

Abstract
Sustained pulmonary vasoconstriction contributes to the elevated pulmonary vascular resistance observed in pulmonary arterial hypertension. A rise in cytosolic Ca2+ in pulmonary artery smooth muscle cells (PASMCs) is major trigger for pulmonary vasoconstriction. One family of drugs currently being pursued as a potential treatment for pulmonary hypertension are the statins, which act by depleting cholesterol and reducing the number of caveolae. This study aimed at investigating the role of caveolae, membrane receptors, and ion channels (that are potentially located in the caveolae) in agonist-mediated pulmonary vasoconstriction in order to gain a greater understanding of the signaling mechanisms involved in the regulation of pulmonary vascular tone. Chronic treatment of PASMCs with the cholesterol-depleting agent, methyl-β-cyclodextrin (MβCD), significantly reduced the number of cholesterol rich caveolae regions in the membrane. This disruption of cholesterol in caveolae significantly inhibited pharmacomechanical (induced by phenylephrine), but not electromechanical (induced by elevated extracellular potassium concentration), rat pulmonary artery contraction. These results indicate that receptors may functionally colocalize in caveolae in PASMCs and coordinate to regulate pulmonary vascular tone.
Keywords: caveolae, pulmonary artery, store depletion, vasoconstriction
Sustained pulmonary asoconstriction and excessive pulmonary vascular remodeling contribute to the elevated pulmonary vascular resistance and pulmonary arterial pressure in patients with idiopathic pulmonary arterial hypertension (IPAH). A rise in cytosolic Ca2+ concentration ([Ca2+]cyt) in pulmonary artery smooth muscle cells (PASMCs) is a major trigger for pulmonary vasoconstriction and an important stimulus for PASMC proliferation and migration (major contributors to pulmonary vascular remodeling). Indeed, enhanced PASMC proliferation [1, 2] and active pulmonary artery contraction [3] are associated with the increased PASMC [Ca2+]cyt in patients and animals with pulmonary hypertension.
Ion channels and signaling pathways may contribute to the enhanced Ca2+entry necessary for pulmonary vasoconstriction. For example, the activity of voltage-gated K+ channels is inhibited in PASMCs from patients with pulmonary hypertension, which increases [Ca2+]cyt by depolarizing the cell membrane and activating L-type voltage dependent calcium channels (VDCCs). Although antagonists of VDCCs (e.g., nifedipine) have been successfully used to treat some patients with IPAH [4, 5], VDCCs may not alone account for the elevated [Ca2+]cyt; other Ca2+-permeable channels such as canonical transient receptor potential channels (TRPCs) may be involved. These form receptor-operated and store-operated Ca2+ channels (ROC and SOC respectively) [6–8] and are up-regulated in PASMCs from IPAH patients, enhancing Ca2+ entry and contributing to sustained pulmonary vaso-constriction [9] and pulmonary vascular medial hypertrophy [1]. The localization of these receptors and ion channels in the membrane may, therefore, be important in the regulation of pulmonary vascular tone.
Caveolae, membrane lipid-raft domains enriched in cholesterol and sph-ingolipids, have been found in various cell types to play a critical role in modulating cell signal transduction by colocalizing receptors, ion channels, and signaling molecules within plasma membrane microdomains [10, 11]. In smooth muscle and endothelial cells, caveolae are important sites for membrane receptors and channels to associate closely with the endo-plasmic/sarcoplasmic reticulum [12] and for ligand-mediated Ca2+ entry through ROC and SOC [13–15]. Intracellular messenger pathways regulated by cAMP and Ca2+ are tightly integrated and functionally colocal-ized in caveolae [16, 17]. In PASMCs from IPAH patients, up-regulated caveolin-1, the main structural protein in caveolae, increases the formation of caveolae, enhances capacitative Ca2+ entry, and accelerates cell proliferation [18]. Disruption of caveolae in PASMCs from IPAH patients with the cholesterol scavenger, methyl-β-cyclodextrin (MβCD) inhibited capac-itative Ca2+ entry and attenuated cell proliferation [13, 18, 19]. Statins are currently being used in clinical studies for the treatment of pulmonary arterial hypertension (PAH). One existing study showed that sim-vastatin reversed established pulmonary hypertension and attenuated the rate of the progression of the disease [20]. For further advancement in the application of statins to treat PAH, more clinical studies need to be carried out and investigation of the cellular and molecular mechanisms requires clarification. Additionally, in caveolin-1 and -2 knockout mice, endothelial cell proliferation and fibrosis resulted in vascular dysfunction and pulmonary defects, highlighting a fundamental role for caveo-lae in organizing multiple signaling pathways in the pulmonary circulation [21, 22].
In pulmonary vascular smooth muscle and endothelial cells, caveo-lae and the signaling cascades localized in the caveolae may have differential effects on pulmonary vascular tone. In this study, we investigated the role of caveolae in electromechanical and pharmacomechanical coupling mechanisms involved in regulating pulmonary vasoconstriction and vasodilation.
