by 2-way ANOVA. TST mRNA levels in subcutaneous (b) and visceral adipose tissue (c) of a Spanish case-control cohort31 from non-obese or obese type 2 diabetic (T2D) subjects. Correlations of adipose TST mRNA with body mass index (BMI) (d) and mRNA levels for markers of adipose insulin sensitivity, including GLUT4 (e), IRS1(f) and PPARG (g). (h) TST mRNA levels in differentiating primary human subcutaneous (black line) and omental (grey line) preadipocytes and in clonal human SGBS adipocytes (i), **, ***=P< 0.01 and 0.001, respectively, significantly different from day 0 by 1 way ANOVA. (j) The effects of 0.1–10mM STS (6 hours) on adiponectin secretion from differentiated human SGBS adipocytes, ***=P<0.001, significantly different from control (C). a–c, h–j data are mean ± SEM. Significance of correlational data for TST and other adipose genes was assessed with Scheffé post hoc tests.
TST mRNA level was higher in differentiated human primary fat cells (Fig. 6h) and in differentiated human SGBS32 clonal adipocytes than in preadipocytes (Fig. 6i). As in rodent adipocytes (Fig. 5f), thiosulfate treatment increased adiponectin release from human SGBS adipocytes (Fig. 6j).
DISCUSSION
Genetic mechanisms that maintain metabolic health and counter excess adiposity have received less attention than those associated with obesity. One example is polymorphism at the melanocortin 4 receptor (MC4R) locus that associated with protection against obesity33. MC4R variants reflected a generality from early obesity genome wide association studies (GWAS) emphasizing the predominance of brain-regulated appetite and energy expenditure pathways33–35. Later GWAS using accurate measures of fat mass such as dual energy x-ray absorptiometry (DEXA) revealed a new class of genes with direct functions in the adipose tissue35–36. Variation at the Insulin Receptor Substrate-1 IRS1 locus35–37 revealed a hub adipose mechanism linking reduced peripheral fat deposition with impaired metabolic health35. From this we hypothesized the existence of adipose-specific mechanisms driving obesity-resistance. However, dedicated GWAS in extreme lean, but healthy, humans distinct from lipodystrophy38 or failure-to-thrive39 syndromes were not reported. To gain insight into this elusive healthy lean genetic category and focus on adipose tissue mechanisms, we exploited the polygenic Lean line of mice4–8, 22. Polygenic Lean and Fat lines resemble the genetic architecture of common human adiposity and do not derive from single adiposity genes with very large effect sizes4–8 such as leptin40, improving our chances of finding novel adiposity genes. Moreover, because Lean mice were selected for extreme low adiposity but maintained lean mass, they are distinct from models selected for obesity relative to a control line, or those selected on overall body weight with adiposity as a correlated trait41. This may explain why the M2 sub-sub-congenic effect size is relatively large for a QTL study of a polygenic trait, supporting a prominent role for Tst as a causal Lean line leanness gene.
Compensatory alleles in the obesity-susceptible C57BL/6N genetic background chosen to model transgenic adipose Tst overexpression may explain the absence of gross baseline leanness in Ad-Tst mice. However, the effect size of obesity-resistance in HFD-fed Ad-Tst transgenic mice supports elevated Tst as an anti-obesity and anti-diabetic mechanism. The Tst effect size is also large compared to human adiposity GWAS hits; the FTO/IRX334 locus has −0.33% body fat per effect allele35. Greater genetic and environmental heterogeneity in human populations is a likely explanation for this. Nevertheless, the Tst allele effect has allowed us to identify a novel driver for reduced adiposity and improved metabolic health in Lean mice that has functional parallels across other mouse strains and humans. Conserved regulation across states of adiposity and metabolic health indicate dynamic expression control by varied mechanisms. Further, although common variant human adiposity GWAS have not identified hits at the TST locus, we may infer biological significance from our observed parallels in human TST expression and function. The Tst effect is a metabolically protective reduction of fat mass across all depots in mice. TST mRNA levels are comparable in subcutaneous and visceral fat in humans, suggesting protection in both depots.
Obesity-resistance to a HFD does not involve WAT beigeing42–43 in Lean8 or Ad-Tst mice. Indeed, Fat mice exhibited WAT beigeing with HFD, which failed to prevent exaggerated obesity8. Consistent with novel WAT-derived leanness mechanisms in Lean mice, beigeing genes (e.g. Ppargc1, Ucp1) do not map to the major adiposity QTLs5–6. Such alternative mechanisms have a precedent, with some parallels and distinctions, compared to Tst elevation. Adipocyte-specific knockout of the mitochondrial transcription factor Tfam (F-TFKO) caused loss of complex-I proteins, a compensatory increase of nuclear-encoded SDH activity and increased adipose and whole-body energy expenditure without canonical beigeing17. Notably, F-TFKO showed a confounding increase of mitochondrial ROS with lower adiponectin release17, whereas elevated TST may quench ROS and increase adiponectin levels. In contrast, adipocyte-specific mitoNEET-overexpression caused pronounced obesity but with elevated adiponectin-associated lipogenic WAT expansion and reduced mitochondrial ROS19. TST-mediated leanness likely derives from altered mitochondrial substrate preference combined with effective ROS and H2S quenching, which maintains adipose tissue insulin-sensitization, lipolytic responsiveness and adiponectin release that ultimately drives peripheral oxidative disposal of excess fat.
Fortuitously, thiosulfate, a TST activator, existed for cyanide detoxification10, and oral thiosulfate administration had ameliorated heart failure in mice27. Thiosulfate ameliorated diabetes, but did not consistently reduce fat mass, suggesting TST activation is predominantly anti-diabetic. This is supported by our findings that transgenic manipulation of Tst predominantly affected glucose homeostasis over fat mass per se and that human TST is associated with metabolic status after adjustment for BMI. Elevation of TST protein levels may have broader beneficial effects that are not recapitulated by increasing availability of a single substrate. It is also likely that thiosulfate efficacy was limited by low oral bioavailability and confounding lower target adipose TST found in obese mice. Notably, millimolar thiosulfate concentrations were needed to elicit beneficial effects in vitro and in vivo. Similar compound ranges were found for other anti-diabetic drugs including salicylate44 and metformin45. Development of more efficacious and specific TST activators would be desirable to realize the therapeutic potential of this novel anti-diabetic target.
ONLINE METHODS
Experimental Animals
All experiments were performed according to guidelines set out by the ethical committees of The University of Edinburgh, The University of Cambridge or the University of Ljubljana and were carried out within the framework of the Animals (Scientific Procedures) Act (1986) of the United Kingdom Home Office or related laws from the European Union (Slovenia).
