

Summary
The CFTR inhibitory factor (Cif) is an epoxide-hydrolase virulence factor from Pseudomonas aeruginosa, with catalytic activity that perturbs essential host-defense networks. Its targets are largely unknown, but include an epoxy-fatty acid. In this class of signaling molecules, chirality can be an important determinant of physiological output and potency. Here we explore the active-site chemistry of this two-step α/β-hydrolase and its implications for an emerging class of virulence enzymes. In combination with hydrolysis data, crystal structures of 15 trapped hydroxyalkyl-enzyme intermediates reveal the stereochemical basis of Cif’s substrate specificity, as well as its regioisomeric and enantiomeric preferences. The structures also reveal distinct sets of conformational changes that enable the active site to expand dramatically in two directions, accommodating a surprising array of potential physiological epoxide targets. These new substrates may contribute to Cif’s diverse effects in vivo, and thus to the success of P. aeruginosa and other pathogens during infection.
Keywords: Epoxide hydrolase, hydroxyalkyl-enzyme intermediate, virulence factor, epoxy-fatty acids, Pseudomonas aeruginosa, X-ray crystallography, structure-function relationships, enzyme stereospecificity
eTOC Blurb
Hvorecny et al. crystallized covalent intermediates of the epoxide-hydrolase virulence factor Cif. The structures collectively illuminate Cif’s stereospecificity and uncover a new set of fatty-acid substrates. Cif can thus target key host immune signals, exacerbating the damage caused by opportunistic pathogens such as P. aeruginosa.
Introduction
Synthetic chemists and biological systems alike exploit the variety of chemistries associated with three-atom cyclic ethers, known as epoxides. Chemists take advantage of epoxide reactivity, employing epoxides as synthetic intermediates. Biological systems have evolved various uses for epoxides, modulating epoxide reactivity and endogenous activity by varying the substituents that emanate from the moiety. In particular, mammalian systems employ the more chemically stable epoxy-fatty acids (EpFAs) as intermediates or direct signals in a number of circuits controlling critical physiological processes, including inflammation (Serhan et al., 2008). While chemists finely tune reaction conditions to control their syntheses, mammalian systems employ enzymes to regulate the signaling networks that involve epoxides.
Evolution has generated a wide variety of epoxide hydrolases (EHs), the enzymes that convert epoxides into their cognate vicinal diols. For example, mammalian microsomal EH (mEH) defuses damaging epoxide by-products of cellular metabolism, while soluble EH (sEH) controls the levels of key signaling epoxides (Morisseau and Hammock, 2005). As a reflection of its physiological importance, sEH is an active therapeutic target in multiple applications, including mitigating pain and inflammation and regulating vascular tone (Imig and Hammock, 2009; Lazaar et al., 2015; Morisseau and Hammock, 2013). In addition, other organisms, including plants, animals, and bacteria, express a plethora of EHs that perform a wide range of biochemical functions (Arand et al., 2005; van Loo et al., 2006). EHs also offer intriguing synthetic advantages, including reduced environmental impact (“green chemistry”) and enhanced control of reaction stereochemistry (Carlsson et al., 2012; Reetz et al., 2009; Ribeiro et al., 2015; Zhao et al., 2004). However, the structural factors that dictate the stereoselectivity and regioselectivity of epoxide hydrolysis are understood in only a handful of cases (Bauer et al., 2016; Horsman et al., 2013; Morisseau and Hammock, 2005).
Our groups have characterized an EH from Pseudomonas aeruginosa that serves as a trigger for degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) (MacEachran et al., 2007; Swiatecka-Urban, 2005). CFTR plays a key role in fluid and ion homeostasis in epithelial tissues, and its loss of function leads to cystic fibrosis. In airway epithelial cells treated with the CFTR inhibitory factor (Cif), the Ras GTPase-activating protein-binding protein 1 (G3BP1) inhibits the ubiquitin-specific protease USP10. Inhibition of USP10 suppresses post-endocytic de-ubiquitination of CFTR, and the cell targets the ion channel to the lysosome rather than recycling it into the apical membrane (Bomberger et al., 2011).
Cif hydrolyzes both industrial epoxides and 14,15-epoxy-eicosatrienoic acid (14,15-EET), an EpFA that generates pro-resolving signals in human inflammatory responses (Flitter et al., 2016). Like many other EHs, Cif is a homodimeric, α/β-hydrolase that employs a conserved catalytic triad (Bahl et al., 2010a). However, based on its active-site motifs, Cif is the founding member of a distinct subclass of EHs, sharing several features with haloacetate dehalogenases. Notably, in phylogenetic studies, Cif segregates to the boundary between EHs and haloacetate dehalogenases, rather than with the canonical EHs (Bahl and Madden, 2012; van Loo et al., 2006). Furthermore, EHs that share these Cif-like signatures are found in other opportunistic pathogens, including Acinetobacter nosocomialis (Bahl and Madden, 2012; Bahl et al., 2014).
Despite these non-canonical sequence motifs, X-ray crystallography revealed active-site adaptations that enable Cif to preserve the two-step mechanism of epoxide hydrolysis described for canonical EHs (Bahl et al., 2010a). According to the model, coordination by His177 and Tyr239 facilitates the epoxide opening via a nucleophilic attack of the aspartate (Asp129). This process creates a covalent hydroxyalkyl-enzyme intermediate. A water molecule, activated by Cif’s histidine-acid pair (His297 and Glu153), then hydrolyzes the ester intermediate to form the corresponding vicinal diol (Bahl et al., 2015a, 2016). For other well-studied EHs, product release is the rate-limiting step, leading to accumulation of the covalently modified enzyme (Elfström and Widersten, 2005; Morisseau and Hammock, 2005). Although only trace quantities of corresponding adduct are observed for WT Cif, the mutant CifE153Q was expected to permit substrate binding and initial nucleophilic attack, but not hydrolysis. As predicted, this mutation traps 2-(bromomethyl)oxirane (rac-epibromohydrin, EBH) or 2-butyloxirane (rac-1,2-epoxyhexane, EpH) in the active site, as hydroxyalkyl-enzyme intermediates covalently attached to the Asp129 nucleophile (Bahl et al., 2016).