Methods and Materials
Tension Measurement in Isolated Pulmonary Artery Rings
Pulmonary arteries, 2nd to 3rd divisions with an internal diameter of ∼400 μm, were isolated from male Sprague-Dawley rats (100 to 250 g) in accordance with the animal use protocol approved by the Institutional Animal Care and Use Committee of the University of California, San Diego. Adipose and connective tissues were carefully removed, and the remaining arteries were cut into 1 to 2 mm long rings and mounted on stainless steel wire (100 μm in diameter) to a force transducer (Harvard Apparatus, Holliston, MA) in an organ bath (the volume is approximately 0.75 mL). Isometric tension was continuously monitored and recorded using DATAQ data acquisition software (DATAQInstruments). Isolated PA rings were superfused (2 to 2.5 mL/min) with modified Krebs solution (MKS; at 37°C) consisting of (in mM): 138 NaCl, 1.8 CaCl2, 4.7 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 5 HEPES, and 10 glucose (pH 7.4, with 2 M NaOH). For Ca2+-free (0 Ca) MKS, CaCl2 was replaced by equimolar MgCl2 and 1 mM EGTA was added to chelate residual Ca2+. In the high-K+ solution, NaCl was replaced by equimolar KCl to maintain osmolarity.
Resting passive tension was set at 600 mg, an optimal passive tension that gives rise to maximal active tension in rat pulmonary artery rings superfused with 40 mM K+ solution. The vessels were equilibrated for 60 minutes and then challenged with 40 mM K+-containing MKS 3 times to establish the viability of the vessels. In some experiments, endothelium was removed by gently rubbing the lumen of pulmonary artery with a wooden stick. Loss of a functional endothelium was confirmed by abolished relaxation of the rings to acetylcholine (10 μM). In same experiments, results are expressed as absolute tension, measured in mg. After each experiment, the rings were weighed using a fine balance and the active tension induced by agonists was normalized by wet tissue weight and expressed as mg tension per mg wet tissue weight (mg/mg).
Electron Microscopy of PASMCs
Primary cultured PASMCs from patients with idiopathic pulmonary arterial hypertension were prepared from transplant lung tissues and fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer for 2 hours and postfixed in 1% OsO4 in 0.1 M cacodylate buffer for 1 hour; all at room temperature. Sections were stained by uranyl acetate and lead citrate to be observed by electron microscopy (EM). Cells were either treated with MβCD 5 mM for 2 hours or used as untreated controls.
Solutions and Chemicals
All chemicals were purchased from Sigma (St. Louis, MO). Acetylcholine and phenylephrine were dissolved in water to make stock solutions of 10 and 20 mM. All stock solutions were aliquotted and stored at −20°C. On the day of the use, the stock solutions were diluted into the perfusate to make the appropriate final concentrations, as indicated in Results. MβCD was dissolved directly in the MKS on the day of use to a final concentration of 7 mM. The pH was measured in all final solutions with various drugs and reset to 7.4 before experiments.
Statistical Analysis
Data are expressed as mean ± SEM. Statistical analysis was performed using the paired or unpaired Student's t test and values of P < .05 were considered statistically significant.
Results
Acute Application of MβCD Reversibly Enhanced Phenylephrine-Induced Active Tension in Endothelium-Intact Pulmonary Arteries
Previous observations by McDaniel et al. (2001) showed that phenylephrine-induced pulmonary artery contraction occurred via multiple pathways regulating [Ca2+]cyt, whereas high K+-induced pulmonary artery contraction depended wholly upon membrane depolarization and activation of L-type VDCCs [9]. To investigate the role of caveolae in regulating pulmonary artery contractility, we used MβCD, a drug known to deplete membrane cholesterol and disrupt the caveolae, to treat acutely (<2 minutes) or chronically (>1 hour) pulmonary artery rings and then determine the effect of MβCD.
As shown in Figure 1, acute application of MβCD (7 mM) induced a rapid and reversible contraction in the endothelium-intact pulmonary artery rings on top of the tension induced by phenylephrine (2 μM). The MβCD-mediated contraction occurred within 1 to 2 minutes and reached the maximal level after 7 to 8 minutes. Washout of MβCD decreased the tension to the control level within 10 minutes. In pulmonary artery rings preconstricted by phenylephrine, acute superfusion with MβCD increased the active tension by 25.5% ± 4.8% (from 1987.4 ± 193.0 to 2467.4 ± 190.4 mg/mg; P < .001). These results indicate that acute application of MβCD causes a rapid and reversible effect on the endothelium-intact pulmonary artery rings that are preconstricted.
FIGURE 1.
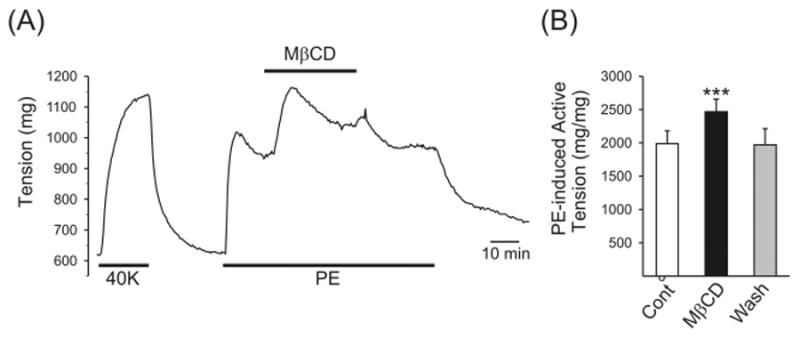
Contractile effect of acute application of methyl-β-cyclodextrin (MβCD) on isolated rat pulmonary artery rings with an intact endothelium preconstricted with phenylephrine (PE). (A) Representative tension record showing a rapid increase in tension subsequent to 7 mM MβCD applied on top of 2 μM PE-induced contraction. (B) Summarized data (mean ± SE; n = 5) showing the amplitude of PE-induced active tension before (cont), during (MβCD), and after (wash) acute application of MβCD. ***P < .001 versus control.