In all studies animals within genotype cohorts were randomly assigned to diet or intervention groups. All animals were maintained in standard housing with 12 hour light dark cycles (7a.m.–7p.m.) and ad libitum access to the appropriate diet. In vivo experiments (e.g. GTT, ITT, euglycemic clamps, CL-injection) were operator/animal handler blinded with data generated by a second individual blinded to treatment until code breaking. All studies, with the exception of F2 crosses of M and M2 lines with parental F lines and the RNA-Seq analyses of adipose tissue of founder strains (see below) used exclusively male mice to avoid confounding effects of the female hormonal cycle. Male C57BL/6J, C57BL/6N, C57BL/6Jlep/lep (Lepob, 10 weeks old) and C57BL/Ksjlepr/lepr (~ 8 weeks old) were purchased from certified distributors Charles River laboratories or Harlan (UK). C57BL/Ksjlepr/lepr were housed in groups of 3 and singly housed 3 days before ITT to determine water intake by weighing the water bottle daily and recording the 3rd day as representative of stable intake. Tst−/− mice were generated by the UCSD knockout mouse project https://www.komp.org/geneinfo.php?geneid=85272, project VG13928, model Tsttm1(KOMP)Vlcg representing a definitive null allele. Sperm from viable heterozygote mice were used to fertilize C57BL/6N embryos at the University of Edinburgh GIST facility. Tst−/− mice and age-matched littermate controls were placed on experimental diets at approximately 5–6 weeks of age. Ad-Tst mice were generated by cloning the mouse Tst cDNA downstream of the adiponectin promoter24 and micro-injecting into C57BL/6J blastocysts using standard cloning techniques (University of Turku, Finland). In general, Ad-Tst and age-matched control littermate mice were placed on experimental diets at approximately 5–6 weeks of age.
High fat diets (D12331, Research Diets, New Brunswick, US) and control low fat high cornstarch diets (D12328) or standard control chow were used throughout the studies.
The following studies had specific subject exclusion. In the thiosulfate treatment study, one C57BL/Ksjlepr/lepr mouse assigned to the control (water) group rapidly and inexplicably lost weight and was humanely culled. All data was removed from the study. In the euglycemic clamp studies, one animal from each genotype (6N and Ad-Tst) failed to recover pre-operation body weight and their data was excluded from the study. In one of the high fat diet cohorts (Fig. 2e) comparing 6N to Ad-Tst (for organ weights), a 6N control mouse failed to gain any weight and was culled due to ill-health during the study. In the gene expression and western blot analyses reported in Fig. 2e–h and Fig. 5d, prior sample analysis limited available group sizes to below the optimal n = 6 for some targets.
F and L polygenic selection lines
The long term selection and further development of the F, L and congenic lines are described elsewhere5–6. Animals were fed pelleted diet (Special Diets Services, Witham, Essex, UK) or defined low (11% calories as fat with sucrose; D12329) or high fat (58% calories as fat with sucrose; D12331) diets (Research Diets, New Brunswick, New Jersey).
Experiments using the congenic mouse lines
Polygenic mouse models: From a previous F2 cross6 a recombinant individual within Fob3b QTL region was used as a founder for development of a subcongenic line M used here. A total of 11–12 backcrosses to the Fat line with additional marker-assisted elimination of other QTL regions were performed. F2 cross between the Fat line and the congenic line M was developed and phenotyped (M-F2 cross). Within this M-F2 cross (Supplementary Table 1) another recombinant individual within the Fob3b2 QTL region was identified carrying a smaller donor segment from the Lean line (Supplemental Fig. 1) – this recombinant was a founder for development of a subsubcongenic line M2 and a subsequent F2 cross with the Fat line (M2-F2 cross). Positions of Lean donor segments in congenic lines were defined based on high density SNP genotyping (Supplemental Fig. 1). For all experiments, food chow (1324 maintenance diet, Altromin, Germany) and acidified water were offered ad libitum except for M2-F2 cross where a high fat diet (HFD; Research Diets, USA, D12108) was used. M2-F2-3-week-old mice were adjusted to the HFD for two weeks and were maintained on it for additional 11 weeks. All the procedures involving animals were performed according to local ethical and regulatory guidelines, which are all in compliance with the EU regulations regarding research on experimental animals (project license number 34401-3/2007/4). All mice were weaned at 3 weeks of age and were group-housed (four mice per cage) in individual ventilated cages (Techniplast Inc., Italy) in a controlled environment (temperature 21°C, humidity 40–70%, and a 12h light/12h dark cycle). Body weights were recorded at 3, 5, 6, 8, 10, 12, 14, and finally at 16 weeks, when adipose depots were dissected: abdominal (ABD), gonadal (epididymal, EPI), subcutaneous-femoral (SC), mesenteric (MES). The sum of all collected fat depot weights was used to calculate the adiposity index (ADI).
Empirical distributions of collected phenotypic data suggested a normal distribution for each analysed variable. Therefore, a statistical model (Eq [1]) with multivariate normal distribution was fitted: [1] y|b,s,R ~ MVN(Xb + Zs, R) where y is an ny×5 matrix of phenotypic values; b is the vector of location parameters for effects that differed between the data sets; s is the vector of location parameters for ns seasons of dissection defined as year-month interaction, and R=Iny×R0 is residual covariance matrix. For F2 congenic intercrosses (M-F2 and M2-F2) the vector b involved the effect of sex (males and females), parity (1, 2, and 3+), the number of pups per litter (1–2, 3, 4, 5, 6, and 7+), line (M and M2), and genotype within line (homozygotes Fat/Fat, Fat/M, M/M and heterozygotes Fat/M or Fat/M2). For the latter the additive and dominance effect was tested using the DIC statistic, which revealed that the dominance model had a better fit that the additive model. Posterior distributions were summarized with mean and standard deviation for each line or genotype as evaluated at the first parity and five pups in the litter. Posterior probabilities that the congenic lines or homozygous genotypes from F2 congenic intercrosses differ were computed. High probability, e.g. 0.95, would suggest a significant difference and presence of a Fob3b2 QTL effect.