CifE153Q not only confirms the two-step model. In combination with our crystallographic toolkit, it also provides an unprecedented opportunity to investigate the enzyme’s specificity. Other active-site mutations within Cif do not afford us this possibility; they either alter the active-site volume (D129S, H177A, Y239F), prevent protein folding (H297A), or regain catalytic activity (D129N) (Bahl et al., 2010a, 2015a, 2016). In this study, we leverage the CifE153Q mutant to investigate active-site constraints within this emerging class of pathogenic EH virulence factors. This work provides a window into the regioisomeric and enantiomeric preferences of Cif. Our results also reveal how the interplay of active-site constraints and substrate properties co-determine specific activity. In addition, we identify a number of new epoxide targets, which may contribute to Cif’s virulence effects in vivo.
Results and Discussion
Stereochemistry of Attack for Monosubstituted Substrates
We first compared the adduct structures formed upon incubation with either EBH or EpH. Notably, we observed only one enantiomer for each substrate trapped in the active-site pocket, even though we incubated the enzyme with racemic mixtures of each substrate (Bahl et al., 2016). To inspect the positioning of the covalently linked intermediates, we aligned all main-chain atoms within 10 Å of the Cα atom of Asp129 from our previous structures: CifE153Q (PDB ID 4DMC), CifE153Q-EBH (4DNF), and CifE153Q- EpH (4DNO) (Figure S1). For each enantiomer, the carbon attacked by Asp129 occupies the same position, and the remaining atoms of the opened epoxide rings overlay (Figures 1A and 1B, shadowed bonds in 1B). Based on the geometries and positions of the covalently linked intermediates, we surmised that they originate from an SN2 attack on the unsubstituted carbon (C-1) of S-EBH and substituted carbon (C-2) of S-EpH, respectively (Figure 1C), consistent with mechanisms seen in other α/β epoxide hydrolases (O’Hagan and Schmidberger, 2010). We also compared specific activities of these two racemic substrates in the same reaction conditions, noting that Cif hydrolyzes EpH faster (Figure 1D).
Figure 1. Comparison of Cif’s interactions with EBH and EpH.
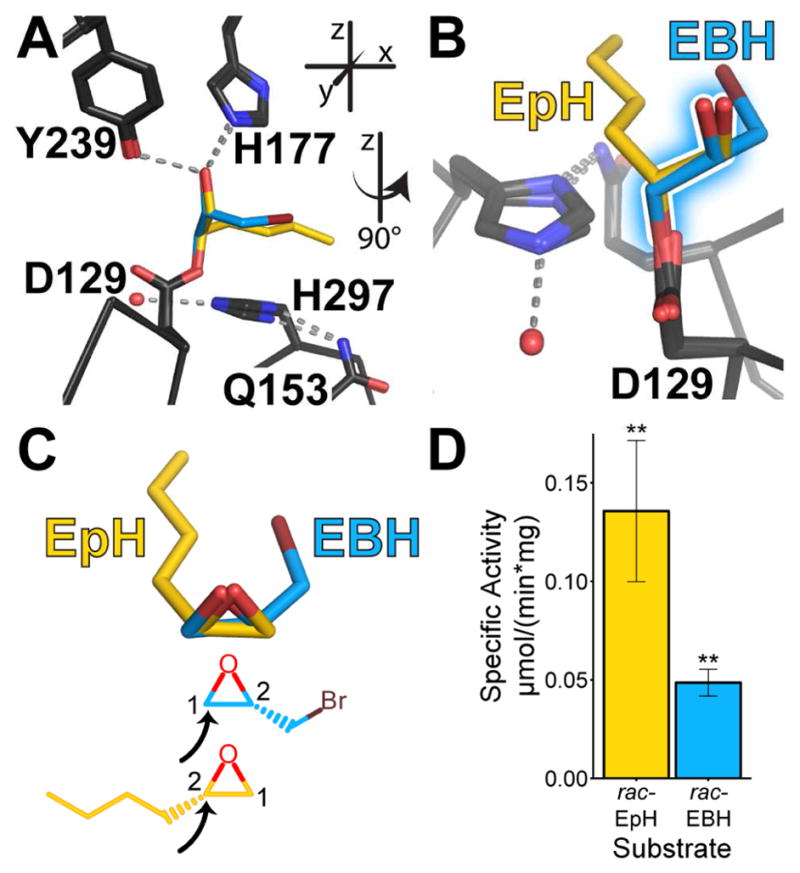
A. View of the active-site residues of the aligned structures, showing the overlay of the CifE153Q intermediates formed with EpH (yellow carbons) and EBH (blue carbons). Inset indicates a descriptive coordinate system used throughout this manuscript. B. View after 90° rotation of the structures, with the proto-diol bonds shadowed. C. Superposition and chemical diagrams of the proposed bound epoxides as inferred from these crystal structures. The black arrows indicate the carbon attacked by Asp129. D. Specific activity of rac-EpH and rac-EBH at a 2 mM concentration. Mean ± S.D.; **, p < 0.01. See also Figure S1.
The alignments also highlight differences between the adducts. As Figure 1C shows, CifE153Q traps S- EBH and S-EpH by attacking differentially substituted carbons. This generates adducts with proto-diol O-C-C-O atoms that superimpose, but aliphatic alkyl “tails” that do not (Figure 1B). Except for the catalytic side chains, the residues lining the binding pocket are largely hydrophobic. They form a surface that is compatible with the alkyl chain of S-EpH, but not with the larger bromine substituent of R-EBH, which would overlay with S-EpH. This may explain why CifE153Q preferentially captures S-EBH. The S enantiomer avoids clashing while also potentially forming a halogen bond in solution with sulfur atom of Met272, located 5 Å from the bromine. In addition, the hydrophobic character of the pocket may dictate Cif’s preference for EpH, which agrees with our general observation that Cif prefers hydrophobic, monosubstituted or cis-disubstituted substrates (Figure S2).