Acute MβCD Treatment-Mediated Contractile Effect Is Dependent of the Endothelium
To examine whether acute application of MβCD causes further pulmonary artery contraction by an endothelium-dependent mechanism, we compared this contractile effect in pulmonary artery rings with an intact endothelium to that where the endothelium was functionally removed. As shown in Figure 2, functional removal of endothelium, confirmed by the absence of 10 μM acetylcholine–induced relaxation and the presence (or enhancement) of 5-hydroxytryptamine–induced contraction (Figure 2A), abolished the rapid MβCD-induced contraction in 25 mM K+– preconstricted pulmonary artery rings and significantly inhibited the contraction in phenylephrine-preconstricted pulmonary artery rings (Figure 2B, lower panel). In endothelium-denuded pulmonary artery rings, the acute M/3CD treatment–induced active tension was reduced from 149.7 ± 26.3 to −4.3 ± 21.4 mg (n = 5; P < .01) in rings preconstricted with 25 mM K+, and reduced from 285.6 ± 38.3 to 54.0 ± 35.6 mg (n = 5; P < .01) in rings preconstricted with 2 μM phenylephrine (Figure 2C). These results indicate that MβCD-mediated acute contractile effect on pulmonary artery rings is endothelium dependent. However, as the response was rapidly reversible, such acute exposure of cells to MβCD may be unrelated to its more chronic effects on cholesterol.
FIGURE 2.
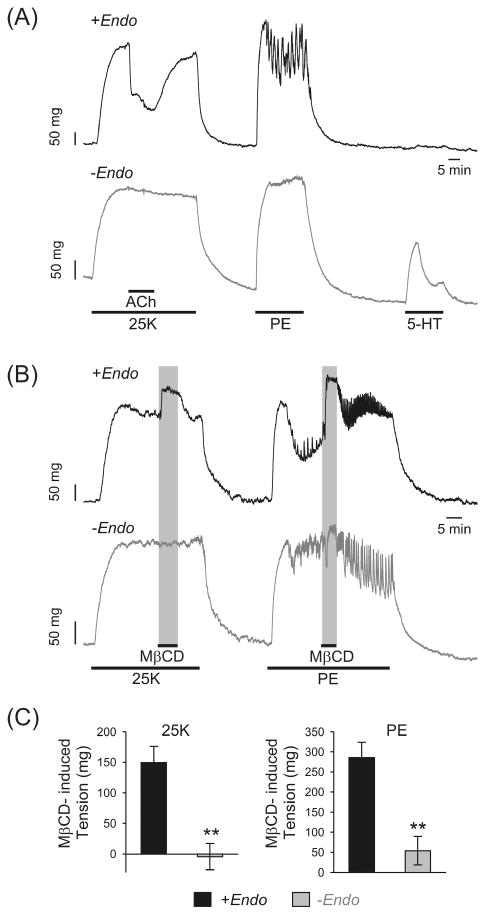
Functional removal of endothelium abolishes the contractile effect of acute application of MβCD on isolated rat pulmonary artery rings. (A) Representative tracings showing the changes of tension in rat pulmonary artery rings with endothelium (+Endo) and with endothelium denuded (−Endo) in response to 25 mM K+ (25 K), PE (2 μM), and 5-hydroxytryptamine (5-HT; 5 μM). Acetylcholine (ACh; 10 μM) induced relaxation in pulmonary artery rings with endothelium (+Endo), but had no effect on tension in pulmonary artery rings with endothelium removed (−Endo). (B) Representative traces showing the effect of acute (5-minute) application of MβCD (7 mM) on 25 K- and PE-induced contraction in pulmonary artery rings with (+Endo) and without (−Endo) endothelium. (C) Summarized data (mean ± SE; n = 5–6) showing changes in 25 K- or PE-induced active tension in response to acute exposure to MβCD. **P < .01 versus +Endo.
Prolonged (or Chronic) Treatment With MβCD Preferentially Inhibited Agonist-Induced Pulmonary Artery Contraction
In contrast to the acute (<2 minutes) contractile effect of MβCD, when pulmonary artery rings were incubated for 1 hour with 7 mM MβCD, the 40 mM K+–induced contraction was negligibly affected (Figure 3), but the 2 μM phenylephrine–induced contraction was significantly inhibited (Figure 4).
FIGURE 3.
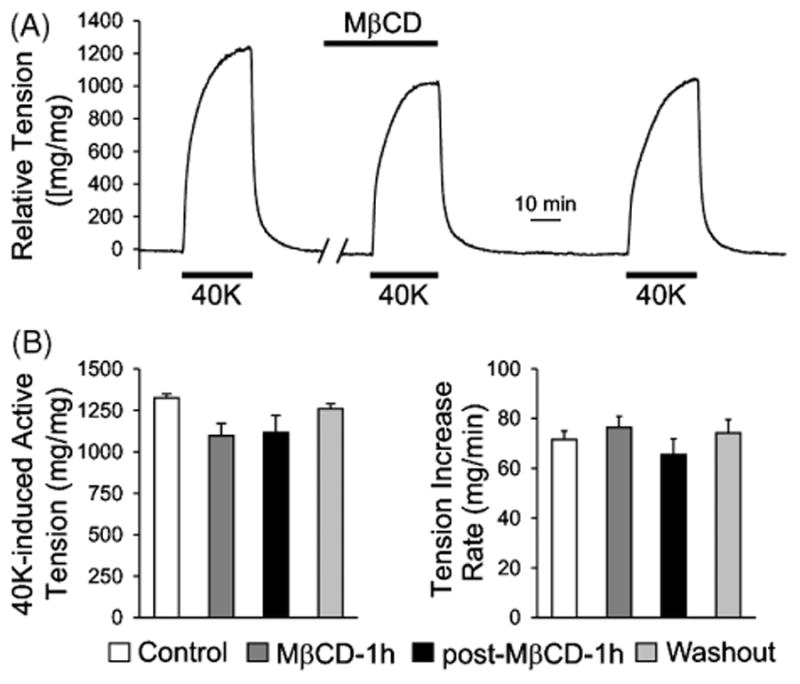
Depletion of cholesterol and disruption of caveolae by prolonged treatment with MβCD has no significant effect on high K+-induced, endothelium-intact pulmonary artery contraction. (A) A representative tension record showing the change of tension in response to 40 mM K+ (40 K) before, during, and after prolonged (1 hour) exposure to MβCD (7 mM). (B) Summarized data (mean ± SE; n = 6-9) showing the amplitude (left) and kinetics (or rate of increase, right) of the 40 K-induced active tension in pulmonary artery rings before (Control), during (MβCD-1 h), after (post-MβCD-1 h), and recovery from (Washout; for 2 hours) treatment with MβCD.
