

SUMMARY
Dorsal root ganglia (DRG) relay sensory information to the brain, giving rise to the perception of pain, disorders of which are prevalent and burdensome. Here, we mapped expression quantitative trait loci (eQTLs) in a collection of human DRGs. DRG eQTLs were enriched within untranslated regions of coding genes of low abundance, with some overlapping with other brain regions and blood cell cis-eQTLs. We confirm functionality of identified eQTLs through their significant enrichment within open chromatin and highly deleterious SNPs, particularly at the exon level, suggesting substantial contribution of eQTLs to alternative splicing regulation. We illustrate pain-related genetic association results explained by DRG eQTLs, with the strongest evidence for contribution of the human leukocyte antigen (HLA) locus, confirmed using a mouse inflammatory pain model. Finally, we show that DRG eQTLs are found among hits in numerous genome-wide association studies, suggesting that this dataset will help address pain components of non-pain disorders.
Graphical abstract
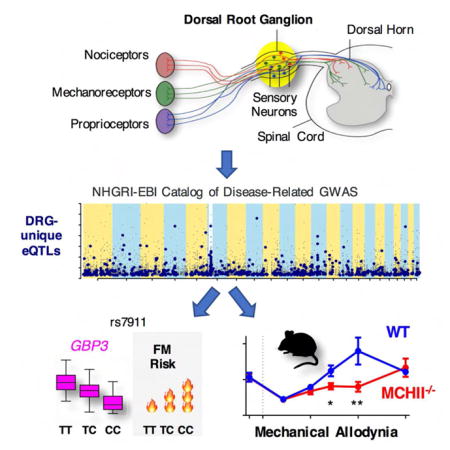
INTRODUCTION
Pain is the primary reason for seeking medical assistance, but its underlying causes remain difficult to identify and remediate. The societal cost of chronic pain is substantial; estimated at around $635 billion in the U.S., and affecting more than 25% of the population (Gereau et al., 2014). Associated sensory pathologies including itch, dysesthesia and numbness also represent a major clinical burden. About half of the inter-individual variability in the experience of pain, risk of development of chronic pain, and responses to treatment are attributable to genetic factors (Diatchenko et al., 2013). Identifying these genetic elements will improve diagnosis and facilitate the development of personalized therapies for people afflicted with chronic pain.
Genome-wide association studies (GWAS) have successfully identified genetic variants affecting risk for a wide range of human psychiatric and neurological conditions (Hardy and Singleton, 2009). The assessment of human chronic pain conditions using GWAS is an emerging but rapidly growing field. The major difficulty in the interpretation of GWAS results derives from the fact that most genetic variants, majority of which are single nucleotide polymorphisms (SNPs), are not protein-coding, thus seldom provide a simple, straightforward molecular-level explanation. Other than producing coding changes, alternative alleles can affect gene function through their effect on mRNA expression level or splicing. SNPs associated with the expression level of a gene or an exon are called expression quantitative trait loci (eQTLs). Both SNPs that produce local expression effects (cis-eQTLs) and SNPs that affect expression levels of distant genes (trans-eQTLs) have been found to be functional. More than half of the known eQTLs are tissue-specific, making the identification of eQTLs in tissues relevant to disease pathogenesis a crucial matter (Ramasamy et al., 2014).
The perception of pain, and other somatosensory experience, results from the relay of neural signals from the periphery into the central nervous system (CNS). Primary afferent neurons, which have their cell bodies located in dorsal root ganglia (DRG), convey sensory information (pain, itch, touch, pressure, vibration and temperature; Figure 1a). DRG neurons are heterogeneous and can be classified according to their size, neurochemical markers, stimulus modality and gene expression profiles (Usoskin et al., 2015). DRG are situated at the junction between the spinal nerve and the dorsal root. They are maintained by a network of permeable capillaries lined by endothelial cells (Jacobs et al., 1976) and are held together by a connective tissue capsule. In addition, resident T-lymphocytes and macrophages are found in DRG, with increasing in number following injury (Zhu et al., 2016). The perception of acute pain results from the activation of specialized DRG neurons referred to as “nociceptors”. Increased nociceptor excitability following nerve injury and/or exposure to inflammatory mediators contributes to the hyperalgesia and allodynia often observed in chronic pain states (Woolf and Ma, 2007). Due to their critical role in initiating, detecting, and perpetuating pain and related sensations, DRGs are a primary tissue target for analgesics, anesthetics, antipruritics and neuro-stimulation procedures.
Figure 1.

Comparative analysis of DRG transriptome and cis-eQTLs. (a) Spinal cord and DRG anatomy. (b) Unsupervised hierarchical clustering of DRG (red) cis-eQTLs or (c) DRG (red) transcriptome with 10 brain regions (braineac dataset). (d) Unsupervised hierarchical clustering of transcriptomes between various tissue types of the central and peripheral nervous system (Harvard dataset), including dorsal root ganglia (red). (e) 3-way Venn diagram showing the extent of shared eGenes across DRGs, whole blood and a mixture of 10 brain tissues. See also Figures S1 to S5, and Tables S1 to S3.
Here, we present a study mapping eQTLs in a collection of human DRGs in order to gain an understanding of the architecture of gene expression and locus-specific regulation, focusing our functional analysis on pain phenotypes. Our dataset presents a valuable resource for others: it is an exhaustive survey of eQTLs in this unique and important human tissue, which is freely available online (http://diatchenko.lab.mcgill.ca/DRG-eQTLs/) for future interpretation of GWAS focused on pain and other sensory phenotypes.
RESULTS
eQTL discovery
A total of 214 human DRGs specimens that passed quality control on DNA genotyping (Figure S1, Tables S1a, b) and on RNA expression (Figure S2) were used in this study (Figure S3). The cohort size is comparable with other eQTL studies involving human neuronal tissue (GTEx_Consortium, 2015). We searched for eQTL associations of ~4.88 million independent SNPs with 36,552 gene-level and 386,507 exon-level expression points. We considered an eQTL to be any SNP that is significantly associated with the mRNA level of a gene or an exon, and we defined such a gene or exon to be an eGene or eExon. We found a total of 1927 gene-level and 6364 exon-level cis-acting eQTL-gene/-exon pairs, and 607 gene-level and 2646 exon-level trans-acting pairs (Tables S1c–i). About 77% of the cis-eQTL signals and 81% of the trans-eQTL signals were found only from exon-specific expression. The sex-specific composition of identified DRG eQTLs also has been estimated (Figure S4, Tables S1e, f).
Global characterization of DRG eQTLs
Our dataset was used to compare DRG to other tissues at the eQTL and transcriptome levels. This analysis identified similarities of DRG transcriptomics and eQTLs with other tissues, thus confirming that the discovered set represents true eQTL signal, while the same time illustrated the uniqueness of the dataset. The recently published data on genetic variability in gene regulation in 10 different human brain regions (Ramasamy et al., 2014) was considered representative of the CNS, for contrast with DRG, part of the peripheral nervous system (PNS). In comparisons across brain regions, DRG eQTLs displayed resemblance to some brain regions, particularly cerebellum, whereas greater differences were observed compared to subcortical regions (Figure 1b). However, when we compared the DRG transcriptome with the 10 brain tissues, we found that DRGs displayed a unique transcriptome that stood out from brain tissues (Figure 1c). An additional analysis of mRNA expression obtained from other previously published microarray experiments on ganglia (Figure 1d, Harvard dataset) (Benita et al., 2010) found the most similarity with other ganglia, in particular to ciliary, trigeminal and superior cervical ganglia. In line with the previous result, the ganglia group altogether diverged from brain-related tissues. The dissimilarity of ganglia transcription pattern from brain regions is in accordance with its embryonic development stemming from neural crest cells as opposed to the neural tube (Frank and Sanes, 1991).