Our structural data suggest that Cif may hydrolyze only one enantiomer from the racemic mixture. However, it is possible that both enantiomers are substrates, but that the trapping process or the crystal lattice favors the S adduct. To explore this possibility, we first assayed Cif’s ability to hydrolyze each enantiomer of several hydrophobic, monosubstituted substrates, beginning with EpH (Figure 2A). To our surprise, when provided with enantiomerically pure substrates, Cif not only turns over R-EpH, but does so faster than S-EpH, with an estimated enantiomeric ratio (E) of 5. We also crystallized the S-EpH intermediate trapped by CifE153Q (Tables 1 and S1). As expected, the positions of the opened epoxide ring and the hydrocarbon substituent of CifE153Q-S-EpH very closely parallel those seen in the structure generated by incubation of CifE153Q with rac-EpH (Figure 2B, Figures S3 and S4A). We were unable to crystallize an adduct formed with the R-EpH leaving open the formal possibility that lattice constraints led to selective crystallization of the S-EpH adduct from a racemic mixture.
Figure 2. Hydrolysis and trapped intermediates of EpH and EpO.

A. Specific activity of Cif for EpH enantiomers. B. Overlay of intermediates in crystal structures of rac-EpH (grey carbons) and S-EpH (yellow carbons). C. Specific activity of Cif for EpO enantiomers. D,E. Intermediates in the crystal structures of S-EpO (yellow carbons) and R-EpO (blue carbons), respectively. F. Adduct found in the crystal structure of rac-EpO (yellow carbons). G. Chemical diagrams of the proposed carbons attacked based on the intermediates from the structures. The black arrows indicate the carbon attacked by Asp129. Mean ± S.D.; *, p < 0.05; **, p < 0.01; ***, p < 0.001. See also Table S1 and Figures S3 and S4.
Table 1.
Summarized Structure Statistics Ia
| PDB ID | 5TNL | 5TNN | 5TNM | 5TNK | 5TNI | 5TNQ | 5TNP | 5TND | 5TNJ | 5TNE | 5TNS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein | CifE153Q | CifE153Q | CifE153Q | CifE153Q | CifE153Q | CifE153Q | CifE153Q | CifE153Q | CifE153Q | CifE153Q | CifD129S |
| Substrate | S-EpH | S-EpO | R-EpO | rac-EpO | S-SOx | R-SOx | rac-SOx | ECH | VCH | cSO | ECH |
|
| |||||||||||
| Space Group | C2 | C2 | C2 | C2 | C2 | C2 | C2 | C2 | C2 | C2 | C2 |
| Resolution (Å) | 1.80 | 1.95 | 1.70 | 1.65 | 1.80 | 1.50 | 1.85 | 1.55 | 1.65 | 1.75 | 1.75 |
|
| |||||||||||
| Rwork/Rfree (%) | 15.8/18.7 | 14.8/19.0 | 14.6/17.1 | 15.1/17.6 | 14.3/17.4 | 16.1/18.3 | 16.7/20.7 | 15.0/17.3 | 14.5/17.1 | 15.0/18.4 | 15.3/18.3 |
|
| |||||||||||
| Ramachandran plotb (%) | 97.3/2.7/0.0 | 97.7/2.3/0.0 | 97.9/2.1/0.0 | 97.7/2.3/0.0 | 96.7/3.3/0.0 | 97.5/2.5/0.0 | 97.6/2.4/0.0 | 97.6/2.4/0.0 | 97.7/2.3/0.0 | 97.4/2.6/0.0 | 97.4/2.6/0.0 |
| Bond-length RMSD | 0.006 | 0.007 | 0.005 | 0.006 | 0.006 | 0.006 | 0.006 | 0.006 | 0.006 | 0.006 | 0.006 |
| Bond-angle RMSD | 0.799 | 0.800 | 0.806 | 0.804 | 0.814 | 0.837 | 0.799 | 0.846 | 0.818 | 0.807 | 0.827 |
For definitions and full data-collection and refinement statistics, see Table S1.
favored/allowed/outliers
We addressed this question directly in studies with the closely related substrate 2-hexyloxirane (rac-1,2-epoxyoctane, EpO) (Figure 2C). EpO exhibits a hydrolysis pattern similar to that of EpH: Cif hydrolyzes R-EpO more quickly than S-EpO (E ~ 11). In contrast to EpH, using EpO we were able to crystallize adducts formed with rac-EpO as well as each of the enantiomorphs R- and S-EpO (Tables 1 and S1, Figures S3 and S4C–E). The intermediate seen with rac-EpO shows evidence only for trapping of the S enantiomer, in analogy to the adducts formed with rac-EpH (Figure 2F). However, when incubated with either R-EpO or S-EpO, CifE153Q traps each respective enantiomer, attacking C-1 of R-EpO and C-2 of S-EpO (Figure 2D, 2E, and 2G). All three of these structures demonstrate the same geometry of attack on the epoxide ring as seen in the CifE153Q-EBH and CifE153Q-EpH structures (Figure 1C and Figure 2G). All three EpO structures show indistinguishable lattice parameters (Tables 1 and S1, Figure S1). Since both adducts are compatible with the observed crystal lattice, S-EpO most likely outcompetes R-EpO for active-site trapping when the enzyme is provided with the racemic mixture.