FIGURE 4.
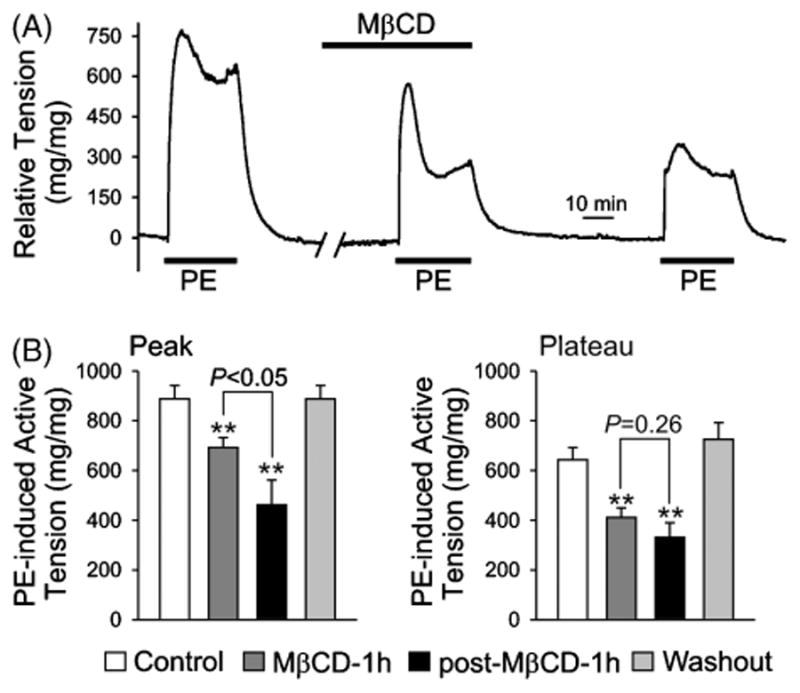
Depletion of cholesterol and disruption of caveolae by prolonged treatment with MβCD reduces 2 μM PE-induced, endothelium-intact pulmonary artery contraction. (A) A representative absolute tension record showing the change of tension in response to 2 μM PE before, during, and after prolonged (1 hour) exposure to MβCD (7 mM). (B) Summarized data (mean ± SE; n = 6–12) showing the amplitude of peak (left) and plateau (right) phase of the PE-induced active tension in PA rings before (Control), during (MβCD-1h), after (post-MβCD-1h), and recovery from (Washout; for 2 hours) treatment with MβCD. **P < .01 versus Control. The amplitude of PE-induced peak contraction 1 hour after MβCD treatment (post-MβCD-1h; black bar) is significantly lower (P < .05) than the amplitude during the 1-hour MβCD treatment (grey bar).
In isolated pulmonary artery rings, a biphasic contraction in response to phenylephrine was observed (Figure 4A), the transient contraction, as previously reported by McDaniel and colleagues [9], was mainly due to Ca2+ release from intracellular stores in PASMCs, whereas the sustained contraction was mainly due to Ca2+ influx through ROC/SOC in PASMCs [9]. As shown in Figure 4B, both the peak (transient contraction) and plateau (sustained contraction) tension induced by phenylephrine in the presence of extracellular Ca2+ were significantly reduced by prolonged treatment with of MβCD. However, the decline of the sustained contraction appeared to take place prior to the decrease in the transient contraction during the 1-hour treatment with MβCD.
As shown in Figure 4B, the phenylephrine-induced peak and plateau tension was decreased from 887.7 ±54.1 and 642.7 ± 49.9 mg/mg to 692.5 ± 39.3 (P < .01) and 410.7 ± 39.0 mg/mg (P < .01), respectively, during the 1-hour MβCD treatment. After approximately 1 hour of washout of MβCD, the phenylephrine-induced contraction did not recover and a further decrease in both the peak (from 692.5 ± 39.3 to 461.8 ± 100.0 mg/mg; P < .05) and plateau (from 410.7 ± 39.0 to 330.7 ± 59.1 mg/mg; P = .2596). After an additional 2 hours of washout, the response to phenylephrine was fully recovered (Figure 4B). There is a significant difference between the peak tension during the MβCD treatment and the tension ∼1 hour after the MβCD treatment (P < .05), whereas there is no significant difference between the plateau tension during the MβCD treatment and the tension ∼1 hour after the MβCD treatment (P = .26). Additionally, at a much higher concentration of PE (20 μM), the effect of MβCD was completely attenuated (Figure 5). It is plausible that, at such a high concentration of PE, maximal activation of surface receptors occurs causing a strong contraction, overpowering the loss of receptors and localized signaling in caveolae disrupted by MβCD. Although the prolonged MβCD treatment significantly reduced the amplitude of both the phenylephrine-induced peak and plateau tension, it did not significantly affect the kinetics of tension development (Figure 6).