To investigate the global biological function of DRG eGenes that were associated with cis-acting eQTLs, we compared them with eGenes from ten brain regions (Ramasamy et al., 2014), and from whole blood (GTEx_Consortium, 2015) (Figure 1e, Table S2). We chose whole blood because it has been proven to be a useful surrogate for gene expression in the CNS and it is easily available from human subjects for genetic marker assessment (Sullivan et al., 2006). To gain insights into pathways potentially affected by eQTLs, we performed gene ontology (GO) analyses for DRG eGenes alone, and for those that intersect with other tissues using Pathway Studio (Nikitin et al., 2003) (Table S2). For DRG alone, GO biological processes identified as “gamma-delta T cell activation” (N=4/4 eGenes, P=1.77e-6), “antigen processing and presentation” (N=11/56 eGenes, P=4.77e-6), and “glutathione derivative biosynthetic process” (N=8/27 eGenes, P=3.66e-6) passed correction for multiple testing. For eGenes shared between DRG and blood, fourteen GO biological processes were found significant, of which eleven are related to immune response and three are related to glutathione derivative biosynthesis. For genes shared between DRG and brain, we found 3 pathways; two were related to glutathione and one was related to dihydrofolate metabolic process. Hence, “glutathione derivative biosynthetic process” related pathways eGenes showed the strongest contribution at the intersection of DRG, blood and brain.
To understand the level of inter-species conservation of eGenes, we estimated overlap between human DRG eGenes and mouse DRG expressed genes. We calculated that 51%–77% of human eGenes are also expressed in mouse DRGs. From microarray data, we found 3rd quartile gene expression above 1st quartile overall expression for 333 (77%) out of 432 matched eGenes (Figure S5a, GEO set GSE65997) (Wieskopf et al., 2015). From single-cell mouse RNA-Seq data, we found expression of 3rd quartile above zero for 376 (51%) out of 731 matched eGenes (Usoskin et al., 2015). Hence, most human eGenes are expressed in mouse DRG. To assess the neuronal versus non-neuronal origin of eQTL signals in human DRG we compared our human DRG eGenes list with gene expression data of single-cell RNA-Seq from mouse DRG (Usoskin et al., 2015). In this dataset, cells were classified into five categories: four neuronal and one non-neuronal. Only one eGene, out of the 376 matched eGenes expressed in any of five cell types, had higher expression in non-neuronal cell types than in any one of the four neuronal types: RPSA (Table S3). Therefore, virtually all (375 out of 376) of identified eGenes in human DRG show characteristics of the neuronal type. We should note that this expression is not exclusive for neuronal cells and does not guarantee biological relevance of any particular molecular pathway.
Functional characterization of the eQTLs in DRGs
To characterize eQTLs identified in DRG tissue, we first analyzed cis-eQTL signals as they have been shown to have a higher confidence of association with eGenes across datasets (Ramasamy et al., 2014) compared to trans-eQTL signals. We tested if the distribution of the DRG cis-eQTLs throughout the genome is non-random with respect to the eQTL’s location. At the gene level, a histogram of the distance between the cis-eQTL and the TSS shows that 55% of cis-eQTLs (N=1055) are located around 100 Kb from the TSS (Z=21.4, P=3.1e-102, Figure 2a), consistent with previous reports (GTEx_Consortium, 2015; Ramasamy et al., 2014). The eQTL signal is still detectable as far as 1 million nucleotides away from the TSS, suggesting that either this low eQTL signal represents the background of the analytical methods or that the true eQTLs can be situated far from the TSS. At the exon level, cis eQTL signals were found most predominantly within 200 bp of the intronic boundaries inside exons. In contrast, we observed strong and consistent eQTL enrichment throughout the flanking introns (Figure 2b).
Figure 2.

Characterization of cis-eQTLs in DRGs. (a) Histogram of the distance between cis-eQTL and the transcription start site (TSS) of the associated eGene. Grey is intergenic and purple is intragenic regions. (b) Histogram of the distance between cis-eQTL and the associated exon. Grey is intronic and purple is exonic regions. (c) Functional annotation of gene-level or (d) exon-level cis-eQTLs. Horizontal barplots track enrichment as log-ratios of observed to expected counts. Positive enrichment (yellow) signifies counts that are higher than expected, while negative enrichment (brown) signifies counts that are lower than expected. Observed counts less than ten may not be reliable (grey). Counts are binned by categories, defined by the CADD web resource. Categories are synonymous (SYN), non-synonymous (NON_SYN), 5′UTR (5P_UTR), 3′UTR (3P_UTR), splice site (SPLICE), regulatory (REG), intron (INTRON), upstream (UPSTRM), downstream (DWNSTRM), intergenic (INTERGEN), and non-coding change (NON_CHNG). Pie charts (green shades) track the relative counts per categories, labeled from 1 to 5, and others “0” combining the remaining categories. Categories marked with a star * pass Bonferroni correction following a binomial test for enrichment of observed over expected. Open chromatin evidence at gene-level (e) or exon-level (f) cis-eQTLs from the ENCODE project. A cumulative distribution function tracks the fraction of SNPs as a function of phred-based P-values for evidence of open chromatin. DRG eQTLs (red) are contrasted against 10 discrete SNPsnap background distributions (grey), and their combined averaged background (black). In inset, a box-and-whisker plot shows the distributions of the phred-based scores for DRG (red) and averaged background (grey). U-test P-value between these distributions is also shown (upper left). A star * indicates Bonferroni-corrected statistical significance. (g) SNP’s deleterious index for gene-level or (h) exon-level cis-eQTLs. A cumulative distribution function tracks the fraction of SNPs as a function of phred-based CADD scores. Deleteriousness is estimated to be proportional to phred-based CADD scores. The background distribution is the same as in e or f, respectively.
We next characterized genetic locations of identified cis-eQTLs. Our analysis showed that for both gene-level and exon-level mRNAs, most eQTLs are “intronic”, followed by “regulatory” (Figure 2c–d, pie). After normalization for minor allele frequencies and gene densities (SNPsnap (Pers et al., 2015), cis-acting eQTLs at the gene level were found significantly enriched in non-synonymous (3.1-fold), regulatory (3-fold), 3′UTR (2-fold) and intergenic (1.4-fold) regions (Figure 2c, bar plot), while depleted in intronic (0.7-fold), upstream (0.6-fold) and downstream (0.5-fold) categories. At the exon level, we observed that non-synonymous (17.5-fold) and regulatory (1.9-fold) regions are likewise significantly enriched, as well as synonymous (16-fold), 5′UTR (4.9-fold) and 3′UTR (6.5-fold), and splice site (5-fold) changes, whereas eQTLs in intergenic (0.1-fold), upstream, downstream and intronic (0.9-fold) regions are depleted.
To examine if there is enrichment for identified cis-eQTLs in open chromatin as an evidence of their functionality, we used the Encyclopedia of DNA Elements (ENCODE) (Birney et al., 2007) database to compare the cumulative distribution functions of these values for cis-eQTLs in DRGs with those of SNPs previously obtained by the SNPsnap server (Pers et al., 2015) as a background. Both gene- and exon-level cis-acting eQTLs displayed significantly lower P-values (or higher phred-scaled values) than the SNPsnap background sets (Figure 2e–f; P=2.2e-4 and 3.1e-5 for gene- and exon-level, respectively). These data are in line with other evidence of substantial statistical enrichment for eQTLs across all tissues within active chromatin (Croteau-Chonka et al., 2015).
As further confirmation of DRG cis-eQTLs functionality, we used the Combined Annotation Dependent Depletion (CADD) database to test if they are predicted to be more deleterious than the population of randomly matched SNPs. Consistently with eQTL data from other tissues (Ionita-Laza et al., 2016), CADD scores of cis-acting eQTLs in DRGs significantly differed from the background distribution for both gene-level (Figure 2g; P=2.4e-2) and exon-level (Figure 2h; P=1.6e-33). Interestingly, this difference is by far more noticeable at the exon-level of cis-eQTLs. If this particular enrichment for pathogenicity within exon-level cis-eQTLs is a reflection of analytical tools allowing identifying the enrichment at exon-level with higher statistical power or there are many more deleterious changes of the substantial impact within alternative exons, it remains to be understood.
Functional characterization of eGenes in DRGs
To further characterize identified eQTLs in DRGs, we next analyzed DRG eGenes: genes for which mRNA or specific exon levels were associated strongly with eQTLs (Figure 3). At the gene level, almost 54% of eGenes code for proteins (Figure 3a; left pie). Among the non-coding genes, the long intergenic non-coding RNAs (lincRNA) class represents the biggest family, at almost 39% (Fig 3a; right pie). Antisense RNAs are also highly represented (14%). The exon-level eGene distribution is substantially different, wherein coding genes represent more than 77% of the total number (Figure 3b, left pie). If we remove eQTLs that are already associated at the gene-level from the analysis, the coding fraction increases up to almost 81% (Fig 3b, right pie). These data suggest that cis-eQTLs contribute more substantially to the expression of specific alternatively-spliced forms of coding genes than overall gene expression. Though only some non-coding RNAs have introns and are thus subject to splicing, most coding genes are expressed as multiple alternately spliced forms (Pal et al., 2011).