Bioinformatics analysis of candidate genes in Fob3b2 interval
(Supplemental Fig. 2): Haplotype analysis was performed to identify haplotype blocks that are not identical by descent (non-IBD). These regions are likely to carry the causal polymorphism(s) that are responsible for the observed phenotypic difference of the congenic F2 progeny. Comparative genomic analyses within the Fob3b2 was performed in human (http://www.genome.gov/gwastudies/), cattle, pig and chicken (http://www.genome.gov/gwastudies/). If an QTL associated with adiposity traits was found to overlap with a Fob3b2 Tst-containing region this was considered as a hit. A set of 1980 SNPs (JAX SNP IDs JAX00405958-JAX00407532) located within the Fob3b2 congenic region of the M line was used to genotype F, L and congenic M or M2 lines. Interval-specific haplotype analysis was carried out to identify haplotype blocks that are not identical by descent (non-IBD) between congenic lines. Such regions are likely to contain the causal polymorphism(s) that cause differences in phenotype between the observed lines6. 20 genes mapped to non-IBD regions (See section Custom RT-PCR array below). A tool within the Mouse Phenome database (SNP Wizard) was used for SNP-strain comparisons. Using the Bio GPS Expression database (http://biogps.gnf.org), genes were selected based on their expression levels in metabolic tissues in two independent microarray platforms (Affymetrix probe sets MOE430 and GNF1M). If a positional candidate gene showed expression level 3 times above the median expression in at least one of the metabolic tissues (pituitary, brain, white and brown adipose, liver, muscle, intestines stomach, pancreas, adrenal) this was considered as a hit. Molecular function annotations for the non-IBD candidates were retrieved from the Gene ontology (GO, http://amigo.geneontology.org) database - any function that could be connected to adipose tissue biology and/or growth was considered a hit. Candidate genes were also searched for any impact on body weight, obesity, food intake or metabolism phenotypes of the knockout and transgenic models using the Mouse Genome Informatics (MGI) (http://www.informatics.jax.org). Differential expression of all positional non-IBD candidates was verified using two microarrays, whole genome expression Affymetrix Genechip 2.0 array and Affymetrix Exon chip array and various tissues of F and L lines, Ref. 8, and this study for M2-F2 microarray) using expression differences of ≥2-fold. Genes with the largest number of bioinformatics hits (highest priority candidates) were also separately assayed by qRT-PCR in WAT of F, L and M2 lines. The expression of all positional candidate genes within the core non-IBD region was measured in WAT using custom RT-PCR array (Life Technologies) – details are in the section Custom RT-PCR array analysis below.
Custom RT-PCR array analysis of positional candidates in the Fob3b2 segment
The expression of all positional candidate genes within the core non-IBD region was measured in WAT using custom RT-PCR array (Life Technologies) comparing homozygotes from the F2 cross between the M2 and Fat line. All TaqMan® assays were custom ordered from Applied Biosystems (Assay IDs below). Real-time PCR was performed using a standard TaqMan® PCR kit protocol on an Applied Biosystems ViiA™ 7 Real-time PCR System. The 10 µl PCR included 2 µl cDNA sample, 2× TaqMan® Universal Master Mix II, 20× TaqMan® Gene Expression Assay and RNase-free water. The reactions were incubated in a 384-well plate at 50°C for 2 min, followed by polymerase activation at 95 °C for 10 min and 40 cycles of 95 °C for 15 s and 60 °C for 1 minute. All reactions, including a no-template control, were run in duplicate. For each sample, average Cq was calculated and by normalizing on three endogenous controls (B-actin, Tbp and Gapdh), ΔCq was determined. The 2−ΔΔCq method was used to quantify the relative change in expression of the target group, homozygous for genotype LL in the Fob3b2 segment. Student’s t-test analysis was performed and differential expression of a gene between the F2 homozygote carrying two Fat-line derived alleles and homozygotes carrying two Lean-line alleles was considered as a hit. Probes used in the study were (Gene, Assay ID): B-Actin, Mm00607939_s1, Gapdh, Mm99999915_g1, Tbp, Mm00446971_m1, Arhgap39, Mm01197504_m1, Zfp251, Mm02342310_m1, Zfp7, Mm00524080_m1, Mb, Mm00442968_m1, Apol6, Mm03990658_m1, Rbm9, Mm00612735_m1, Apol7a, Mm01200950_m1, Apol9a, Mm04206749_gH, Apol7b, Mm01616698_m1, Apol10a, Mm04214065_g1, Apol7c, Mm01628124_s1, Apol10b, Mm04212537_m1, Apol7e, Mm01616699_m1, Myh9, Mm01197036_m1, Pvalb, Mm00443100_m1, Ncf4,, Mm00476300_m1, Csf2rb2, Mm00655763_m1, Csf2rb, Mm00655745_m1, Tst, Mm01195231_m1, Mpst, Mm00460389_m1.
Quantification of Tst-allelic mRNA expression
To examine if the Tst allele from the Lean (L) line is expressed at a higher level than that from the Fat (F) line in heterozygotes from the M2-F2 congenic cross, allele-specific TaqMan probes were designed at the SNP in the 3’ UTR (rs31534689). Sequences of the PCR primers were 5’CCTGCTGTAGGTTCACCTTTTAGG (forward), GGAGGCACCAAGAGCAATTCTAAA (reverse) and the TaqMan probes at the SNP site (underlined) were CCCTGTCAATCTCCGT (Lean-allele-specific) ACCCTGTCAATATCCGT (Fat-allele-specific). To determine the calibration curve, genomic DNA isolated from two individuals, homozygous for genotype FF and LL in the Fob3b2 segment, was mixed in various molar ratios of F allele/L allele (VIC allele/FAM allele and dye swapped): 8:1, 4:1, 2:1, 1:1, 1:2, 1:4 and 1:8. By calculating the log of (ΔRnFAM/ΔRnVIC) for each mixing ratio, a standard curve, with linear regression line equation: log2 (ΔRnFAM/ΔRnVIC) = a + b • log2 (VICallele/FAMallele) was generated. Allele-specific expression of the gene was measured on isolated RNA samples from eight heterozygotes in the Fob3b2 segment. Through intercepting measured fluorescence intensities on the standard curve, allele ratio of heterozygous individuals was extrapolated and relative expression of Lean: Fat line allele determined (Supplementary Fig. 1f–g).
Correlation analyses between Tst expression and other genes or phenotypes
Datasets of gene expression in adipose tissue from different mouse strains were downloaded from BioGPS (http://biogps.org/dataset/8/eqtl-adipose-moe430-v2/) and phenotype data for the same set of strains were obtained from the Mouse Phenome Database (http://phenome.jax.org/; CGDpheno1, 2009). A Pearson’s correlation analysis was performed in SAS/STAT® software. Strong and highly statistically significant correlations (P value < 0.0001) between Tst and genes expressed in adipose tissue and Tst and phenotypes are presented (Fig. 1e–g).
RNA-seq analysis and eQTL mapping
Methods for RNA-seq library preparation, analysis, and eQTL mapping in Diversity Outbred (DO) mice are described in detail in Munger et al. (2014). Briefly, liver or inguinal fat pad adipose tissue were dissected from 26-week old DO or founder strain mice and stored in RNAlater® solution (Life Technologies) at −80°C. Total RNA was extracted using the Trizol Plus RNA extraction kit with on-column DNase digestion (Life Technologies), indexed mRNA-seq libraries were generated from 1µg total RNA using the TruSeq kit (Illumina), and 100bp single-end reads were sequenced on the HiSeq 2000 sequencer (Illumina) according to Illumina protocols. Samples were multiplexed and replicated across 2–4 sequencing lanes to minimize technical variation. Raw RNA-seq fastq files and processed gene-level abundance estimates for the DO liver and founder strain adipose samples are archived at GEO under the accession numbers GSE45684 and GSE80162, respectively. Individualized genome and transcriptome sequences were constructed for every DO and strain sample using Seqnature software46. RNA-seq reads were aligned to their respective individualized transcriptome using the Bowtie aligner, and gene expression was quantified from the read alignment profiles using RSEM software as described in46. Expression quantitative trait loci (eQTL) in the DO liver samples were identified with an additive liner regression model46.