FIGURE 5.
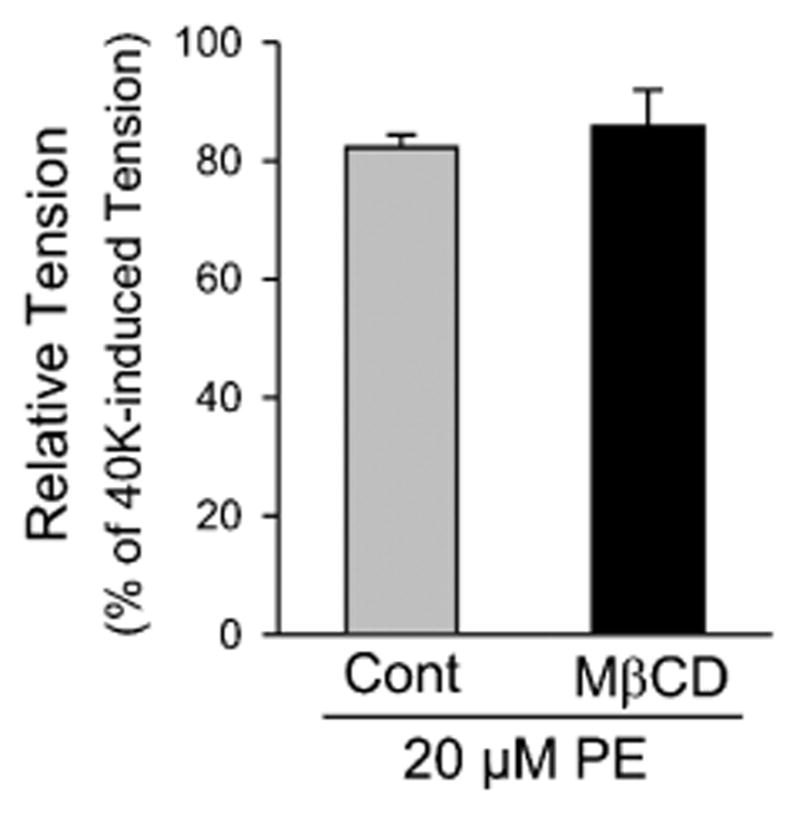
Depletion of cholesterol and disruption of caveolae by prolonged treatment with MβCD has no effect on 20 μM PE–induced, endothelium-intact pulmonary artery contraction. Summarized data (mean ± SE; n = 5) comparing the amplitude of peak of the PE-induced active tension in PA rings before (Control) and after 1-hour pretreatment with MβCD (7 mM).
FIGURE 6.
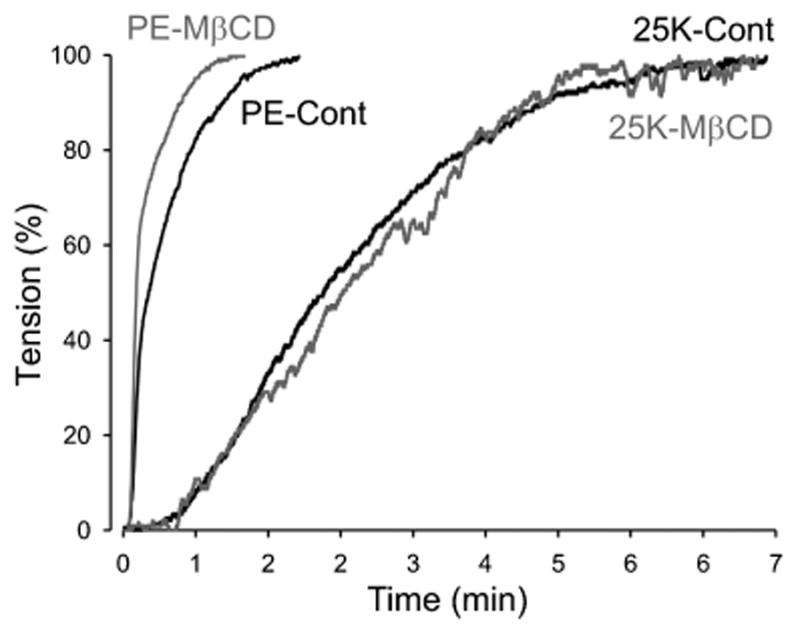
The rate of contraction to PE or KCL is not affected by MβCD. Comparison of the rate of increase in PE- or 25K-induced active tension in endothelium-intact PA rings before (Cont) and during (MβCD) prolonged treatment with MβCD.
Prolonged (or Chronic) Treatment With MβCD-Disrupted Caveolae Domains in the Membrane
In PASMCs from patients with IPAH, we have previously shown that cave-olae are increased due to up-regulation of caveolin-1 and caveolin-2 [18]. Treatment of PASMCs with MβCD significantly decreased the number of caveolae (Figure 7). Although this experiment was performed in isolated cells from IPAH patients using 5 mM MβCD for 2 hours and not rat pulmonary artery rings using 7 mM MβCD, they do prove that MβCD is an effective drug to decrease caveolae structures.
FIGURE 7.