Figure 3.

Characterization of eGenes in DRGs. (a) Classes of genes associated at gene-level or (b) exon-level. Pie charts track the relative counts per category. sQTL refers to splicing eQTL; total sQTLs genes classes (left) are compared with the sQTLs classes resulting from the removal of those that are also eQTLs (right). (c) Gene class enrichment for gene-level associations. Horizontal barplots represent enrichment of log-ratios of observed counts to expected counts. (d) Exon class enrichment for exon-level associations. Classes are 5′UTR, 3′UTR, coding sequence (CDS) beginning (CDS beg) and ending (CDS end), intron, transcription start site (TSS), pre-TSS, and non-coding. (e) Expression levels for all genes versus eGenes at gene-level or (f) exon-level. Shown insets are box-and-whisker plots for all genes (grey) or eGenes (red). U-test P-value between the two distributions is shown. (g) Relationship between expression level and cis-eQTL effect size (beta) for gene-level or (h) exon-level associations. Histograms show the distribution of scattered points along the two main axes. Lines show correlations between expression levels and beta (positive beta, green; negative beta, red). Pearson’s correlation coefficient values are indicated at the top. In all panels, a star * indicates statistical significance after Bonferroni correction for gene classes following a binomial test for enrichment of observed over expected.
We then conducted a comparative analysis for enrichment of observed gene categories relative to those found on the expression platform. It shows that protein coding transcripts are statistically significantly enriched (1.1-fold) in the cis-acting eGenes set, while non-coding RNAs are depleted (0.9-fold, Figure 3c, top). A breakdown of non-coding RNA shows that categories that remarkably depart from expected counts are piwi-interacting RNAs (piRNAs; 0.5-fold) and pseudogenes (0.8-fold, Figure 3c, bottom). Thus, though piRNAs lack sequence conservation and display immense complexity (Seto et al., 2007), we found them less frequently associated to eQTLs than expected.
At the exon-level, both 3′ and 5′ UTRs display enrichment (1.7-fold and 1.4-fold, respectively) (Figure 3d), as well as the exons that embed coding sequence ends (1.1-fold) or beginnings (1.2-fold) (Figure 3d). On the contrary, exons that embed exclusively coding regions (CDS; 0.9-fold) or the transcription start site (TSS; 0.8-fold) are associated less often than expected (Figure 3d).
Last, we analyzed how eGene expression levels compare to tissue-wide levels. We found that eGene expression is generally lower at both the gene level (Figure 3e, P=4.1e-4, Bonferroni threshold is 0.05/2 = 2.5e-2), and the exon level (Figure 3f, P=8.3e-45). Furthermore, the expression levels of eGenes and eExons and effect sizes of eQTLs are not correlated, at either gene level (Figure 3g, Pearson’s R=0.05 for beta < 0), or exon level (Figure 3h, R=0.02 for beta < 0, R=0.06 for beta > 0). Only a weak correlation was found for gene-level with positive effect size (Figure 3g, R=0.25, beta > 0; P=4.9e-13). One noticeable difference between gene and exon level was found in the range of effect sizes; for gene level, half the density of beta values lay between −0.2 and +0.2, while for exon level, half the density of beta values lay between −0.4 and +0.4. Only 11% of eGenes are associated with |beta| > 0.5 at the gene-level (Figure 3g), whereas we find almost 36% for exon level (Figure 3h). To summarize, our results suggest that DRG eQTLs regulate in general lower abundant genes, and the effect size of the regulation is more substantial at the level of alternatively spliced forms.
NHGRI catalog for human diseases
One of the primary practical uses for the DRG eQTL dataset is to confer functionality to the results of human association studies, especially in the field of pain-related clinical conditions. To do so, we looked at the enrichment of DRG eQTLs among existing genetic association result hits.
We started our analysis by using the NHGRI catalog (Welter et al., 2014), since it provides a unified list of GWAS results. We analyzed gene-level cis-acting DRG eQTLs and found that some concur with SNPs already associated with human diseases or conditions (Figure 4, Table S4). Given that multiple tissues frequently share eQTLs, an enrichment of DRG eQTLs among known hits from human association studies further validates potential functionality of identified eQTLs. To analyze eQTLs that coincide with GWAS-associated SNPs, we examined how these coincident DRG SNPs (Figure 4a) are also shared by brain eQTLs (Figure 4b) or blood eQTLs (Figure 4c), or by brain and blood (Figure 4d). We observed several coincident SNPs in DRG that were both brain and blood eQTLs (for example, rs1142287), or only brain (rs885224) or only blood (rs11191676) eQTLs. Finally, we also observed a rich set of eQTLs unique to DRG (Figure 4e), not shared by other tissues’ eQTLs. These signals represent additional DRG eQTLs and provide a basis for the functional characterization of future GWAS risk loci.
Figure 4.

Contribution of DRG eQTLs to human diseases. Manhattan plots show association for cis-acting DRG eQTLs at gene- and exon-level. SNPs from the NHGRI catalog reported to be associated with human diseases are highlighted. Strips of alternative yellow/blue colors bring out chromosome loci, while white strips emphasize the Human Leukocyte Antigen locus (HLA; 10x magnification). (a) NHGRI SNPs overlapping with DRG eQTLs are highlighted in red. (b–d) NHGRI SNPs overlapping with shared eQTLs between DRG and other tissues are highlighted in green where eQTLs are shared by (b) DRG and brain, (c) DRG and blood, (d) DRG, brain and blood. (e) Non-overlapping eQTLs unique to DRG in blue. See also Table S4.
Association studies of human pain conditions
The diseases listed in the NHGRI catalog do not include pain-centric conditions, and, currently, there are very few high-powered GWAS available in the field of chronic pain (Chasman et al., 2011; Peters et al., 2013). However, a fair number of candidate gene-based studies have reported genes associated with various chronic pain conditions, including temporomandibular disorders (TMD) (Aneiros-Guerrero et al., 2011; Smith et al., 2011), fibromyalgia (FM) (Smith et al., 2012), and lower back pain (LBP) (Guo et al., 2011). To assemble a list of SNPs currently associated with human pain conditions, we used the Human Pain Genetics Database (http://diatchenko.lab.mcgill.ca/hpgdb/), a hand-curated literature survey of genetic associations related to human pain phenotypes. The database currently contains around 150 genetic variants associated with pain in both gene-candidates and GWAS association studies. For each featured SNP, a full scan at gene and exon levels, and at cis- and trans-acting correspondences was performed. We tested if SNPs that have been associated with human pain phenotypes are also DRG eQTLs, thus pointing to a functional molecular pathophysiology that underlies the original association results. Eleven loci have been identified (Table S5) at FDR 5%. Four loci were found to be eQTLs by more than one test.
We first assessed candidate gene-targeted association studies. The lowest eQTL association P value was identified for SNP rs7911 (Table S5a), whose minor allele was shown to be associated with increased risk of FM from among a large panel of 350 pain-related genes (Smith et al., 2012). This SNP is situated in the 3′UTR of the guanylate binding protein 1 (GBP1) gene (Figure 5a) and is rated fairly inconsequential by the CADD database (phred-scaled score of ~5). In DRG, association of SNP rs7911 with mRNA level of GBP1 is weak (P=1.9e-2). However, SNP rs7911 is also upstream of another guanylate binding protein family member, GBP3 (Figure 5a), with which it is associated very strongly (P=2.5e-20). On the other hand, rs7911 is also associated with GBP1 at the exon level (P=4.89e-07). In both human and mouse DRGs, matching genes from the GBP cluster are found to be expressed (Figure S5b). As GBP-1 and -3 proteins are interferon-inducible contributors to T-cell activation (Forster et al., 2014), viral response (Nordmann et al., 2012), and actin cytoskeleton remodeling factors (Ostler et al., 2014), the minor allele-dependent decrease in expression levels for both GBP3 and GBP1 is a potential indicator of a protective role of one or both proteins in a pathophysiology of FM.
Figure 5.