Fat cell size estimation
Fat beds were collected from the mouse shortly after cull. The beds were transferred to a 4% PFA based fixing solution, and sent to histology for paraffin embedding. The paraffin blocks were cut to a central core (estimated as closest to midway through the fat pad) then cut to 4µm sections and mounted onto microscope slides for haemotoxylin/eosin staining. Images were captured using an Eclipse E800 camera (Nikon): QICAM Fast1394 (QImaging) supported by an automated Stage (ProScan II, PRIOR Scientific) for random selection. Images were generated using Image Pro Plus 7.0 software. Twenty high magnification images (10× to 40× depending on cell size) of the tissue were randomly selected using the ProScanII random selection facility. From 6 individual imaged sections cut across a fat pad from a given mouse, 6 of the largest visible whole cells in each field of known area were selected and used to estimate the average cross sectional area of the cells from that fat bed for each mouse as µm2/cell (average of 36 random cell bodies).
Glucose and insulin tolerance tests, in vivo NEFA release
Glucose tolerance was determined by oral or intraperitoneal administration of D-glucose (2 mg/g bodyweight) after a 6 hour fast. Insulin tolerance was determined by intraperitoneal injection of Humulin S (0.5 up to 2 mU/g [for insulin-resistant Lepr mutants] bodyweight) after a 4 hour fast. Lipolytic responsiveness was determined by injection of 1 µg/g bodyweight CL316,243 after a 5hour fast. In all cases blood was sampled before administration of the test substance (0 time) and then generally at 15, 30, 60 and up to 120 minutes later (GTT). Glucose was measured from a tail venesection using a hand held glucometer (OneTouch, Lifescan, Milpitas, USA) and insulin (Crystalchem ELISA, Chicago, USA) or NEFA (Wako, Richmond VA, USA) was measured in plasma prepared from blood collected in EDTA-coated microtubes (Sarsted, Leicester, UK).
Euglycemic hyperinsulinemic clamps
Male C57BL/6N or Ad-Tst littermates were fed high fat diet for 2 weeks. Mice were anesthetized with isofluorane (5% induction, 2% maintenance in 2% oxygen) and a mirocrenathane cannula implanted in the right exterior jugular vein and exteriorised at the back of the neck. Mice were injected with 0.05 mg/kg buprecare subcutaneously (buprenorphine) at the start of surgery and were given Rimadyl (0.001% v/v) in their drinking water overnight on the first day after surgery. Clamps were performed 3 days after jugular cannulation. Mice were fasted overnight and body weight, lean and fat mass was determined by TD-NMR (Bruker LF50, Bruker.com). All mice were infused for 2 hours with 0.05 µCi/min D3-3H-glucose (NET331C005MC, Perkin Elmer, perkinelmer.co.uk) to assess endogenous glucose production by sampling shortly before the clamp procedure (−5 minutes). The clamp was started with infusion of higher specific activity tracer (0.1 µCi/min) and a prime of insulin (HumulinS, Eli Lily) at 9.0 mU/kg lean mass/minute for 3 minutes followed by a constant infusion of insulin at 2.5 mU/kg lean mass/minute and a variable infusion of 20% glucose. Blood glucose was sampled from 20 minutes until 60 minutes to achieve a steady state blood glucose level at ~7.6 mmol/L. A bolus of 10 µCi 2-14C-deoxyglucose (NEC495001MC, Perkin Elmer) was administered at 75 minutes and samples were taken from the cut tail for tracer and glucose analysis at 80, 85, 90, 100, 110 and 120 minutes. Insulin levels were measured from basal (−5 minutes) and clamp (100 minutes) time points. Mice were euthanized with euthatal administered down the cannula and tissue (quadriceps muscle and epididymal fat) rapidly excised. Tissues were weighed, extracted in 10 volumes of water and the phosphorylated 2-14C-deoxyglucose was recovered using Poly-Prep® Columns, AG 1-X8 anion exchange columns (Bio-Rad, biorad.com). Tracers were used to calculate various aspects of glucose metabolism47. Tissue glucose clearance was (Kg) calculated by dividing the column-recovered tissue phosphorylated 2-14C-deoxyglucose by the area under the curve of plasma 2-14C-deoxyglucose (trapezoidal method). Kg was used to calculate a metabolic index (Rg), a measure of tissue-specific glucose uptake, and is the product of Kg and the mean glucose concentration over the clamp period47. Plasma tracers were measured after deproteinization with Ba(OH)2 and ZnSO4. Plasma glucose/tracers were measured after drying the sample and used to calculate specific activity and rates of disposal (Rd)/endogenous production (EndoRa)47. The wet sample containing 3H2O was used to estimate glycolytic rates. Tracers were counted using Prosafe FC+ High Efficiency LSC cocktail (Meridian Biotechnologies Ltd) in a TriCarb 2100TR β-scintillation counter (Perkin Elmer) with a dual 3H/14C programme.
Antibodies (dilutions) used for this study
Donkey anti Goat IR Dye 680RD (1: 10,000), 926-68074, Donkey anti Mouse IRDye 680RD (1: 10,000), Goat anti Rabbit IRDye 800CW (1: 10,000), 926-32211 (All Licor Biosciences Ltd), Goat anti Aconitase 2, Ab99467 (1: 10000), Rabbit anti Aconitase 2, Ab129069 (1: 50,000), Goat anti Aconitase 1, Ab10694 (1: 500), Complex II Ab Cocktail, Ab110410 (1: 250), Rabbit anti Glut4, Ab654 (1: 2,000), Rabbit anti SDHB, Ab84622 (1: 1,000), Rabbit anti TST, Ab60128, (1: 2,000)(All Abcam), Rabbit anti SDHB (1: 7,000), NBP1-54154, Rabbit anti SOD2 (1; 2,000), NBP1-40422, Goat anti SOD1 (1: 10,000), all Novus Biologicals, NB100-60944, Rabbit polyclonal anti-TST antibody, GTX 114858 (1: 1,000), Source Biochemicals.