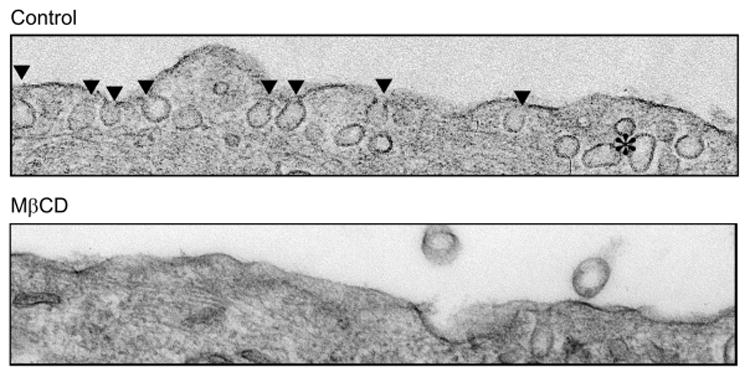
MβCD depletes IPAH PASMCs of cholesterol rich membrane domains. Electron micrographs comparing the membrane morphology from control (top) and MβCD-treated (bottom) PASMCs from IPAH patients. ▾indicates individual caveolae structures in the membrane.
Discussion
Two major excitation-contraction coupling mechanisms are involved in regulating pulmonary vascular tone: electromechanical and pharmacome-chanical. An increase in [Ca2+]cyt in PASMCs is required for both mechanisms to trigger pulmonary vasoconstriction. In electromechanical coupling, membrane depolarization-mediated Ca2+ influx through VDCCs is the major resource for raising [Ca2+]cyt in PASMCs, whereas in pharma-comechanical coupling, agonist-mediated increases in [Ca2+]cyt in PASMCs are due to Ca2+ release from the sarcoplasmic/endoplasmic reticulum and Ca2+ influx through ROC and SOC.
The results from this study demonstrate that (a) acute (2 minutes) application of the cholesterol extracting drug, MβCD caused a reversible contraction in pulmonary artery rings preconstricted with vasoconstrictor, phenylephrine, or high K+, whereas functional removal of endothelium abolished the contractile effect of MβCD; (b) prolonged (≥1 hour) treatment with MβCD significantly inhibited agonist-induced pulmonary artery contraction, but negligibly affected high K+-induced pulmonary artery contraction; and (c) chronic treatment of the cells with MβCD decreased the number of caveolae structures visible in the cell membrane.
Acute Contractile Effect of MβCD on Phenylephrine- and High K+–Induced Pulmonary Artery Contraction: Dependence on the Endothelium
During agonist-induced pulmonary artery contraction, endothelial nitric oxide synthase (eNOS) in endothelial cells is also activated, especially the eNOS in caveolae. Activation of eNOS in endothelial cells may contribute as a somewhat “compensatory” mechanism against excessive contraction. Rapid depletion of the cholesterol and caveolae may inhibit the “compensatory” activation of eNOS and attenuate the subsequent release/production of nitric oxide from endothelial cells, thus leading to the rapid increase in pulmonary artery tension. This may account for the additional endothelium-dependent contraction caused by MβCD and, indeed, MβCD treatment has previously been shown to correlate with reduced eNOS activity and nitric oxide production. As the relationship between caveolin-1 and eNOS is dynamic and a prerequisite to homeostasis, it is also plausible that MβCD may reversibly disrupt eNOS localization by altering the membrane organization. These results also support a recent new perspective on the interrelationship between caveolae and eNOS [23] that endothelial dysfunction observed in hypertension is associated with eNOS dysfunction. A tight eNOS–caveolin-1 association attenuates the ability of caveolin to inhibit signaling moieties and enhances cell proliferation [24]. It is, however, unclear whether the effects of MβCD are directly related to its depletion of caveolae in endothelial cells.
Inhibitory Effect of Prolonged MβCD Treatment on Agonist-Induced Pulmonary Artery Contraction
An opposing effect with a prolonged treatment of MβCD was observed, where the phenylephrine-induced pulmonary vasoconstriction was significantly inhibited. Although some inhibition of the KCl contraction was observed, it was insignificant, suggesting that any functional colocalization of VDCCs and caveolae in these cells is not essential to the control of membrane depolarization and subsequent contractile responses. These results contrast with those reported recently by Bailey and colleagues [25] where alpha-1 stimulation was not affected by MβCD. It is important to note that this study had significant differences to those in the current manuscript using: mice instead of rats, tail arteries of the systemic circulation and not the highly specialized pulmonary arteries, a lower concentration (2 mM) of MβCD, and different experimental techniques [25]. Other studies, in ferret aorta and rat mesenteric arteries do show similarity in the effects of MβCD, with attenuation of the contraction to PE but no significant effects on the KCl-induced increase in tension [12, 26]. These, and our, results indicate that, regardless of the location of VDCCs or whether or not VDCCs are colocalized into caveolae, membrane depolarization (e.g., induced by downregulated Kv channel expression and inhibited Kv channel function) is sufficient to induce adequate Ca2+ influx for efficiently triggering pulmonary vasoconstriction. The agonist-mediated Ca2+ influx or pulmonary vasoconstriction, however, is greatly regulated by the location of receptors, ROC/SOC, signaling molecules, and, perhaps, the sarcoplasmic or endo-plasmic reticulum.