Contribution of DRG eQTLs to gene-candidates association results with pain phenotypes. (a) Manhattan plot of DRG eQTLs in the GBP genes cluster. The minor allele for SNP rs7911 has been previously associated with increased risk for FM. The same SNP is also an eQTL at gene-level for GBP3 (magenta), and at exon-level for GBP1 (blue). Color-coded insets show gene and exon expression level change as a function of minor allele count. A star (*) indicates statistical significance for reported P-values. (b) Cumulative distribution function of cis-eQTL P-values for all genes (grey) is compared with that of reported pain genes (red) at gene-level or (c) at exon-level. Box-and-whisker plots show difference in P-value distributions. See also Tables S5 and S6.
Although only a handful of GWAS association studies on pain have been published to date (Anttila et al., 2010; Chasman et al., 2011; Cook-Sather et al., 2014; Docampo et al., 2014; Kim et al., 2009; Peters et al., 2013), they have identified many significant and suggestive SNPs. Among the GWAS hits passing FDR correction for multiple testing, the minor allele of rs13361160 (Table S5b) linked with a higher risk of chronic widespread pain (Peters et al., 2013) (CWP) showed association with expression sequence tag AK092568 (P=7.37e-03). AK092568 is not the gene suggested by the original publication, which speculated on the role of chaperonin-containing-TCP1-complex-5 gene (CCT5, P=1.83e-01) and/or family with sequence similarity 173, member B (FAM173B, P=4.36e-01). Interestingly, AK092568 is situated within death-associated protein (DAP) gene, which is a positive mediator of programmed cell death induced by interferon-gamma, and can potentially regulate expression of DAP, which can be found in mouse DRG (GEO set GSE65997 (Wieskopf et al., 2015)), in both neuronal and non-neuronal DRG cell types (2.5 and 1.0 rpkM respectively (Usoskin et al., 2015), Figure S5c). As such, the potential role of AK092568 in pain is at least as likely as CCT5 or FAM173B.
Finally, we compared the distribution of P-values of DRG eQTLs for all genes with the distribution of P-values of DRG eQTLs corresponding to the eGenes previously associated with pain phenotypes (Table S6). We found that at both the gene (Figure 5b) and exon (Figure 5c) levels, the P-values in the eGenes previously associated with pain conditions are lower than in the genome-wide case. This result suggests that a significant proportion of genetic variability contributing to pain phenotypes may act by modifying corresponding RNA levels in DRGs.
Next, we conducted genome wide association tests that evaluated the contribution of DRG eQTLs to pain phenotypes in humans (Figure 6). Data were from the 3,014 participants in the OPPERA (Orofacial Pain Prospective Evaluation and Risk Assessment) GWAS study (Maixner et al., 2011). The phenotypes represented either clinical pain conditions (TMD and low back pain (LBP)) or responses to quantitative sensory testing (QST; mechanical pain, pressure pain threshold (PPT) and heat pain). For all pain phenotypes, significant enrichment for lower P-values within DRG eQTLs was detected (Figure 6a–f). Then, tissue-specific eQTLs’ contribution to tested phenotypes was quantitatively assessed by enrichment measures using either our DRG eQTL dataset, blood eQTL dataset (GTEx_Consortium, 2015), or brain eQTL datasets, grouped as cortical or sub-cortical (Ramasamy et al., 2014). First, we took the sum of observed to expected ratio for P-values of the top 50 contributing eQTLs from QQ plots (Figure 6, QQ bar plot). When comparing the relative contribution of other tissues’ eQTLs to each of the six phenotypes, DRG eQTLs contributed most to LBP (Figure 6a), while in TMD, DRG eQTLs were preceded by cortical brain structures (Figure 6b). Interestingly, PPT showed greatest enrichment for cortical eQTLs followed by DRG eQTLs (Figure 6c) whereas mechanical pain showed greater enrichment for sub-cortical brain eQTLs than DRG eQTLs (Figure 6d). Finally, for heat pain tolerance and threshold, brain-specific and blood eQTLs made the greatest contribution (Figure 6e–f). Another enrichment analysis was concerned with over-representation of observed eQTLs in the top 1, 10 or 100% GWAS hits compared to chance (Figure 6, GWAS curve plot). From the perspective of eQTL over-representation, both LBP (Figure 6a) and PPT (Figure 6c) phenotypes display more observed DRG eQTLs than expected in top 1% GWAS hits among all tissues. For TMD, DRG eQTLs still show significant enrichment as well, following the cortical brain eQTLs. The mechanical pain threshold phenotype seems to be sub-cortical in character.
Figure 6.

Contribution of DRG eQTLs to GWAS results with pain phenotypes. QQ plots (left) depict that DRG eQTLs (red) show significant enrichment for SNPs associated with pain phenotypes relative to all SNPs (gray). Barplots (right, QQ) depict relative contributions of tissue-specific eQTLs to associated phenotype. Tissue contribution is measured as the sum of log2 of QQ plot’s observed to expected ratio for the best 50 eQTLs in that tissue. Curves (right, GWAS) depict enrichment of tissue-specific eQTLs found in the top-ranking GWAS hits. The enrichment is the log2 of observed q/n ratio to the expected Q/N ratio, where q is the observed number of tissue-specific eQTLs in the top n GWAS hits, out of a total Q tissue-specific eQTLs in the total N GWAS hits. Horizontal grey line show no enrichment threshold (i.e. O ~ E, or log2(1)=0). The tested phenotypes are: (a) Low Back Pain; (b) Temporomandibular Disorders; (c) pressure pain threshold; (d) mechanical pain threshold; (e) heat pain threshold; (f) heat pain tolerance.
To identify DRG eQTLs that would potentially interpret association results with pain phenotypes, we searched for alignment of the DRG eQTL signals with that of GWAS associations. Since association strength varies substantially in magnitude between GWAS and eQTL studies, we first converted P-values of association into rank-based scores in their respective association studies (GWAS and eQTL). We assigned a score S from 1 to 0 to all association results in GWAS and eQTL datasets, where a score of 1 was the most strongly associated. We then ranked the SNPs by their Scombined = SGWAS * SeQTL scores to identify eQTLs with the highest probability to contribute to chronic pain conditions (Table S7). Combined scores are evaluated genome-wide, and distribution fitted to a negative exponential; P-value for statistical significance of a combined score can thus be evaluated. One region of the human genome that was systematically statistically significantly associated (FDR 1%) with multiple pain phenotypes was the human HLA region expressing MHCII class genes (Figure 7, Table S7). This region was not only associated with gene expression levels in DRG (Table S7 SeQTL, Figure 7b), but also with all the selected pain phenotypes from the OPPERA cohort, and especially TMD and LBP (SGWAS, Figures 7c–h). The greater overlap of DRG eQTL signal with LBP and TMD association results suggest a greater contribution of DRG eQTLs from HLA locus to these two chronic pain phenotypes rather than to QSTs (Figures 7c–d).
Figure 7.

Associations of HLA gene locus for DRG eQTLs and pain phenotypes, and role in a mouse inflammatory pain model. Dot plots track strength of the association of each SNP along the HLA locus in chromosome 6, with higher scores show stronger association with selected phenotype. Horizontal lines mark the genome-wide statistical threshold of significance at FDR 1%. (a) HLA genes, with arrows indicating the direction of transcription for each gene (forward, yellow; reverse, blue). Footprints of HLA genes are also underlined in other panels. (b) DRG eQTLs. (c–h) pain phenotypes: (c) LBP; (d) TMD; (e) pressure pain threshold in epicondyle; (f) mechanical pain threshold; (g) heat pain threshold; (h) heat pain tolerance. (i) Replication in the UK BioBank cohort for (I) back pain; and (II) facial pain. Plots show cumulative percentage of SNPs as a function of increasing replicative P-values, for SNPs whose eQTL/GWAS OPPERA discovery score is above statistical significance (FDR 1%; pink) and those below (grey). Kolmogorov-Smirnov test between the two curves; *** P < 2e-16. (j) Prolongation of mechanical allodynia in MHCII−/− mice in the CFA inflammatory pain model. Mechanical threshold was measured as 50% withdrawal threshold with von Frey filaments at baseline (BL) before, and 3, 7, 10, 14 and 21 days after CFA injection (dashed line) in the hindpaw of wild type (WT, blue) and MHCII−/− (red) mice. Data are presented as mean +/− s.e.m. * P ≤ 0.05; ** P ≤ 0.01 compared with WT mice. See also Table S7.