To test the generality of these observations, we also quantified Cif’s ability to hydrolyze other commercially available, enantiomeric substrate pairs. Cif hydrolyzes 2-(4-nitrophenyl)-oxirane (NPO, Figure S5) and 2-phenyloxirane (styrene oxide, SOx, Figure 3A). NPO turnover is faster for the R enantiomer than for the S enantiomer (E ~ 2); however, as with rac-EBH, turnover was comparatively low. We did not obtain crystals of the CifE153Q-NPO complexes. Cif also hydrolyzes the R enantiomer of Sox faster than the S enantiomer (E ~ 4). We crystallized the enantiomers of SOx and the racemic mixture with the CifE153Q mutant (Figures 3B, 3C, and 3D, Tables 1 and S1, Figures S3 and S4F–I). Each demonstrates the same pattern of attack as described above for the enantiomers of EpH and EpO (Figure 3E). However, in contrast to the apparent dominance of the S enantiomer from rac-EpH and rac- EpO substrates, we observe nearly equal occupancies for the hydroxyalkyl-enzyme intermediates of S- SOx and R-SOx following trapping of the racemic mixture with CifE153Q (Figure 3D).
Figure 3. Hydrolysis and trapped intermediates of SOx.
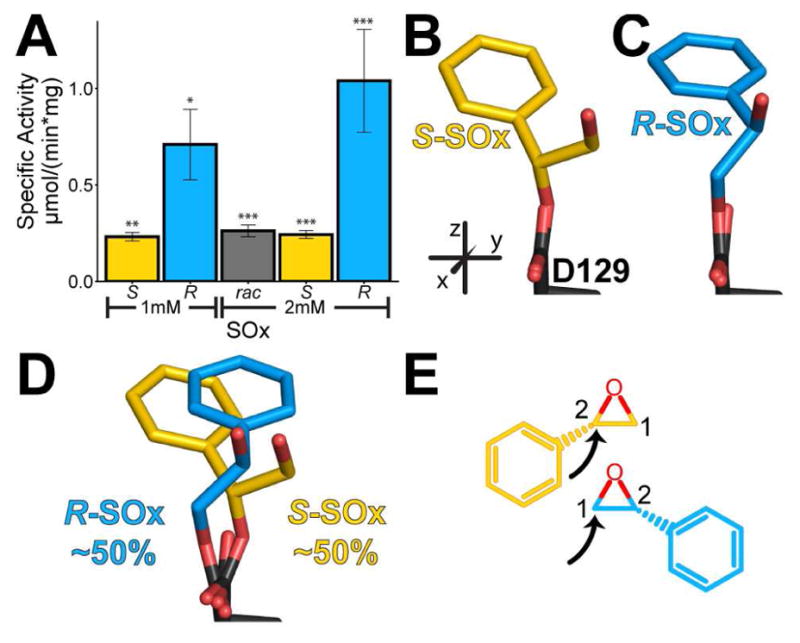
A. Specific activity of Cif for SOx enantiomers. B, C. View of intermediates in the crystal structures with S-SOx (yellow carbons) and R-SOx (blue carbons), respectively. D. View of intermediates in the crystal structures with rac-SOx. E. Chemical diagrams of the proposed carbons attacked by Cif based on the crystal structures. The black arrows indicate the carbon attacked by Asp129. Mean ± S.D.; *, p < 0.05; **, p < 0.01; ***, p < 0.001. See also Table S1 and Figures S3, S4, and S5.
Taken together, our structural data show that Cif attacks distinct carbons on the epoxide moieties of each enantiomeric pair. The initial nucleophilic attack occurs on the more substituted carbons (C-2) of S-EpO, S-SOx, and S-EpH, and on the less substituted carbons (C-1) of R-EpO, R-SOx, and S-EBH. Whichever carbon is targeted, the structures of the resulting adducts suggest that an SN2 attack occurs opposite the bond to the epoxide oxygen. As a result, attack on C-1 of R-EpO, R-SOx, and S-EBH results in adducts that retain the configurations of the substrate epoxide carbons. However, attack on C-2 of S-EpO, S-SOx, and S-EpH inverts the configuration of those stereocenters, flipping from the S configuration in the epoxide to an R configuration in the diol. Assuming hydrolytic release follows the mechanism described for other EHs, Cif hydroxyalkyl-enzyme intermediates will resolve into R-products for either enantiomer of EpO and SOx. Such enantioconvergence has previously been described, e.g., for the EH from Solanum tuberosum, and can potentially be leveraged for stereospecific synthesis (Carlsson et al., 2016; Monterde et al., 2004).
Steric Determinants of Substrate Attack and Product Release
The pattern of attack on the epoxide ring described in the previous section should affect the rate of turnover of each enantiomer. While our assay is not suitable for detailed kinetic analysis, we can make some qualitative observations. Specific-activity assays demonstrate that Cif turns over the R enantiomer more quickly for all of the epoxide pairs tested thus far (Figures 2A, 2C, and 3A). Nevertheless, S is the dominant enantiomer found trapped in the pocket of CifE153Q incubated with the racemic mixtures of EpH or EpO. Furthermore, the predominance of the S enantiomer does not appear to reflect lattice constraints. For EpO, each enantiomer trapped individually leads to interchangeable asymmetric unit assemblies (Figure S1). As a result, the S enantiomers of EpH and EpO presumably are trapped more quickly by the mutant enzyme and thus dominate the intermediates seen in the crystal structures. For SOx, the trapping frequency is presumably similar between the enantiomers.
In measuring the specific activity of racemic mixtures of individual substrates, we allow the enantiomers to compete for the active site of the enzyme. At the same time, we measure substrate turnover, which includes not only formation of the intermediate seen crystallographically, but also its hydrolytic release. For both EpO and EpH the specific activities of Cif for racemates at 2 mM concentration are indistinguishable from the sum of the specific activities for the R and S enantiomers at 1 mM each (Figure 2A and 2C). In contrast, turnover of the 2 mM racemic mixture of SOx is substantially slower than turnover of 1 mM R-SOx (Figure 3A). This observation was confirmed in a mixture of 1 mM R-SOx and 1 mM S-SOx, suggesting that the S enantiomer can inhibit turnover of the R enantiomer under these conditions.