Prolonged treatment of pulmonary artery rings with MβCD disrupts caveolae in PASMCs (and endothelial cells) displaces the colocalized receptors [27], ROC/SOC, signaling proteins, and effectors in caveolae [28]; increases the distance between the plasma membrane and the sarcoplasmic/endoplasmic reticulum membrane; and thus leads to an inhibition of agonist-induced pulmonary artery contraction. As shown in this study using isolated rat pulmonary artery rings, the agonist-induced transient contraction, which is mainly due to Ca2+ release from intracellular stores (e.g., the sarcoplasmic/endoplasmic reticulum), and the agonist-induced sustained contraction, which is mainly due to Ca2+ influx through ROC and/or SOC, were both significantly inhibited by prolonged (≥1 hour) exposure to MβCD. These results indicate that disruption of caveolae functionally disassembles the interaction of receptors (e.g., α-adrenergic receptor) with (a) ROC in the plasma membrane; (b) signaling molecules (e.g., inositol triphosphate [IP3]) that lead to opening Ca2+ release channels (e.g., IP3 receptor) in the sarcoplasmic reticulum membrane; and (c) other ion channels and transporters (e.g., Na+/Ca2+ exchangers) that can mediate inward transportation of Ca2+. Furthermore, disruption of caveolae may also increase the distance between the plasma membrane and the sarcoplasmic reticulum membrane, inhibit store depletion-mediated opening of SOC, attenuates capacitative Ca2+ entry, decrease Ca2+ refilling into the sarcoplas-mic reticulum, and ultimately reduce agonist-induced pulmonary vasocon-striction.
It is important to note that prolonged (≥1 hour) treatment with MβCD had negligible effect on 40 mM K+–induced pulmonary artery contraction, indicating that (a) membrane depolarization-mediated pulmonary artery contraction does not necessarily require colocalization of, for example, VD-CCs in the caveolae; (b) the VDCCs located in caveolae only account for a small portion of total VDCCs in the plasma membrane in PASMCs; (c) membrane depolarization is sufficient to open all VDCCs in the plasma membrane to contribute to Ca2+ influx required for pulmonary vasocon-striction; (d) VDCCs disassembled from the caveolae can still be opened by membrane depolarization induced by high K+; and (e) the localization of VDCCs is not important in the pulmonary artery.
These data further indicate that agonist-mediated pulmonary vasocon-striction relies heavily on the colocalization of receptors with “effectors” (e.g., ion channels and transporters) in caveolae [28]. The close vicinity of the caveolae receptors and channels with the sarcoplasmic reticulum and interaction via signaling molecules such as IP3, diacylglycerol, and protein kinase C is also very important in assuring efficiency of agonist-induced vasoconstriction. Our data also indirectly suggest that receptors, ion channels, enzymes, and signaling molecules may colocalize in caveolae differently in different cell types, such as PASMCs versus pulmonary artery en-dothelial cell. Further study is needed to define what receptor and channel proteins are located in caveolae, and whether proteins and molecules in caveolae are functionally different in cells or arteries that are stimulated by agonists.
In summary, we have demonstrated a complexity in the regulation of agonist-mediated changes in pulmonary vascular tone. Although an exact signaling pathway cannot be precisely established at this stage, this research does highlight, in particular, an essential functional involvement of caveolae in pulmonary vascular contractility. Additionally, potential endothelial dysfunction and alterations in caveolae could interfere with multiple signaling mechanisms including Ca2+ signaling causing proliferation and contraction on PASMCs.
Acknowledgments
This work was supported in part by grants from the National Heart, Lung, and Blood Institute (HL064945, HL054043, and HL66012).
Contributor Information
Christian Schach, Department of Medicine, University of California, San Diego, LaJolla, California, USA.
Amy L. Firth, Department of Medicine, University of California, San Diego, LaJolla, California, USA
Minlin Xu, Department of Medicine, University of California, San Diego, LaJolla, California, USA.
Carmelle V. Remillard, Department of Medicine, University of California, San Diego, LaJolla, California, USA
Hemal H. Patel, Departments of Pharmacology and Anesthesiology, University of California, San Diego, LaJolla, California, USA
Paul A. Insel, Departments of Medicine and Pharmacology, University of California, San Diego, LaJolla, California, USA
Jason X.-J. Yuan, Department of Medicine, University of California, San Diego, La Jolla, California, USA
References
- 1.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistleth-waite PA, Rubin LJ, Yuan JXJ. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA. 2004;101:13861–13866. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JXJ. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol. 2003;284:C316–C330. doi: 10.1152/ajpcell.00125.2002. [DOI] [PubMed] [Google Scholar]
- 3.Madden JA, Dawson CA, Harder DR. Hypoxia-induced activation in small isolated pulmonary arteries from the cat. J Appl Physiol. 1985;59:113–119. doi: 10.1152/jappl.1985.59.1.113. [DOI] [PubMed] [Google Scholar]
- 4.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 5.Rich S, Brundage BH. High-dose calcium channel-blocking therapy for primary pulmonary hypertension: evidence for long-term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation. 1987;76:135–141. doi: 10.1161/01.cir.76.1.135. [DOI] [PubMed] [Google Scholar]