Clonal adipocyte cell experiments
The 3T3-L1 preadipocyte cell line was obtained from the American Type Culture Collection. The Expression Arrest GIPZ lentiviral shRNAmir-mediated plasmid based system (Open Biosystems, Epsom, UK) was used to transduce mature 3T3-L1 adipocytes (days 6–8 of differentiation) to knockdown Tst (Supplementary Fig. 7f). Mitochondrial ROS studies were performed by exposing the transduced 3T3-L1 cells to medium containing 1% H2O2 for 6 hours followed, after careful rinsing, by exposure to Mitotracker Red-CM-H2XROS (M7513, final concentration 250 nM, Molecular Probes, Invitrogen, Paisley, UK). Probe oxidation was quantified using an M1000 fluorescence reader and Magellan software (Tecan Uk Ltd, Reading, UK). Human Simpson-Golabi-Behemel syndrome (SGBS)32 human clonal preadipocytes were maintained at 37 °C, 5% CO2 in DMEM/F12 supplemented with 10% (vol/vol) FBS, 50 U/ml penicillin, 50 µg/ml streptomycin, 33 µM biotin and 17 µM pantothenic acid. And differentiated with addition of 0.5 mM IBMX, 0.25 µM dexamethasone, 20 nM insulin, 2 µM rosiglitazone, 0.01 mg/ml transferrin, 0.1 µM cortisol and 200 pM triiodothyronine (T3). From day 7 to day 21 cells were differentiated without IBMX and Dexamethasone. Neither cell line was tested for mycoplasma during these studies.
TST gene expression during human preadipocyte differentiation
Isolated omental (Om) and subcutaneous (Sc) preadipocytes (Zen-Bio Inc., Research Triangle Park, NC, USA) were cultured as previously described31. The differentiation was routinely monitored by measuring fatty acid synthase and adiponectin expression (FASN, Hs00188012_m1 and Adipoq, Hs00605917_m1).
Preparation of primary adipocytes and lipolysis
Adipocytes were prepared from mice as follows. Mice were culled, and epididymal fat removed into warmed Krebs buffer (118 mM NaCL, 5 mM KCl, 1.2mM MgSO4, 10 mM NaPO4, pH 7.4, 1% BSA, 1 mg/ml Glucose, 1.25 mM CaCl2). The tissue was then cut using sharp scissors into approximately 2–4 mm diameter pieces in Krebs buffer, 2mg/ml Collagenase I (Worthington Lot: 43C14114B). Fat fragments were incubated at 37 °C in a cell culture incubator for between 40 and 50 minutes. The suspension was swirled to mix every 10 minutes. Digestion was stopped when the majority of material was visible as dissociated adipocytes. The cell suspension was passed through a 250 µM mesh into a 50 ml Falcon tube, in order to remove undigested material. An equal volume of Experimental Media (1×DMEM, 4.5 g/L Glucose, 4mM Glutamine, 100 U/ml Penicillin/Streptomycin, 10% Charcoal Dextran stripped Foetal Bovine Serum) was added to the suspension, prior to centrifugation at room temperature for 3 minutes (1500 g). Adipocytes, which accumulate at the top of the suspension, were removed into 5 volumes of fresh Experimental Media, using a cut off P1000 tip. Adipocytes were swirled, and given 5 minutes to settle to the top. Media was removed from below the adipocyte layer, and another 5 volumes were added. Cells were incubated for 60 minutes at 37 °C 5% CO2. Cells were then given a third wash, and after removal of media, the adipocytes were added to their experimental tubes. For basal lipolysis, 35µl of adipocytes were transferred to 300 µl Experimental Media in a 500 µl eppendorf tube with open lids open to maintain oxygenation of adipocytes. Tubes were gently shaken to mix the adipocytes before incubating in a 37 °C incubator (5% CO2). After 90 minutes tubes were gently shaken once more. Tubes were left still for 1 minute to allow resettling of adipocytes, before samples were taken carefully from below the adipocyte layer with a catheter-linked microsyringe. For CL stimulation, CL316,243 was added in a volume of 15 µl of experimental media to the adipocyte suspension to a final concentration indicated for each experiment. Tubes were gently shaken to mix the suspension, and tubes returned to the incubator for 30 minutes. At this time, tubes were gently shaken prior to media sampling. Tubes were returned back to incubator for a further 90 minutes. Tubes were shaken once more prior to final sampling as above.
NEFA assay
Standards and samples of 40 µl were incubated with WAKO NEFA kit with 100 µl of Reagent 1 (5 minutes, 37 °C) in a clear, flat-bottom plastic 96 well assay plate. 50 µl of Reagent 2 was then added, and the sample incubated for a further 5 minutes. Absorbance of each sample was read at 565 nm, with a background correction at 660 nm. A standard curve (which is linear between 5 µM and 250 µM) was prepared from a 1 mM reference sample (Wako).
Lipogenesis assay
Adipocytes were prepared as for the lipolysis assay above. Experimental media (±insulin, ±10µM cytochalasin B glucose uptake inhibitor as a negative control) was prepared by adding 3H Glucose to a final concentration of 0.5 µCi/ml. Typical media/cells ratio was 5/1 by volume. Media (+ 3H-Glucose) was added to a plastic scintillation vial then adipocytes added. Cells and media were mixed and incubated for 2 hours in 5% CO2 37 °C incubator with gently shaking every 15 minutes. 6 M H2SO4 (1 ul/10 ul of Experimental Media) was added and the sample was vortexed for 30 seconds to stop the reaction. Organic Scintillant (2 volumes /1 volumes of Media) was added without mixing, and left to allow the lipids to diffuse into the organic scintillant layer for >4 hours. Incorporation into lipid was calculated using a 3H sensitive scintillation counter and correcting for no cell blank, cell blank and cytochalsin B (3H glucose background) and normalised to lipid mass calculated by extraction of parallel samples in extraction solvent (4:1:1, Isopropanol: heptane: 1 N sulphuric acid). For each 100 µl adipocyte suspension 1.5 ml of water, 2.5 ml of extraction solvent and 1.5 mls of heptane was added followed by solvent evaporation and accurate mass determination on a fine balance.
HbA1c levels
HbA1c levels were measured by reverse-phase cation exchange chromatography using the Evolution HA-8160 automated analyser (A.Menarini Diagnostics Ltd. Wokingham, UK) according to the manufacturer’s instructions. Briefly, 3.4 µL of diluted lysed whole blood was injected onto the stainless steel Arkray column, consisting of a prefilter and an analytical column packed with an ion exchange resin (a hydrophilic polymer of methacrylate ester copolymer). Elution was achieved with a five-step phosphate buffered gradient with increasing ionic strength, utilizing three buffers (80A, 80B and 80CV). Hemoglobin fractions are detected with a dual wavelength (420–500 nm) LED-photodiode. The instrument was calibrated with two calibrators. The reported result is derived from the ratio HbA1c/HbA total, adjusted for calibration. Within-run and between-run CVs were both <3.0%.’