If S enantiomers are trapped at least as rapidly, but R enantiomers are turned over more quickly, it is likely that the hydrolytic step is faster for the R substrates. To investigate the structural underpinnings of potential hydrolysis differences between the enantiomers of each substrate, we aligned the crystal structures of the trapped S-EpO and R-EpO enantiomers (Figure 4A) and the S-SOx and R-SOx enantiomers (Figure 4B), using least-squares fitting of the main-chain atoms within 10 Å of the Asp129 Cα atom. These alignments highlight a pattern. When CifE153Q traps the R or S enantiomers, the Asp129 residues are generally angled either towards or away from the catalytic water, respectively, altering the geometry between the water and Asp129. Averaged over the multiple complexes present in the crystallographic asymmetric unit, this shift creates an average distance between the Oδ atoms of 0.25 ± 0.00 Å for CifE153Q-S-EpO vs. CifE153Q-R-EpO, (arrowheads in Figure 4A), and 0.40 ± 0.09 Å for CifE153Q-S-SOx vs. CifE153Q-R-SOx (Figure 4B). While only a small change in location for the Oδ, this distance is larger than coordinate error (Figure S1) and reflects a shift of multiple linked atoms. It is also associated with a change in the average angle between the catalytic water and carboxylate of Asp129: 6.5 ± 1.9° for the EpO-intermediate structures, and 14.1 ± 0.4° for the SOx-intermediate structures, as highlighted in Figure 4B.
Figure 4. Comparisons of S and R structures of substrates EpH, EpO, and SOx.
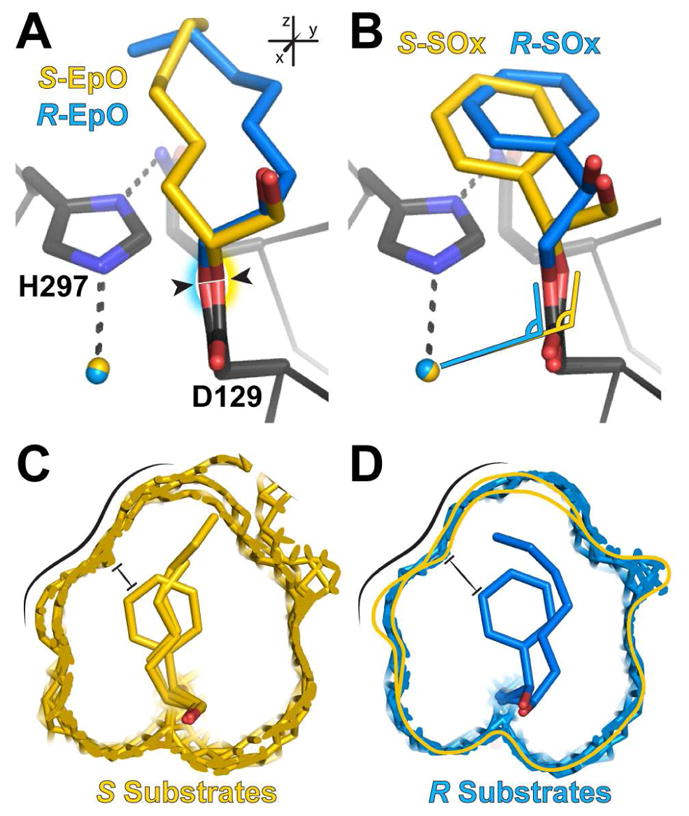
A. Overlay of the S-EpO (yellow carbons) and the R-EpO (blue carbons) adduct structures. Shading highlights the direction of the shift of Asp129. B. Same as A, but with SOx. Angle symbols highlight geometric changes. C. Overlay of adduct S-substrate structures, in which the van der Waals surface of the atoms enclosing the active-site pockets with S adducts are shown in yellow mesh. D. Overlay of adduct R-substrate structures, in which the van der Waals surface of the atoms enclosing the active-site pockets with R adducts are shown in blue mesh and yellow line indicates the active site containing the S enantiomers. Black lines in C and D highlight the clashing wall of the S substrates and the pocket and the black bars indicate the point of closest contact for S-SOx. See also Figure S1.
These differences, although modest, may help to determine the relative rates of hydrolysis, and thus turnover for each enantiomer. The offsets can be explained by the interactions of the intermediates with the walls of the active site, whose overall shape remains similar among the full set of substrates (Figure 4C and 4D). The substituents of the S enantiomers are pressed against the wall of the active site highlighted in Figure 4C, and thus held further away from Asp129. This same surface is not as restrictive for the R enantiomers (Figure 4D), allowing for closer approach.
Disubstituted Substrates as Probes of Attack Position
The adduct structures examined thus far indicate that Cif has defined rules for the carbon it attacks in monosubstituted substrates. All of the substrates described so far occupy most of the active-site volume and do not leave much space for variations in substrate positioning during binding and initial attack (e.g., Figure 4C and 4D). To test whether Cif’s stereochemical requirements relax with smaller substrates, we tested and confirmed hydrolysis of the small, disubstituted epoxides 7-oxabicyclo[4.1.0]heptane (epoxycyclohexane, ECH) and 3-vinyl-7-oxabicyclo[4.1.0]heptane (4-vinyl cyclohexene epoxide, VCH) (Figure 5A).
Figure 5. Hydrolysis and trapped structure of ECH.

A. Hydrolysis of ECH and VCH at a 2 mM concentration. B, C. The two ECH hydroxyalkyl-enzyme intermediates (carbons in purple). The carbon of attack determines the intermediate trapped. D. View of both intermediates in the active site, in which the van der Waals surface of the active site is shown in purple mesh. The double-headed arrow indicates the axis of rotation that allows for either carbon to be attacked. Mean ± S.D; *, p < 0.05; ***, p < 0.001. See also Table S1 and Figures S3, S4, and S6.