In an effort to further confirm association of the HLA locus with pain phenotypes, we sought replication of OPPERA’s discovery results (Figure 7c, d) in the UK BioBank cohort, which is a high-powered prospective study of 150,000 people recruited in the United Kingdom (Sudlow et al., 2015). GWAS on both self-reported chronic (>3 months) back and facial pain show enrichment for lower P-values for SNPs whose combined eQTL/GWAS discovery score is better than the threshold of statistical significance (FDR 1%). There is a significant difference in replication P-values for SNPs above (pink) or below (grey) the threshold for (I) back pain and (II) facial pain (Figure 7i, Kolmogorov-Smirnov test P < 2e-16). For back pain, 90% of high-scored SNPs have P-values lower than 0.2, when only 20% are expected by chance alone. For facial pain, 20% of high-scored SNPs have P-values lower than 0.1, when only 10% are expected by chance alone.
We were then able to examine whether HLA locus region containing MHCII genes contribute to the development and maintenance of inflammatory pain and allodynia in a mouse pain model. MHCII gene expression in human DRGs is mirrored in mouse DRGs by matching genes in the H2 complex (Figure S5d). We compared the mechanical allodynia induced by hind paw injection of complete Freund’s adjuvant (CFA) between wild type (WT) and MHCII−/− mice (Figure 7i). We observed no strain difference in baseline mechanical thresholds prior to CFA injection or peak mechanical allodynia observed 3 days after CFA injection (Figure 7i). However, we observed a significant strain difference over time as MHCII−/− mice maintained their allodynic phenotype at 10 and 14 days, while WT mice returned to baseline values by day 10 post-injection (repeated measures x strain: F5,60=3.8, P=0.03). These data suggest that MHCII is not required for the development of allodynia evoked by CFA, but is important for the recovery phase. The prolonged allodynia observed in MHC−/− mice is consistent with the direction of human association results, in which chronic pain risk alleles correlated with lower MHCII gene expressions (Table S7), and suggests that variable levels of MHCII expression in humans could influence the duration of an inflammatory pain episode.
DISCUSSION
Given the central role of DRGs in pain signal transduction, insights into the molecular genetic architecture of DRGs will be helpful to understand the pathophysiology of human pain conditions. Because much of variability of steady-state protein expression levels can be attributed to mRNA levels (Csardi et al., 2015), any dysregulation of mRNA levels or of alternative splicing patterns can alter the balance of multiple feedback loops and sensory relays intersecting with DRGs. Here, the combination of high-density genotyping arrays with second-generation expression microarrays allowed for a detailed view of the impact of allelic variants on mRNA levels in unique cohort of human DRG. The DRG sample size is among the largest used for eQTL studies of hard-to-obtain human nervous system tissues to date (GTEx_Consortium, 2015; Ramasamy et al., 2014), and thus is highly powered and suitably equipped to detect eQTL signals. Additionally, we used the latest and largest cDNA array expression array platform providing robust hybridization signal detection via resiliency to polymorphic sites, high sensitivity to cost ratio, and fine exon mapping.
First, we compared human DRGs to other tissues. The transcript analysis indicated that DRG expression is very similar to that of sympathetic ganglia; however, it differs significantly from the CNS. On the other hand, DRG eQTLs cluster with brain region eQTL patterns (Figure 1b). Taken together, it seems that although the transcription pattern of DRG substantially differs from CNS, DRG-specific genes are less regulated by common genetic variability in the human population, possibly reflecting their high degree of evolutionary conservation and importance for basic function. On the other hand, the regulatory architecture of the human brain regions and DRG at the level of cis-eQTLs is shared, suggesting that DRG eQTLs could work in concert with eQTLs in other tissues affecting simultaneously multiple functions within the nervous system. Comparison with mouse expression data not only suggests that most eGenes identified in humans are also expressed in mouse (between 51% and 77%), but also that virtually all identified eGenes show more expression in neuronal cell types than in non-neuronal ones. Pathway analyses of DRG eGenes, and eGenes common between DRG and brain regions and blood, show that genetic variability in the human population affects expression of immune response and glutathione derivative biosynthesis genes. The latter is particularly important, as it is shared with blood- and brain-specific gene expression. Given the role of glutathione in neurotoxicity, aging, oxidative stress, and neurodegenerative diseases, our finding provides insights into the pathogenesis of pain conditions.
Next, we undertook a detailed analysis of the identified set of DRG eQTLs to localize eQTLs to functional sites in the human genome (Figure 2) and to distinguish classes of eGenes (Figure 3). These analyses provided converging results and offered a global overview of DRG eQTL architecture that has genome-wide implications outside of the tissue-specific DRG-related expression. Our results support previous discoveries related to basic processes underlying gene regulation driven by genetic variability and provide additional insights (Ramasamy et al., 2014).
The enrichment for 5′ and 3′UTRs, and especially synonymous or non-synonymous changes over regulated regions is in line with previously reported conclusions that internal cis-eQTL signals more substantially change the rate of mRNA degradation than the rate of transcription per se (Ramasamy et al., 2014), though previous studies have only examined the entire coding mRNA category. Particular enrichment for synonymous and non-synonymous changes is possibly an indication of its crucial contribution to mRNA stability, or it may also be a reflection of trans-regulation of transcription through the negative feedback loop by non-synonymous changes rather than direct regulation of mRNA level.
At the level of gene structure, our results suggest that human genetic variability influences the regulation of protein coding genes more than non-coding genes, and preferably low abundant coding genes. Within coding mRNAs, coding nucleotides are conserved while 3′ and 5′ UTRs are under greatest regulation by genetic variability. Interestingly, although there is a strong enrichment for synonymous and non-synonymous changes among eQTLs (Figure 2d), the coding regions of eGenes are depleted. This discrepancy can be either a reflection of generally lower genetic variant density within coding regions or indicate that synonymous and non-synonymous changes mostly control level of nearby 3′ and 5′ UTRs rather than corresponding coding region. Finally, the enrichment for low-abundance genes among eGenes probably suggests easier tuning of lower-abundance genes by common genetic variability.
We also gathered further confirmation of the functionality of our eQTL set through significant enrichment for cis-eQTLs for open chromatin and pathogenicity level. In both cases, exon-level eQTLs showed stronger enrichment than gene-level eQTLs, especially for pathogenicity, providing evidence for substantial contribution of alternative splicing regulation in disease etiology, consistent with reports on other tissues’ eQTLs (Ionita-Laza et al., 2016).
We illustrated the value of our eQTL data set for the interpretation of existing association results (Figure 4), particularly for the field of pain (Table S5, Figure 5). We tested SNP rs7911 of the 3′UTR GBP1 gene, previously associated with FM, in our data set and showed that in fact GBP1 and GBP3 gene expression are associated with corresponding mRNA levels with different strength, and our eQTL data provided stronger evidence for GBP3. The full value of our DRG eQTL resource for the interpretation of GWAS results will be demonstrated as further GWAS associations in the pain field are reported. It appears thus far that none of the multiple migraine GWAS hits seem to be DRG eQTLs. On the other hand, the only significant SNP associated with complex widespread pain by GWAS seems to be in association with EST AK092568, possibly controlling DAP expression.
We also provided evidence for the functional role of identified DRG eQTLs for GWAS-associated loci (Figure 4, 6). Access to a new GWAS dataset from the OPPERA cohort, characterized for a diverse set of pain phenotypes, allowed us to assess the contribution of eQTLs from various tissues, including DRG eQTLs, to clinical and experimental pain (Figure 6). All tested pain phenotypes showed significant enrichment for DRG eQTL top associations. LBP showed the strongest contribution of DRG eQTLs relative to cortical and sub-cortical brain structures and blood by two enrichment analyses, followed by TMD and PPT (Figure 6a–f). Significant enrichment of DRG eQTLs in LBP, TMD and PPT GWAS results supports their critical role in pathogenesis of pain states. Comparison of tissue-specific eQTLs’ contributions to various phenotypes clearly show a distinct DRG eQTL signature despite a noticeable overlap between DRG eQTLs with brain and blood eQTLs (Figure 1e).