Mitochondrial functional tests
Mitochondria were extracted from the livers of fed male adult C57Bl6 or Tst−/− mice. Livers were rapidly excised after mice were humanely killed and placed in ice-cold isolation buffer (IB: 10 mM Tris-MOPS (3-(N-morpholino)propanesulphonic acid), 1 mM EGTA/Tris, 200 mM sucrose, pH 7.4, all Sigma Aldritch). All further steps were performed on ice or at 4 °C during centrifugation. Whole livers were minced using scissors, washed in ice cold IB buffer and then homogenised in 5 ml IB buffer using a glass Potter Elvehjem homogeniser. Homogenates were transferred to 50 ml falcon tubes (Polypropylene Falcon tube, Corning) and centrifuged at 1800 rpm for 10 minutes (Heraeus Megafuge 1.0R). Supernatants were transferred to Beckmann ultra centrifuge glass tubes (Beckman, USA) and further centrifuged at 7800 rpm for 10 minutes in a Beckman J2-MC ultracentrifuge (Beckman, USA). Supernatants were discarded and the remaining pellet was washed with 5 ml ice-cold IB buffer before being centrifuged again at 7800 rpm for 10 minutes. Finally supernatants were again discarded and the pellets resuspended using a glass rod and by gently pipetting. This represented a purified mitochondrial preparation. Protein concentration was determined by a Bradford protein assay and all experiments were performed at a final concentration of 0.2 mg/ml protein. Mitochondrial preparations were injected directly into an Oroboros Oxygraph 2K (Oroboros Instruments, Austria) high-resolution respirometer using Hamilton microsyringes (Hamilton, UK). Prior to this the 2 chambers of the Oxygraph 2K were filled with MiR-05 buffer designed to support optimal respiration (0.5 mM EGTA, 3mM MgCl2.6H2O, 60 mM K-lactobionate, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, 1 g/L BSA, pH 7.1, all reagents from Sigma Aldritch) and allowed to normalise with room oxygen concentration for a minimum of 1 hour minutes before closing the chambers. Oxygen concentration of the chambers and rate of oxygen use (respiration) were recorded using DataLab software (Oroboros). All further substrates or reagents including 2.5 mM malate, 5 mM pyruvate, 2 µM rotenone (complex I inhibition for complex II-specific assessment), 10 mM succinate, 100 µM ADP and 1µg/ml oligomycin (all Sigma Aldritch) were added into the chamber directly using microsyringes (Hamilton). Na2S (from Sigma) pre-incubation was for 2 hours on ice in the neat mitochondrial fraction before assessment of mitochondrial function.
Blood sulfide measurement
Determination of hydrogen sulfide concentration in whole blood samples were performed by derivatization with monobromobimane (mBBr) followed by HPLC separation and detection with a fluorescence detector. Briefly, for each blood sample to be derivatized one 1.5 ml brown glass vial was labelled and filled with 100 µL of 160 mM EPPS/16 mM diethylenetriaminepentaacetic acid (DTPA) pH 8.0, followed by 100 µL of acetonitrile and 10 µL of 46 mM mBBr solution in acetonitrile. To this solution 50 µL of blood (immediately withdrawn from a mouse) was dispensed and the reaction vials were capped tightly and vortexed for 10 minutes. Subsequently, 1 mL ethyl acetate was added, the tube capped and vortexed for another 10 minutes. after this, the reaction vials were centrifuged gently at 350 × g for 7 min to separate aqueous and organic layers. The organic layer was collected from each extraction, transferred to a 1.5 mL brown glass vial and the solvent was evaporated completely under a nitrogen stream. Acetonitrile (200 µL) was added to each vial, and the solvent was again evaporated to remove any traces of ethyl acetate. To quantify sulfide dibimane in a sample, the dried residue was re-suspended in 50 µL of Buffer A (10 mM tetrabutylammonium phosphate aqueous, 10% methanol, 45 mM acetic acid adjusted to pH 3.4). The entire sample was transferred to an HPLC autosampler vial with a 200 µL glass sample insert, and the vial was closed with a penetrable cap. 20 µL of the sample was injected onto a C8 reverse-phase column (LiChrospher 60 RP-select B 5 µm 4.0 × 125 mm LiChroCART 125-4, Merck KGaA) and a guard column (LiChroCART 10-2, Superspher 60 RP-select B cartridge) on an Ultimate 3000 UHPLC+ focused system (Thermo Scientific). Sulfite dibimane was eluted with a linear gradient from 10% buffer B (10 mM tetrabutylammonium phosphate in methanol, 10% water, 45 mM acetic acid) to 100% buffer B over 30 min. The eluent was analysed by fluorescence (λex = 380 nm, λem = 480 nm).
Human adipose tissue analysis
Cohort 1: samples of visceral (omental) and subcutaneous adipose tissue were obtained from white female morbidly obese subjects (BMI >40 kg/m2) that had undergone laparoscopic bariatric surgery and lean to overweight control subjects (BMI <30 kg/m2) undergoing other elective laparoscopic surgery at the General Hospital of Vienna (n = 8). This group size was initially chosen as it had revealed consistent changes between visceral and subcutaneous gene expression in previous analyses. Criteria for exclusion were the presence of any infectious, inflammatory, neoplastic or systemic disease, diabetes (excluded by fasting plasma glucose or the use of anti-diabetic drugs), or other uncontrolled endocrine disease. None of the individuals used antibiotics, anti-inflammatory, anti-obesity or lipid-lowering drugs when participating in the study. The study was approved by the ethics committee of the Medical University and the General Hospital of Vienna. Cohort 2: subcutaneous adipose tissue was obtained from a large cohort30 of Icelandic subjects (n = 670). Cohort size was predefined30. Cohort 3: a group of 180 adipose tissue samples (96 visceral and 84 subcutaneous depots) from participants with a body mass index (BMI) within 20 and 68 kg/m2, who were recruited at the Endocrinology Service of the Hospital Universitari Dr. Josep Trueta (Girona, Spain) were analysed31. Cohort size was predfined31. Ethics related to the sampling in Spain and Iceland are detailed in the respective references30, 31 and all were done with informed consent according to the relevant designations/countries guidelines. Significance of correlational data for TST and other adipose genes was assessed with Scheffé post hoc tests (Supplemental Table 4).
Additional Gene expression
RNA extraction, cDNA synthesis and realtime PCR was performed as described8,31. Probes were mouse Tst: Mm00726109_m1, Gapdh (internal control): Mm99999915_g1 and Tbp (internal control): Mm0000446973_m1. Prx3: Mm00545848_m1, Slc2a4 (Glut4): Mm00436615_m1, Cpt1a: Mm01231183_m1, Adrb3: Mm02601819_g1, Pparg: Mm00440940_m1, Cebpd: Mm00786711_s1, Cebpb: Mm00843434_s1, Cebpa: Mm00514283_s1, Dlk1: Mm00494477_m1, Klf5: Mm00456521_m1, Srebp1: Mm00550338_m1:, Human TST was measured using Hs00361812_m1 and TBP with Hs99999910_m1.