Following incubation with ECH, crystallization of CifE153Q revealed two distinct intermediates linked to Asp129 (Figure 5B and 5C, Tables 1 and S1, Figures S4J and S4K). Occupancy refinement of the two intermediates demonstrates that they are formed by patterns of attack on the epoxide ring similar to those observed with the monosubstituted substrates, confirming that active-site geometry drives selective attack on the carbon positioned closest to the catalytic water (Figure 5B). However, in this case Cif is able to attack either carbon of the epoxide ring in ECH (Figure 5C). This substrate is much smaller than the other tested substrates, and thus able to move and rotate within the active site in a manner not possible for the other substrates (Figure 5D). We observed the same pattern for VCH (Tables 1 and S1, Figures S4L, S4M and S6).
We were also able to capture the ECH molecule in complex with CifD129S (Tables 1 and S1, Figure S6). His177 and Tyr239 coordinate the epoxide oxygen into position, and the cyclohexane ring fills much of the space above Ser129. However, the positioning of ECH in the CifD129S mutant is not compatible with nucleophilic attack, as the bound ECH molecule sterically overlaps with the position occupied by Asp129 in the WT enzyme. In the mutant, the replacement of Asp129 with a serine significantly increases the space available in the active site, allowing ECH to adopt a non-productive orientation. The combination of the CifE153Q and CifD129S structures thus strongly suggests that the snugness of substrate fit within the active site determines the fidelity of carbon attack by Asp129, and that the Asp129 side chain is itself an important component of that fit.
Disubstituted Substrates as Probes of Active-Site Flexibility
Thus far, all of the substrates described have either been compact (ECH and VCH) or monosubstituted (EBH, EpH, EpO, NPO, and SOx). Previous work has shown that Cif can hydrolyze (2R,3S)-2,3-diphenyloxirane (cis-stilbene oxide, cSO), but neither (2S,3S)-2,3-diphenyloxirane nor (2R,3R)-2,3-diphenyloxirane (trans-stilbene oxide, tSO) (Bahl et al., 2010a). Hydrolysis of these two molecules has long been used to probe EH active-site geometry (Morisseau and Hammock, 2005; Oesch et al., 1986). However it remained unclear how a bulky substrate like cSO (Figure 6A) could fit in the small active site of Cif, especially considering the tight pocket constraints for S-SOx (Figures 4C). To explore how the active site accommodates cSO, we crystallized CifE153Q after incubation with cSO (Tables 1 and S1, Figures S3 and S4B).
Figure 6. Residues shifted for accommodation of cSO and 14,15-EET.

A. Schematic representations of cSO (grey bonds), R,R-tSO (blue carbons) and S,S-tSO (yellow carbons). B–E. Slices through Chain A (B–C, E) or Chain B (D) of the CifE153Q-cSO active site. The residues and van der Waals surfaces are shown in red for the atoms lining the active site that shift to accommodate cSO. All others protein residues and surfaces are shown in dark grey, with oxygen atoms in red. B. In the 60% of Chain A containing the cSO intermediate (grey carbons), residues Leu200 and Phe203 (red bonds) shift to accommodate the intermediate, which opens the active site to solvent. C. Top view of the active site containing the cSO intermediate. D. In Chain B, the active site is not open. The grey outline illustrates the position that would be occupied by cSO. E. Top view of the active site in C, in which cSO is replaced with the R,R-tSO (blue outline) and S,S-tSO (yellow outline) enantiomers modeled as described in the text. F. View of tunnel through Cif generated by the presence of the 14R,15S-EET intermediate (grey carbons), with colors as described for B–E. G. Three residues shift to accommodate the α tail at the right-hand side of panel F. Residues from CifE153Q without substrate are shown with dark grey carbons, and residues from CifE153Q-14,15-EET with red carbons. See also Table S1 and Figures S1, S2, S3, and S4.
We were able to place cSO in the active sites of only two of the four molecules in the asymmetric unit of the crystal, and in both cases it exhibits only partial occupancy (~60%), reflecting incomplete labeling most likely associated with limited substrate solubility. Nevertheless, the labeled subset of active sites reveals how Cif accommodates the hydroxyalkyl-enzyme intermediate of cSO through the formation of a new tunnel that leads from the enzyme pocket to bulk solvent (opened, Figure 6B and 6C). This tunnel is created by rotation of two side chains, Leu200 and Phe203 (red in Figure 6), away from the active site. In the monomers without cSO, we observe all residues in a closed conformation (closed, Figure 6D) that resembles CifE153Q without substrate covalently linked (PDB ID: 4DMC; RMSD = 0.12 Å). Modeling of a bound cSO (grey outline, Figure 6D) shows that the opening of the pocket is required: cSO could not fit in the closed CifE153Q active site without clashing sterically with Phe203.
Modeling also reveals why Cif cannot accommodate tSO. We aligned the tSO enantiomers with the hydroxyalkyl-intermediate of cSO, overlaying the oxygen atom of tSO with the oxygen coordinated by His177 and Tyr239, while keeping the epoxide ring in the plane containing the cSO proto-diol atoms (Figure S1). The cSO active-site pocket does not accommodate either tSO enantiomer (Figure 6E). R,R- tSO (Figure 6A, blue) clashes with Asp129, while S,S-tSO (Figure 6A, yellow) clashes with the α-helix ending with Asp129. Thus, hydrolysis requires that the epoxide, the oxyanion hole (His177 and Tyr239), and the catalytic Asp129 be arranged in a geometry that cannot accommodate tSO and other trans substrates (Figure S2).
The steric constraints preventing tSO binding also affect the monosubstituted epoxides. This wall of the active site (highlighted in Figure 6E) helps to establish the positions we observe in our crystal structures for each enantiomer of EpH, EpO, SOx, and EBH. The clashes seen in Figure 6E for R,R-tSO and S,S-tSO would also occur for the mono-substituted epoxides, if the epoxide ring and its substituents were rotated 180° to swap the positions of the oxir ane carbons.