- 6.Lin MJ, Leung GPH, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JSK. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res. 2004;95:496–505. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 7.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 8.Wang M, Bianchi R, Chuang SC, Zhao W, Wong RK. Group I metabotropic glutamate receptor-dependent TRPC channel trafficking in hippocampal neurons. J Neurochem. 2007;101:411–421. doi: 10.1111/j.1471-4159.2006.04377.x. [DOI] [PubMed] [Google Scholar]
- 9.McDaniel SS, Platoshyn O, Wang J, Yu Y, Sweeney M, Krick S, Rubin LJ, Yuan JXJ. Capacitative Ca2+ entry in agonist-induced pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol. 2001;280:L870–L880. doi: 10.1152/ajplung.2001.280.5.L870. [DOI] [PubMed] [Google Scholar]
- 10.Lisanti MP, Scherer PE, Vidugiriene J, Tang Z, Hermanowski-Vosatka A, Tu YH, Cook RF, Sargia-como M. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol. 1994;126:111–126. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sargiacomo M, Sudol M, Tang Z, Lisanti MP. Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J Cell Biol. 1993;122:789–807. doi: 10.1083/jcb.122.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw L, Sweeney MA, O'Neill SC, Jones CJP, Austin C, Taggart MJ. Caveolae and sarcoplasmic reticular coupling in smooth muscle cells of pressurised arteries: the relevance for Ca2+ oscillations and tone. Cardiovasc Res. 2006;69:825–835. doi: 10.1016/j.cardiores.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 13.Bergdahl A, Gomez MF, Dreja K, Xu SZ, Adner M, Beech DJ, Broman J, Hellstrand P, Sward K. Cholesterol depletion impairs vascular reactivity to endothelin-1 by reducing store-operated Ca2+ entry dependent on TRPC1. Circ Res. 2003;93:839–847. doi: 10.1161/01.RES.0000100367.45446.A3. [DOI] [PubMed] [Google Scholar]
- 14.Mathew R, Huang J, Shah M, Patel K, Gewitz M, Sehgal PB. Disruption of endothelial-cell caveolin-1α/raft scaffolding during development of monocrotaline-induced pulmonary hypertension. Circulation. 2004;110:1499–1506. doi: 10.1161/01.CIR.0000141576.39579.23. [DOI] [PubMed] [Google Scholar]
- 15.Beech DJ, Muraki K, Flemming R. Non-selective cationic channels of smooth muscle and the mammalian homologues of Drosophila TRP. J Physiol. 2004;559:685–706. doi: 10.1113/jphysiol.2004.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fagan KA, Graf RA, Tolman S, Schaack J, Cooper DM. Regulation of a Ca2+-sensitive adenylyl cyclase in an excitable cell. Role of voltage-gated versus capacitative Ca2+ entry. J Biol Chem. 2000;275:40187–40194. doi: 10.1074/jbc.M006606200. [DOI] [PubMed] [Google Scholar]
- 17.Rybin VO, Xu X, Lisanti MP, Steinberg SF. Differential targeting of beta-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway J Biol Chem. 2000;275:41447–41457. doi: 10.1074/jbc.M006951200. [DOI] [PubMed] [Google Scholar]
- 18.Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, Swaney JS, Niesman IR, Schermuly RT, Pullamsetti SS, Thistlethwaite PA, Miyanohara A, Farquhar MG, Yuan JX, Insel PA. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J. 2007;21:2970–2979. doi: 10.1096/fj.07-8424com. [DOI] [PubMed] [Google Scholar]
- 19.Bergdahl A, Sward K. Caveolae-associated signalling in smooth muscle. Can J Physiol Pharmacol. 2004;82:289–299. doi: 10.1139/y04-033. [DOI] [PubMed] [Google Scholar]
- 20.Kao PN. Simvastatin treatment of pulmonary hypertension: an observational case series. Chest. 2005;127:1446–1452. doi: 10.1378/chest.127.4.1446. [DOI] [PubMed] [Google Scholar]
- 21.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 22.Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, Kneitz B, Hou H, Jr, Christ GJ, Edelmann W, Lisanti MP. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol. 2002;22:2329–2344. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maniatis NA, Brovkovych V, Allen SE, John TA, Shajahan AN, Tiruppathi C, Vogel SM, Skidgel RA, Malik AB, Minshall RD. Novel mechanism of endothelial nitric oxide synthase activation mediated by caveolae internalization in endothelial cells. Circ Res. 2006;99:870–877. doi: 10.1161/01.RES.0000245187.08026.47. [DOI] [PubMed] [Google Scholar]
- 24.Mathew R, Huang J, Gewitz MH. Pulmonary artery hypertension: caveolin-1 and eNOS interrelationship: a new perspective. Cardiol Rev. 2007;15:143–149. doi: 10.1097/01.crd.0000249381.49138.b9. [DOI] [PubMed] [Google Scholar]
- 25.Bailey SR, Mitra S, Flavahan S, Bergdall VK, Flavahan NA. In vivo endothelial denudation disrupts smooth muscle caveolae and differentially impairs agonist-induced constriction in small arteries. J Cardiovasc Pharmacol. 2007;49:183–190. doi: 10.1097/FJC.0b013e318031d5dd. [DOI] [PubMed] [Google Scholar]
- 26.Je HD, Gallant C, Leavis PC, Morgan KG. Caveolin-1 regulates contractility in differentiated vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2004;286:H91–H98. doi: 10.1152/ajpheart.00472.2003. [DOI] [PubMed] [Google Scholar]
- 27.Schwencke C, Okumura S, Yamamoto M, Geng YJ, Ishikawa Y. Colocalization of beta-adrenergic receptors and caveolin within the plasma membrane. J Cell Biochem. 1999;75:64–72. doi: 10.1002/(sici)1097-4644(19991001)75:1<64::aid-jcb7>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- 28.Schwencke C, Yamamoto M, Okumura S, Toya Y, Kim SJ, Ishikawa Y. Compartmentation of cyclic adenosine 3′,5′-monophosphate signaling in caveolae. Mol Endocrinol. 1999;13:1061–1070. doi: 10.1210/mend.13.7.0304. [DOI] [PubMed] [Google Scholar]