TST activity assays
The adapted TST assay48 was performed in a 96 well plate with tissue homogenates. The reaction was started by the addition of 10 µL 500 mM KCN (final concentration 50 mM). The reaction proceeded for 20 min and was stopped by the addition of formaldehyde (63 µL of reaction mix into 7 µL formaldehyde). The colourmetric change was achieved by the addition of 30 µL 300mM Fe(NO3)3 (final concentration 30 mM) which quickly underwent ligand exchange with thiocyanate to give the consequent colour change from yellow to red. The absorbance was measured at 460 nm and TST activity corrected for non–enzymatic activity. A standard curve was produced by addition of SCN (1.8 mM to 0.025 mM) to Fe(NO3)3. From the standard curve the relative concentrations of −SCN produced could be calculated and therefore the turnover of TST activity with the compounds could be determined. A recombinant human TST preparation was tested in parallel to infer non TST-mediated substrate turnover.
Statistical analyses and approaches
For gene expression, hormone, protein levels, generally group sizes of 6 were calculated to allow detection of differences in these variable parameters to a threshold of 15% (there is sufficient power to detect smaller differences in certain parameters with this cohort size) with a power of at least 0.8. In some studies, limitations in animal numbers, or fewer remaining samples from larger group sizes resulting from their use for multiple end-points, precluded the desired minimum of n = 6 per group. Protein or mRNA differences in validation studies with 2 parameters (e.g. diet with line or genotype) were analysed using 2-way ANOVA for line and diet effects followed, where appropriate, by post-hoc Tukey tests or Holm-Sidak multiple comparison tests using Sigmastat version 3.5 (Systat Software) or Prism (Graphpad Software). For simple 2 condition comparisons, t-test was used. For simple control versus treated (including different treatments or concentration response curves) data were analysed by 1-way ANOVA. For longitudinal measures (e.g. GTT, ITT, bodyweight gain) repeated measures (RM) ANOVA was used and multiple comparisons determined. The statistical model for the analysis of F2 mouse cross data were as described above and in previous studies6. Parameters and other statistical quantities were inferred using MCMC methods as employed in the MCMCpack R package. Data were normally distributed. For all main in vivo studies, a blinding strategy was used where the operator (e.g. for injections of glucose, or administration of drug) was blind to the genotype of the subject during the experiment. Similarly, for analysis of images (e.g. fat cell size) the scorer was blind to genotype and the data coded, with the code broken by a second individual. Downstream analysis of e.g. tissue mRNA and protein content was not generally blinded to allow appropriate data arrangement on e.g. representative western blots.
Supplementary Material
Acknowledgments
N. Morton was supported by a Career Development Fellowship, Institutional Strategic Support Fund award and a New Investigator Award from the Wellcome Trust (100981/Z/13/Z), a Research Councils UK Fellowship and a British Heart Foundation Centre of Research Excellence exchange award. We thank the Slovenian Research Agency (core funding P4-0220, project N5-0003 Syntol and J4-6804 to S.H.); and a Young Scientist Fellowship to J. Beltram. We acknowledge support of the British Heart Foundation Research Excellence Award in support of the Bioinformatics Core contribution. T. Stulnig received funding from the Federal Ministry of Economy, Family and Youth and the Austrian National Foundation for Research, Technology and Development. G. Churchill was supported by the US National Institutes of Health grant R01GM 070683. J.M. Fernandez-Real acknowledges funding from FIS PI11/00214. A. Vidal-Puig was funded by the UK Medical Research Council (MRC) MDU, an MRC Programme grant, MRC DMC Core and MITIN (HEALTH-F4-2008-223450). We thank Martin Wabitsch (University of Ulm, Germany) for the gift of the SGBS human preadipocyte cell line.
Footnotes
ACCESSION CODES
The F and L microarray data can be found at GEO E-MEXP-3094.
The RNA Seq data for the DO mice liver can be found at GEO GSE45684 and for the founder strain adipose tissue at GSE80162.
AUTHOR CONTRIBUTIONS
N.M.M. and S.H. conceived the experiments. N.M.M., J.B., R.N.C., Z.M., G.G., S.C.M., S.R-C., C. Mc F., M.B–L, R.E.A, L.R., A.F.H. and S.H. performed experiments on in vivo models or samples. N.M.M., R.N.C., J.M.M.-N., M.T.G.G., C. Mc F. and A.G. performed experiments on in vitro models. J.M.M.-N., V.G., J.M.F–R. and V.E. provided and analysed human adipose gene expression data. M.Z., T.M.S. provided human adipose tissues. G.N. generated the TST inhibitor. A.S. and P.S. generated the Ad-Tst mice. Z.V.W. generated the adiponectin promoter DNA vector. D.R.D. performed bioinformatics analyses. S.C.M., K.L.S. and G.A.C generated the Diversity Outbred mouse resources and data. S.R–C., C.J.K., J.R.S., B.R.W, S.P.W., T.V–P., J.M.F–R, V.E and S.H. discussed results and commented on the manuscript. N.M.M. and S.H. wrote the paper.
COMPETING FINANCIAL INTERESTS
N.M.M. and S.P.W. hold a target patent WO2012/104589 for TST in weight-related disorders.