Epoxy-Polyunsaturated Fatty Acids
In addition to cSO, we also observed significant active-site side-chain shifts for another previously identified substrate, racemic cis-14(15)-epoxy-5Z,8Z,11Z-eicosatrienoic acid (14,15-EET, PDB ID: 5JYC) (Flitter et al., 2016). 14R,15S-EET opens the active site to bulk solvent in two locations, creating a tunnel through the enzyme (Figure 6F). Cif accommodates the ω end of 14,15-EET via the same residue shifts that allow cSO to fit, namely Leu200 and Phe203 (Figure 6F). The carboxylic acid of the molecule protrudes through the same opening that is stabilized by inhibitor binding, but is closed in all the other CifE153Q structures (Figure 6G) (Bahl et al., 2015a; Kitamura et al., 2016).
The expansion of the active site seen with 14,15-EET should enable Cif to accommodate other cis EpFAs. To explore this possibility, we assayed Cif’s ability to convert epoxides derived from a number of poly-unsaturated fatty acids (PUFAs) by surveying a broad population in a mixed-substrate format (Figure 7A). Compared to human sEH (HsEH), which prefers the epoxide moiety at mid-chain positions (n-6, n-9, and n-12), Cif exhibits different specificity, hydrolyzing epoxides with rates that decrease from the n-3 to the n-6 positions.
Figure 7. Hydrolysis and structures of EpFAs.

A. Mixed substrate hydrolysis assay. Bar colors for Cif correspond to crystal structures listed below. B. Hydrolysis of the EDP series at a 1 mM concentration. C. Hydrolysis of the EEQ series at a 1 mM concentration. D. Aligned structures of hydroxyalkyl-enzyme intermediates of 16,17-EDP (carbons and mesh surface in cyan) and 14,15-EEQ (carbons and mesh in pink). E. Aligned structures of hydroxyalkyl-enzyme intermediates of 19,20-EDP (carbons and mesh in teal) and 17,18-EEQ (carbons and mesh in purple), with CifE153Q without substrate (carbons and mesh in black). For D and E, the proposed trapped epoxides as inferred from the crystal structures are depicted as chemical diagrams, with the arrows indicating the carbon attacked by Asp129. F. Assay of enantiomeric excess for rac-cis-19,20-EDP. G. Examples of residue shifts near the -COOH of the EpFA intermediates, with colors as listed above. H. Residue changes between the EpFA trapped structures and the CifE153Q unoccupied structure occur primarily within the cap domain, as mapped onto a Cif monomer. The cap domain residues are shown in grey (no shift), yellow (loop shift in CifE153Q unoccupied protein), red (main chain shifts of more than 0.5 Å or side chains with conformational differences), and blue (catalytic residues). Mean ± S.D; *, p < 0.05; ***, p < 0.001. See also Table S2 and Figures S3, S5, and S7.
Cif also appears to have a much narrower preference range when compared to HsEH, showing hydrolysis for only 5 of the 14 epoxides in the assay conditions, as compared to the 9 molecules hydrolyzed by HsEH. Cif hydrolyzes a number of epoxides derived from the epoxy-docosapentaenoic acid family (EDPs), as well as 14,15-EET. The preference for the more terminal epoxides holds true when Cif hydrolyzes the EDP family members in separate reactions (Figure 7B).
Surprisingly, given Cif’s ability to hydrolyze the EDPs and 14,15-EET, we did not observe detectable hydrolysis of epoxides from the epoxy-eicosatetraenoic acid family (EEQs) in the competing format (Figure 7A). When tested in individual reactions, however, Cif hydrolyzes the n-3 and n-6 EEQs in a similar pattern as compared to the EDP family (Figure 7C). This pattern indicates that Cif must have higher substrate specificity for the EDP molecules and 14,15-EET, then it does for any of the EEQ molecules. To explore the structural basis for the different relative rates of turnover of the EpFAs, we trapped intermediates of the n-3 and n-6 EDPs and EEQs using the CifE153Q mutant (Figures 7D and 7E, Tables 2 and S2, Figures S3 and S7). Cif trapped the R,S epoxide enantiomers of both 19,20- and 16,17-EDP in the active site, as was seen for 14,15-EET. The substrate intermediates are oriented so that the ω-end occupies space deep within the active-site pocket. The ω-tails of the n-6 substrates, 16,17-EEQ and 14,15-EEQ, require the additional space created by the Leu-Phe shift (Figure 8D). The n-3 epoxides, on the other hand, do not need to access the additional tunnel space: the short carbon tails fit into the pocket without the movement of residues Leu200 and Phe203 (Figure 8E).
Table 2.
Summarized Structure Statistics IIa
| PDB ID | 5TNR | 5TNF | 5TNG | 5TNH |
|---|---|---|---|---|
| Protein | CifE153Q | CifE153Q | CifE153Q | CifE153Q |
| Substrate | 16,17-EDP | 19,20-EDP | 14,15-EEQ | 17,18-EEQ |
|
| ||||
| Space Group | C222 | C2 | C2 | C2 |
| Resolution (Å) | 1.80 | 1.75 | 1.75 | 2.10 |
|
| ||||
| Rwork/Rfree (%) | 16.1/18.9 | 14.7/17.4 | 15.0/18.1 | 18.7/23.6 |
|
| ||||
| Ramachandran plotb (%) | 98.1/1.9/0.0 | 98.0/2.0/0.0 | 98.1/1.9/0.0 | 97.5/2.5/0.0 |
| Bond-length RMSD | 0.007 | 0.005 | 0.006 | 0.007 |
| Bond-angle RMSD | 0.855 | 0.804 | 0.843 | 0.826 |
For definitions and full data-collection and refinement statistics, see Table S2.