The distinct DRG eQTL signature is also observed among SNPs from the NHGRI catalog that matches eQTLs in DRG not shared by other tissues eQTLs (Figure 4e). This suggests that our DRG eQTL collection contains additional functional SNPs associated with human disease, including human pain conditions. Importantly, we found numerous SNPs in the NHGRI catalog that uniquely match eQTLs in DRG, hence promising that our dataset will be useful outside of the pain field, for example to address the pain component of non-pain disorders such as multiple sclerosis or Crohn’s disease (Table S4).
When we searched for an eQTL locus with the strongest association with chronic pain conditions, the eQTLs in the HLA region through MHCII genes consistently showed the best scores, SNPs within the HLA region often demonstrate strong association in many GWAS, including psychiatric and immune disorders (Wang et al., 2015). Replication in the UK BioBank cohort of OPPERA’s findings for back and facial pain further strengthens the evidence for HLA’s role in pathophysiology of chronic pain.
The function of MHCII is to present peptides derived from extracellular sources to CD4+ T helper cells as part of the adaptive immune response. The normal development of peak allodynia at day 3 in MHC−/− mice suggests that the innate immune response to CFA was not affected. However, the extended allodynia that followed suggests that MHCII complex contributes to successful pain resolution after injury. At the clinical level this finding suggests that individuals with lower MHCII expression levels may be at higher risk to develop chronic pain following acute inflammatory injury.
In conclusion, here we present a characterized set of 8,551 DRG eQTLs, a driving force of the genetic susceptibility and maintenance of chronic pain and related somatosensory disorders. The identification of the genes and pathways in which these DRG eQTLs are enriched is essential to understand the functional genotypic structure and pathophysiology of pain perception, and to identify therapeutic targets and biomarkers for pain.
EXPERIMENTAL PROCEDURES
The DRG Cohort
Dorsal root ganglia were collected from human subjects with the consent of first-tier family members. They consist of snap-frozen bilateral lumbar L4 and L5, collected from brain-dead subject following asystole. All procedures were approved by the University of Pittsburgh Committee for Oversight of Research and Clinical Training Involving Decedents and the Center for Organ Recovery and Education, Pittsburgh, PA (http://www.core.org). A total of 214 samples had matched genotype/phenotypes.
DNA Genotyping
Genomic DNA was isolated from frozen and homogenized DRG tissue using a QIAamp DNA Mini kit (Qiagen, Austin, TX, US). The genotyping was done on Illumina’s Infinium Human Omni Express Exome-8 v1.2 chip (~1M probes). Arrays were scanned using Illumina iScan, and data analyzed using Illumina Genome Studio 2011.1 with Genotyping module 1.9.4 (Illumina Inc., San Diego, CA, USA). Imputation of the data followed: pre-phasing was performed with SHAPEIT version v2.r790 (O’Connell et al., 2014) with phase 3 data of the 1,000 genomes project, followed by imputation with impute2 version 2.3.2 (Howie et al., 2012), again with the 1,000 genomes project phase 3 data.
mRNA Expression
RNA was isolated from DRG samples frozen in TRIzol® reagent (Qiagen, Austin, TX, US). Total RNA levels were measured with Affymetrix’ Human Transcriptome Array 2.0 (~70K gene-level probes, ~900K isoform-level probes), scanned with Affymetrix’ GeneChip 3000 G7 Instrument System. Normalization of the microarray data was done in the R statistical software (R Core Team, 2014), with the help of the oligo (Carvalho and Irizarry, 2010) the Platform Design Info for Affymetrix HTA-2_0 (MacDonald, 2015), and the Robust Multi-Array Average (RMA) algorithm (Irizarry et al., 2003). As a default post-processing step, the RMA algorithm performs quantile normalization of the probe intensities.
GWAS of DRG eQTLs
We considered an eQTL to be cis-acting when the distance separating the eQTL to the transcription start site (TSS) of the associated gene was ≤ 1M nucleotides (GTEx_Consortium, 2015; Ramasamy et al., 2014), and trans-acting otherwise. For exons, cis-acting eQTLs were set to be found within 5K nucleotides of the exon-intron boundaries (GTEx_Consortium, 2015), and trans-acting otherwise. All association tests have been performed using Matrix eQTL (Shabalin, 2012), with knowledge of cis- and trans- boundaries. All statistical tests were performed with age, gender, sample’s average expression and genotype’s first two racial eigenvectors per chromosome as covariates. eQTL discovery was performed on 36,552 gene-level (52% of probesets) and 386,507 exon-level (42% of probesets) expression points of intensity level above 4.
Correction for multiple testing was done using the Benjamini-Hochberg procedure, considering false discovery rates (FDR) of 5% for cis-acting, and 1% for trans-acting eQTLs. Estimated number of independent statistical tests was 4.7M for gene-level and 1M for exon-level cis-acting eQTLs, while 6.6M for gene-level and 70M for exon-level trans-acting eQTLs, following linkage-disequilibrium with r2 ≥ 0.5 dependence. List of eQTLs as well as input files for matrix_eQTL can be found on our web portal at http://diatchenko.lab.mcgill.ca/DRG-eQTLs/.
Animal Pain Model
Animal testing was approved by the Downtown Animal Care Committee at McGill University, and conformed to ethical guidelines of the Canadian Council on Animal Care. Eight homozygous 10-week-old mice (4 male; 4 female) with a deletion targeting the region of the H2 locus encoding all genes for MHC class II in mice (Madsen et al., 1999) (MHCII−/−) and age-matched C57BL/6J mice (3 males; 4 females) were purchased from The Jackson Laboratory (stock #003584; B6.129S2-H2dlAb1-Ea/J). All mice received 20-μl unilateral injections of 50% complete Freund’s adjuvant (CFA, Sigma) into the plantar surface of the left hind paw. Mice were tested for mechanical sensitivity using von Frey fibers before at baseline and 3, 7, 10, 14 and 21 days’ post-injection to quantify mechanical allodynia. Statistical analyses were conducted using an α level of 0.05. Data were analyzed by repeated-measures two-way ANOVA followed by a posthoc Sidak’s test with reported P values adjusted for multiple comparisons.
Supplementary Material
eQTL quality control, imputation, and discovery. Related to Figure 1. (a) Pre-imputation statistics. The total number of SNPs is broken down into four classes: those which did not pass QC filters, those with high missing genotyping rate, those with low minor allelic counts and those which depart from the Hardy-Weinberg equilibrium. (b) Post-imputation statistics. After imputation, total of ~82 million SNPs was detected (column B). After applying QC filters, there were 4.9 million SNPs left (column C). For comparison, post-imputation (column E) and pre-imputation (column F) SNP counts, corrected for linkage-disequilibrium, are compared to assess the gain (column G) in genotyping density following the imputation. (c) Statistics for DRG eQTLs. The number of tests that were performed to detect gene-level cis-acting, gene-level trans-acting, exon-level cis-acting and exon-level trans-acting eQTLs in DRG. The threshold p-values calculated are shown at 5% and 1% FDR for cis-acting and trans-acting eQTLs, respectively. (d) Cis-acting, gene-level DRG eQTLs. FDR 5%. (e) Gender-specific, female-only. (f) Gender-specific, male-only. (g) Cis-acting, exon-level DRG eQTLs. FDR 5%. (h) Trans-acting, gene-level DRG eQTLs. FDR 1%. (i) Trans-acting, exon-level DRG eQTLs. FDR 1%.
eGenes in DRGs, blood and brain. Related to Figure 1. eGenes for each tissue can be found in 1-way lists. eGenes lists shared by two (two 2-way) or common to all three (one 3-way) tissues are also included. Pathway Studio analyses for enrichment of GO term “biological process” are adjoined. Statistically significant P-values (Bonferroni threshold of 0.05/number of rows) are highlighted in yellow.
(a) Neuronal versus non-neuronal (NoN) mouse gene expression corresponding to human DRG eGenes. Related to Figure 1. Neuronal cell types include: NF, NP, PEP and TH cluster of cells, where the NF cluster of cells expresses neurofilament heavy chain and parvalbumin, NP - expresses nonpeptidergic nociceptors, PEP expresses peptidergic nociceptors, and TH expresses tyrosine hydroxylase. (b) Master eGenes that are also DRG eGenes. The master eGenes list is derived from a previous analysis of eQTLs in 53 tissues. DRGs master eGenes are indicated.
Lists of DRG eQTLs corresponding to SNPs reported to be associated with diseases or related pathological phenotypes using the NHGRI catalog. Related to Figure 4. The lists also include DRG eQTLs overlapping with those in blood and/or brain, as well as excluding blood and/or brain eQTLs.