References
- 1.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–41. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 2.Yanovski SZ, Yanovski JA. Obesity prevalence in the United States – up, down, or sideways? N. Eng. J. Med. 2011;364:987–989. doi: 10.1056/NEJMp1009229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ljungvall A, Zimmerman FJ. Bigger bodies: long-term trends and disparities in obesity and body-mass index among U.S. adults, 1960–2008. Soc Sci Med. 2012;75:109–119. doi: 10.1016/j.socscimed.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Morton NM, et al. A polygenic model of the metabolic syndrome with reduced circulating and intra-adipose glucocorticoid action. Diabetes. 2005;54:3371–3378. doi: 10.2337/diabetes.54.12.3371. [DOI] [PubMed] [Google Scholar]
- 5.Horvat S, et al. Mapping of obesity QTLs in a cross between mouse lines divergently selected on fat content. Mamm. Genome. 2000;11:2–7. doi: 10.1007/s003350010002. [DOI] [PubMed] [Google Scholar]
- 6.Prevorsek Z, Gorjanc G, Paigen B, Horvat S. Congenic and bioinformatics analyses resolved a major-effect Fob3b QTL on mouse Chr 15 into two closely linked loci. Mamm. Genome. 2010;21:172–185. doi: 10.1007/s00335-010-9252-z. [DOI] [PubMed] [Google Scholar]
- 7.Bünger L, et al. Long-term divergent selection on fatness in mice indicates a regulation system independent of leptin production and reception. FASEB J. 2003;17:85–7. doi: 10.1096/fj.02-0111fje. [DOI] [PubMed] [Google Scholar]
- 8.Morton NM, et al. A stratified transcriptomics analysis of polygenic fat and lean mouse adipose tissues identifies novel candidate obesity genes. PLoS One. 2011;6:e23944. doi: 10.1371/journal.pone.0023944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westley J. Rhodanese. Adv Enzymol Relat Areas Mol Biol. 1973;39:327–68. doi: 10.1002/9780470122846.ch5. [DOI] [PubMed] [Google Scholar]
- 10.Hall AH, Saiers J, Baud F. Which cyanide antidote? Crit Rev Toxicol. 2009;39:541–552. doi: 10.1080/10408440802304944. [DOI] [PubMed] [Google Scholar]
- 11.Bonomi F, Pagani S, Cerletti P, Cannella C. Rhodanese-Mediated sulfur transfer to succinate dehydrogenase. Eur J Biochem. 1977;72:17–24. doi: 10.1111/j.1432-1033.1977.tb11219.x. [DOI] [PubMed] [Google Scholar]
- 12.Pagani S, Galante YM. Interaction of rhodanese with mitochondrial NADH dehydrogenase. Biochim. Biophys. Acta. 1983;742:278–284. doi: 10.1016/0167-4838(83)90312-6. [DOI] [PubMed] [Google Scholar]
- 13.Nandi DL, Horowitz PM, Westley J. Rhodanese as a thioredoxin oxidase. Int. J. Biochem. Cell Biol. 2000;32:465–473. doi: 10.1016/s1357-2725(99)00035-7. [DOI] [PubMed] [Google Scholar]
- 14.Wang R. Physiological implications of hydrogen sulfide: a whiff of exploration that blossomed. Physiol. Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 15.Tiranti V, et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- 16.Smirnov A, et al. Mitochondrial enzyme rhodanese is essential for 5 S ribosomal RNA import into human mitochondria. J Biol Chem. 2010;285:30792–30803. doi: 10.1074/jbc.M110.151183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vernochet C, et al. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab. 2012;16:765–776. doi: 10.1016/j.cmet.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tormos KV, et al. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011;14:537–544. doi: 10.1016/j.cmet.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kusminski CM, et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 2012;18:1539–1549. doi: 10.1038/nm.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geng B, Cai B, Liao F, Zheng Y, Zeng Q, Fan X, Gong Y, Yang J, Cui QH, Tang C, Xu GH. Increase or decrease hydrogen sulfide exert opposite lipolysis, but reduce global insulin resistance in high fatty diet induced obese mice. PLoS One. 2013;8(9):e73892. doi: 10.1371/journal.pone.0073892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng X, et al. Hydrogen sulfide from adipose tissue is a novel insulin resistance regulator. Biochem Biophys Res Commun. 2009;27(1):380. 153–9. doi: 10.1016/j.bbrc.2009.01.059. [DOI] [PubMed] [Google Scholar]
- 22.Simoncic M, et al. Divergent physical activity and novel alternative responses to high fat feeding in polygenic fat and lean mice. Behav Genet. 2008;38:292–300. doi: 10.1007/s10519-008-9199-y. [DOI] [PubMed] [Google Scholar]
- 23.Svenson KL, et al. High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics. 2012;190:437–447. doi: 10.1534/genetics.111.132597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang ZV, Deng Y, Wang QA, Sun K, Scherer PE. Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology. 2010;151:2933–2939. doi: 10.1210/en.2010-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu A, et al. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herman MA, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012;484:333–338. doi: 10.1038/nature10986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sen U, et al. Cardioprotective role of sodium thiosulfate on chronic heart failure by modulating endogenous H2S generation. Pharmacology. 2008;82:201–213. doi: 10.1159/000156486. [DOI] [PubMed] [Google Scholar]
- 28.Sabelli R, et al. Rhodanese-thioredoxin system and allyl sulfur compounds. FEBS J. 2008;275:3884–3899. doi: 10.1111/j.1742-4658.2008.06535.x. [DOI] [PubMed] [Google Scholar]
- 29.Koh EH, et al. Essential role of mitochondrial function in adiponectin synthesis in adipocytes. Diabetes. 2007;56:2973–2981. doi: 10.2337/db07-0510. [DOI] [PubMed] [Google Scholar]
- 30.Emilsson V, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 31.Moreno-Navarrete JM, et al. Decreased RB1 mRNA, protein, and activity reflect obesity-induced altered adipogenic capacity in human adipose tissue. Diabetes. 2013;62:1923–1931. doi: 10.2337/db12-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wabitsch M, et al. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int J Obes Relat Metab Disord. 2001;25:8–15. doi: 10.1038/sj.ijo.0801520. [DOI] [PubMed] [Google Scholar]
- 33.Loos RJ. The genetic epidemiology of melanocortin 4 receptor variants. Eur. J. Pharmacol. 2011;660:156–164. doi: 10.1016/j.ejphar.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 34.Smemo S, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kilpelainen TO, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet. 2011;43:753–760. doi: 10.1038/ng.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heid IM, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–960. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rung J, et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet. 2009;41:1110–1115. doi: 10.1038/ng.443. [DOI] [PubMed] [Google Scholar]
- 38.Vigouroux C, Caron-Debarle M, Le Dour C, Magré J, Capeau J. Molecular mechanisms of human lipodystrophies: from adipocyte lipid droplet to oxidative stress and lipotoxicity. Int J Biochem Cell Biol. 2011;43:862–876. doi: 10.1016/j.biocel.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 39.Jacquemont S, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478:97–102. doi: 10.1038/nature10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 41.Mathes WF, Kelly SA, Pomp D. Advances in comparative genetics: influence of genetics on obesity. Br J Nutr. 2011;106:S1–10. doi: 10.1017/S0007114511001905. [DOI] [PubMed] [Google Scholar]
- 42.Soloveva V, et al. Transgenic mice overexpressing the beta 1-adrenergic receptor in adipose tissue are resistant to obesity. Mol. Endocrinol. 1997;11:27–38. doi: 10.1210/mend.11.1.9870. [DOI] [PubMed] [Google Scholar]
- 43.Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat. Med. 2013;10:1252–1263. doi: 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- 44.Hawley SA, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawley SA, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munger SC, et al. RNA-Seq alignment to individualized genomes improves transcript abundance estimates in multiparent populations. Genetics. 2014;198:59–73. doi: 10.1534/genetics.114.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altszuler N, et al. Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am J Physiol. 1956;187:15–24. doi: 10.1152/ajplegacy.1956.187.1.15. [DOI] [PubMed] [Google Scholar]
- 48.Hildebrandt TM, Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.