SNPs associated with pain phenotypes that are eQTLs in DRGs. Related to Figure 5. (a) In targeted association studies. (b) In genome-wide association studies.
Genes commonly implicated in pain pathways. Related to Figure 5. The list is made from several sources: Algynomics’ Pain Panel V2, Pain Research Forum’s pain gene resource, and Amigo’s GO term 0019233. DRG eQTLs corresponding to pain genes. The eQTLs are cis-acting, at the gene- and at the exon-level.
Comparison of DRG eQTL and OPPERA cohort GWAS results. Related to Figure 7. The lists represent SNPs that show combined P-value-weighted ranked-based scores between SNPs of selected phenotypes and eGenes for which these SNPs are eQTLs for in DRG. Combined scores highlighted in yellow are genome-wide statistically significant at FDR 1% level. Phenotypes are: 1). TMD - Temporomandibular Disorders; 2). LBP- Low Back Pain; 3). PPT- pressure pain threshold; 4). Mech- mechanical pain threshold; 5). Heat Pain Thresh - QST heat pain threshold; 6). Heat Pain Toler - QST heat pain tolerance. Column abbreviations are: PHENO_B, the association’s beta with the GWAS’ phenotype; PHENO_P, the association’s P-value with the GWAS’ phenotype; PHENO_S, the association’s S-score with the GWAS’ phenotype; EQTL_B, _P, and S are same as for PHENO_B, _P and _S, respectively, for DRG eQTLs. COMBINED_S is the combined association’s S-score for the GWAS and the eQTL studies.
Acknowledgments
Funding for this work is kindly provided by the US Cancer Pain Relief Committee Career Development Award “Neurochemistry and Physiology of Human Pain-Processing Nuclei” (IB), by the Canadian Excellence Research Chairs (CERC) Program, Grant CERC08 (LD), and by NIDCR U01-DE017018 grant. This project used the University of Pittsburgh HSCRF Genomics Research Core services. We thank Drs. Simon Gravel and Shamil Sunyaev for fruitful discussions.
ABREVIATIONS
- CNS
Central nervous system
- PNS
peripheral nervous system
- DRG
dorsal root ganglion
- GWAS
genome-wide association study
- SNP
single nucleotide polymorphism
- eQTL
expression quantitative trait loci
- QST
quantitative sensory testing
- HLA
human leukocyte antigen
- MHCII
major histocompatibility complex class II
Footnotes
ACCESSION NUMBERS
The accession number for the datasets reported in this paper is GEO: GSE78150.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
M.P.: eQTL bioinformatics, statistical and computer analyses, manuscript preparation; S.K.: statistical analyses for OPPERA pain cohort; A-J.C-D.: interpretation of eQTL for pain phenotypes, and design, execution, analysis and interpretation of the mouse behavior experiment; S.S.: execution and analysis of mouse behavior experiment; J.S.M.: interpretation of eQTL for pain phenotypes, and design, analysis and interpretation of the mouse behavior experiment; I.B.: funding acquisition, study design, collection of human DRGs; results interpretations; L.D.: funding acquisition, study design, statistical analyses, results interpretation; G.D.S., S.B.S., R.B.F., R.O., J.D.G. and W.M.: OPPERA pain cohort study design and data collection; M.P., S.K., A-J.C-D., G.D.S., S.B.S., R.B.F., R.O., J.D.G., W.M., J.S.M., I.B., and L.D. all participated in the manuscript preparation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aneiros-Guerrero A, Lendinez AM, Palomares AR, Perez-Nevot B, Aguado L, Mayor-Olea A, Ruiz-Galdon M, Reyes-Engel A. Genetic polymorphisms in folate pathway enzymes, DRD4 and GSTM1 are related to temporomandibular disorder. BMC medical genetics. 2011;12:75. doi: 10.1186/1471-2350-12-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila V, Stefansson H, Kallela M, Todt U, Terwindt GM, Calafato MS, Nyholt DR, Dimas AS, Freilinger T, Muller-Myhsok B, et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet. 2010;42:869–873. doi: 10.1038/ng.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benita Y, Cao Z, Giallourakis C, Li C, Gardet A, Xavier RJ. Gene enrichment profiles reveal T-cell development, differentiation, and lineage-specific transcription factors including ZBTB25 as a novel NF-AT repressor. Blood. 2010;115:5376–5384. doi: 10.1182/blood-2010-01-263855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26:2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasman DI, Schurks M, Anttila V, de Vries B, Schminke U, Launer LJ, Terwindt GM, van den Maagdenberg AM, Fendrich K, Volzke H, et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet. 2011;43:695–698. doi: 10.1038/ng.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook-Sather SD, Li J, Goebel TK, Sussman EM, Rehman MA, Hakonarson H. TAOK3, a novel genome-wide association study locus associated with morphine requirement and postoperative pain in a retrospective pediatric day surgery population. Pain. 2014;155:1773–1783. doi: 10.1016/j.pain.2014.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau-Chonka DC, Rogers AJ, Raj T, McGeachie MJ, Qiu W, Ziniti JP, Stubbs BJ, Liang L, Martinez FD, Strunk RC, et al. Expression Quantitative Trait Loci Information Improves Predictive Modeling of Disease Relevance of Non-Coding Genetic Variation. PLoS One. 2015;10:e0140758. doi: 10.1371/journal.pone.0140758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G, Franks A, Choi DS, Airoldi EM, Drummond DA. Accounting for experimental noise reveals that mRNA levels, amplified by post-transcriptional processes, largely determine steady-state protein levels in yeast. PLoS Genet. 2015;11:e1005206. doi: 10.1371/journal.pgen.1005206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diatchenko L, Fillingim RB, Smith SB, Maixner W. The phenotypic and genetic signatures of common musculoskeletal pain conditions. Nat Rev Rheumatol. 2013;9:340–350. doi: 10.1038/nrrheum.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docampo E, Escaramis G, Gratacos M, Villatoro S, Puig A, Kogevinas M, Collado A, Carbonell J, Rivera J, Vidal J, et al. Genome-wide analysis of single nucleotide polymorphisms and copy number variants in fibromyalgia suggest a role for the central nervous system. Pain. 2014;155:1102–1109. doi: 10.1016/j.pain.2014.02.016. [DOI] [PubMed] [Google Scholar]
- Forster F, Paster W, Supper V, Schatzlmaier P, Sunzenauer S, Ostler N, Saliba A, Eckerstorfer P, Britzen-Laurent N, Schutz G, et al. Guanylate binding protein 1-mediated interaction of T cell antigen receptor signaling with the cytoskeleton. J Immunol. 2014;192:771–781. doi: 10.4049/jimmunol.1300377. [DOI] [PubMed] [Google Scholar]
- Frank E, Sanes JR. Lineage of neurons and glia in chick dorsal root ganglia: analysis in vivo with a recombinant retrovirus. Development. 1991;111:895–908. doi: 10.1242/dev.111.4.895. [DOI] [PubMed] [Google Scholar]
- Gereau RWt, Sluka KA, Maixner W, Savage SR, Price TJ, Murinson BB, Sullivan MD, Fillingim RB. A pain research agenda for the 21st century. J Pain. 2014;15:1203–1214. doi: 10.1016/j.jpain.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx_Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo TM, Liu M, Zhang YG, Guo WT, Wu SX. Association between Caspase-9 promoter region polymorphisms and discogenic low back pain. Connect Tissue Res. 2011;52:133–138. doi: 10.3109/03008207.2010.487621. [DOI] [PubMed] [Google Scholar]
- Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med. 2009;360:1759–1768. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016;48:214–220. doi: 10.1038/ng.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jacobs JM, Macfarlane RM, Cavanagh JB. Vascular leakage in the dorsal root ganglia of the rat, studied with horseradish peroxidase. Journal of the neurological sciences. 1976;29:95–107. doi: 10.1016/0022-510x(76)90083-6. [DOI] [PubMed] [Google Scholar]
- Kim H, Ramsay E, Lee H, Wahl S, Dionne RA. Genome-wide association study of acute post-surgical pain in humans. Pharmacogenomics. 2009;10:171–179. doi: 10.2217/14622416.10.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JW. pd.hta.2.0: Platform Design Info for Affymetrix HTA-2_0. 2015. [Google Scholar]
- Madsen L, Labrecque N, Engberg J, Dierich A, Svejgaard A, Benoist C, Mathis D, Fugger L. Mice lacking all conventional MHC class II genes. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:10338–10343. doi: 10.1073/pnas.96.18.10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maixner W, Diatchenko L, Dubner R, Fillingim RB, Greenspan JD, Knott C, Ohrbach R, Weir B, Slade GD. Orofacial pain prospective evaluation and risk assessment study--the OPPERA study. J Pain. 2011;12:T4–11. e11–12. doi: 10.1016/j.jpain.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitin A, Egorov S, Daraselia N, Mazo I. Pathway studio--the analysis and navigation of molecular networks. Bioinformatics. 2003;19:2155–2157. doi: 10.1093/bioinformatics/btg290. [DOI] [PubMed] [Google Scholar]
- Nordmann A, Wixler L, Boergeling Y, Wixler V, Ludwig S. A new splice variant of the human guanylate-binding protein 3 mediates anti-influenza activity through inhibition of viral transcription and replication. FASEB J. 2012;26:1290–1300. doi: 10.1096/fj.11-189886. [DOI] [PubMed] [Google Scholar]
- O’Connell J, Gurdasani D, Delaneau O, Pirastu N, Ulivi S, Cocca M, Traglia M, Huang J, Huffman JE, Rudan I, et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 2014;10:e1004234. doi: 10.1371/journal.pgen.1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostler N, Britzen-Laurent N, Liebl A, Naschberger E, Lochnit G, Ostler M, Forster F, Kunzelmann P, Ince S, Supper V, et al. Gamma interferon-induced guanylate binding protein 1 is a novel actin cytoskeleton remodeling factor. Mol Cell Biol. 2014;34:196–209. doi: 10.1128/MCB.00664-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC, Dahmane N, Davuluri RV. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res. 2011;21:1260–1272. doi: 10.1101/gr.120535.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pers TH, Timshel P, Hirschhorn JN. SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics. 2015;31:418–420. doi: 10.1093/bioinformatics/btu655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MJ, Broer L, Willemen HL, Eiriksdottir G, Hocking LJ, Holliday KL, Horan MA, Meulenbelt I, Neogi T, Popham M, et al. Genome-wide association study meta-analysis of chronic widespread pain: evidence for involvement of the 5p15.2 region. Ann Rheum Dis. 2013;72:427–436. doi: 10.1136/annrheumdis-2012-201742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A language and environment for statistical computing. 2014. [Google Scholar]
- Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, De T, Coin L, de Silva R, Cookson MR, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014;17:1418–1428. doi: 10.1038/nn.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto AG, Kingston RE, Lau NC. The coming of age for Piwi proteins. Mol Cell. 2007;26:603–609. doi: 10.1016/j.molcel.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SB, Maixner DW, Fillingim RB, Slade G, Gracely RH, Ambrose K, Zaykin DV, Hyde C, John S, Tan K, et al. Large candidate gene association study reveals genetic risk factors and therapeutic targets for fibromyalgia. Arthritis Rheum. 2012;64:584–593. doi: 10.1002/art.33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SB, Maixner DW, Greenspan JD, Dubner R, Fillingim RB, Ohrbach R, Knott C, Slade GD, Bair E, Gibson DG, et al. Potential genetic risk factors for chronic TMD: genetic associations from the OPPERA case control study. J Pain. 2011;12:T92–101. doi: 10.1016/j.jpain.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Fan C, Perou CM. Evaluating the comparability of gene expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:261–268. doi: 10.1002/ajmg.b.30272. [DOI] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci. 2015;18:145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yang C, Gelernter J, Zhao H. Pervasive pleiotropy between psychiatric disorders and immune disorders revealed by integrative analysis of multiple GWAS. Hum Genet. 2015;134:1195–1209. doi: 10.1007/s00439-015-1596-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieskopf JS, Mathur J, Limapichat W, Post MR, Al-Qazzaz M, Sorge RE, Martin LJ, Zaykin DV, Smith SB, Freitas K, et al. The nicotinic alpha6 subunit gene determines variability in chronic pain sensitivity via cross-inhibition of P2X2/3 receptors. Sci Transl Med. 2015;7:287ra272. doi: 10.1126/scitranslmed.3009986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Ma Q. Nociceptors--noxious stimulus detectors. Neuron. 2007;55:353–364. doi: 10.1016/j.neuron.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eQTL quality control, imputation, and discovery. Related to Figure 1. (a) Pre-imputation statistics. The total number of SNPs is broken down into four classes: those which did not pass QC filters, those with high missing genotyping rate, those with low minor allelic counts and those which depart from the Hardy-Weinberg equilibrium. (b) Post-imputation statistics. After imputation, total of ~82 million SNPs was detected (column B). After applying QC filters, there were 4.9 million SNPs left (column C). For comparison, post-imputation (column E) and pre-imputation (column F) SNP counts, corrected for linkage-disequilibrium, are compared to assess the gain (column G) in genotyping density following the imputation. (c) Statistics for DRG eQTLs. The number of tests that were performed to detect gene-level cis-acting, gene-level trans-acting, exon-level cis-acting and exon-level trans-acting eQTLs in DRG. The threshold p-values calculated are shown at 5% and 1% FDR for cis-acting and trans-acting eQTLs, respectively. (d) Cis-acting, gene-level DRG eQTLs. FDR 5%. (e) Gender-specific, female-only. (f) Gender-specific, male-only. (g) Cis-acting, exon-level DRG eQTLs. FDR 5%. (h) Trans-acting, gene-level DRG eQTLs. FDR 1%. (i) Trans-acting, exon-level DRG eQTLs. FDR 1%.
eGenes in DRGs, blood and brain. Related to Figure 1. eGenes for each tissue can be found in 1-way lists. eGenes lists shared by two (two 2-way) or common to all three (one 3-way) tissues are also included. Pathway Studio analyses for enrichment of GO term “biological process” are adjoined. Statistically significant P-values (Bonferroni threshold of 0.05/number of rows) are highlighted in yellow.
(a) Neuronal versus non-neuronal (NoN) mouse gene expression corresponding to human DRG eGenes. Related to Figure 1. Neuronal cell types include: NF, NP, PEP and TH cluster of cells, where the NF cluster of cells expresses neurofilament heavy chain and parvalbumin, NP - expresses nonpeptidergic nociceptors, PEP expresses peptidergic nociceptors, and TH expresses tyrosine hydroxylase. (b) Master eGenes that are also DRG eGenes. The master eGenes list is derived from a previous analysis of eQTLs in 53 tissues. DRGs master eGenes are indicated.
Lists of DRG eQTLs corresponding to SNPs reported to be associated with diseases or related pathological phenotypes using the NHGRI catalog. Related to Figure 4. The lists also include DRG eQTLs overlapping with those in blood and/or brain, as well as excluding blood and/or brain eQTLs.
SNPs associated with pain phenotypes that are eQTLs in DRGs. Related to Figure 5. (a) In targeted association studies. (b) In genome-wide association studies.
Genes commonly implicated in pain pathways. Related to Figure 5. The list is made from several sources: Algynomics’ Pain Panel V2, Pain Research Forum’s pain gene resource, and Amigo’s GO term 0019233. DRG eQTLs corresponding to pain genes. The eQTLs are cis-acting, at the gene- and at the exon-level.
Comparison of DRG eQTL and OPPERA cohort GWAS results. Related to Figure 7. The lists represent SNPs that show combined P-value-weighted ranked-based scores between SNPs of selected phenotypes and eGenes for which these SNPs are eQTLs for in DRG. Combined scores highlighted in yellow are genome-wide statistically significant at FDR 1% level. Phenotypes are: 1). TMD - Temporomandibular Disorders; 2). LBP- Low Back Pain; 3). PPT- pressure pain threshold; 4). Mech- mechanical pain threshold; 5). Heat Pain Thresh - QST heat pain threshold; 6). Heat Pain Toler - QST heat pain tolerance. Column abbreviations are: PHENO_B, the association’s beta with the GWAS’ phenotype; PHENO_P, the association’s P-value with the GWAS’ phenotype; PHENO_S, the association’s S-score with the GWAS’ phenotype; EQTL_B, _P, and S are same as for PHENO_B, _P and _S, respectively, for DRG eQTLs. COMBINED_S is the combined association’s S-score for the GWAS and the eQTL studies.