favored/allowed/outliers
In these experiments, the EpFAs incubated with CifE153Q were racemic mixtures of the cis enantiomers. Notably, for all of our EpFA hydroxyalkyl-enzyme intermediate structures, we only find one of the epoxide enantiomers, the R,S molecule, trapped in the active site. This echoes what we see for CifE153Q incubated with racemic mixtures of either EpH or EpO substrates, as discussed above. Using 19,20-EDP, the racemic EpFA with the highest turnover, we assayed Cif’s preference for either enantiomer (Figure 7F). Cif turns over the R,S molecule much more rapidly than the S,R molecule, and by 120 minutes, only the 19S,20R epoxide remains detectable in the mixture. Overall, this produces an enantiomeric ratio (E) of 8.1 ± 0.9 in favor of 19R,20S-EDP. Compared to other EHs, Cif displays a modest preference for one enantiomer over the other. sEH shows much lower enantioselectivity (E ~ 1), while mEH and the EH from the soybean plant demonstrate much greater enantioselectivity (E ~ 200) against 9,10-epoxysteric acid (Bellucci et al., 1996; Summerer et al., 2002; Zeldin et al., 1995).
A comparison across the structures containing the trapped EpFAs demonstrates trends among the shifts caused by all four substrates (Figure 7G). In order to accommodate the α-carboxylate tails, all of the trapped EpFAs open the active site at residues Met272, Phe164, and Leu174, as we see for 14,15- EET. Phe164 and Leu174 show considerable differences in movement among the PUFAs. Val175 also shifts in all structures. There is evidence for a pucker flip of Pro165 and a 180° rotation of His207. His207 in particular is intriguing, as mutation of this residue decreases hydrolysis of EBH in a previous study, implicating it in hydrolysis (Bahl et al., 2015a).
The cap domain, which contains residues 155 through 242, contains the major substrate-associated steric rearrangements within the active site, including resides at both ends of the tunnel (Figures 6 and 7) and other amino acids we have not specifically cited (Figure 7H). We observe this even though flexibility within the cap domain is partially restricted by lattice contacts. As noted by other researchers, cap domains contribute to EH substrate selectivity, and this appears also to be true for Cif, permitting accommodation of a wider range of substrates than originally predicted (Bahl et al., 2010a; Barth et al., 2004; Lindberg et al., 2010; Schiøtt and Bruice, 2002).
Conclusions
Through a combination of X-ray structures and specific-activity measurements, we have outlined the substrate preferences for a bacterial epoxide hydrolase that sabotages human signaling networks (Flitter et al., 2016; MacEachran et al., 2007; Swiatecka-Urban, 2005). While the active site is constrained, it also exhibits a surprising degree of localized conformational flexibility that greatly expands the range of substrates it can accommodate. Based on these active-site expansions, it is likely that additional EpFA substrates exist for Cif. To test this hypothesis, we incubated Cif with an EpFA from the epoxy-eicosadienoic acid family (cis-14(15)-epoxy-8Z,11Z-eicosadienoic acid, 14,15-EED) (Figure S5). Cif hydrolyzes this molecule. While much remains to be learned about the four EpFA categories evaluated in Figure 7, even less is known about 14,15-EED (Deline et al., 2015). Cif may be employed as a molecular scalpel, helping to dissect yet-to-be-described EpFA signaling pathways.
Based on this study, we can extrapolate a number of rules concerning Cif. This EH is likely enantioconvergent for the monosubstituted epoxides and potentially enantioconvergent for the EpFAs. This is a direct result of steric restrictions within the active site. A combination of substrate chemistry and the constraints of the active site conspire to slow S-enantiomer turnover. In fact, because of tight steric packing, the S enantiomers may need to access the additional space observed when cSO and the n-6 EpFAs are bound (Figures 6B, 6F, and 7D). Proper geometry for catalysis may only be achieved through rearrangement of the active site that allows the substituents of the S substrates to move into the space, properly aligning the Asp129 with the catalytic water and thus raising the free-energy barrier for the S substrates.
The Cif active site in the fully open formation contains a sharp turn at the catalytic Asp129 (Figures 6F and 7D). Despite the expanded volume, the turn prevents accommodation of trans substrates and forces the conformations adopted by the monosubstituted substrates (Figure 1B and 6E). This active-site bend explains why classic sEH inhibitors do not block Cif catalytic activity (Bahl et al., 2015a). The sEH inhibitors function by inserting themselves into the active site tunnel, where the catalytic Asp and the oxyanion hole residues coordinate the 1,3-disubstituted amide and urea moieties of the inhibitors (Gomez et al., 2006). As the groups are planar, the bent active site of Cif cannot accommodate them. However, terminal amide and urea moieties enhance the effectiveness of Cif inhibitors, as they engage active-site residues by reaching into the enzyme from one end of the tunnel only (Kitamura et al., 2016).
Cif’s substrate preferences differ from the list of epoxides effective in inducing Cif expression (Bahl et al., 2016; Ballok et al., 2012). In addition, when deployed by P. aeruginosa in the human body, Cif will encounter many substrates in parallel. Therefore, the turnover of substrates in situ is more closely approximated by our racemic mixtures (Figures 2A, 2C, and 3A) and mixed-substrate experiments (Figure 7A), in which turnover rates differ based on the population of substrates present in a given niche, as well as Cif’s proclivity to hydrolyze them. In an extreme case, a substrate that is effectively hydrolyzed in isolation, such as 17,18-EEQ (Fig. 7C), may exhibit almost undetectable hydrolysis in the presence of competitors (Fig. 7A). This increases the importance of identifying the entire population of epoxides in the lungs, where Cif is deployed, as has been completed for enzymes outside of the EH family (Bishop-Bailey et al., 2014; Ringholz et al., 2014; Schäuble et al., 2013; Yandek et al., 2013).
Employing our strategy with other enzymes using two-step reactions could help probe the constraints of the active site as well as the kinetics of turnover. This may be especially pertinent for other EHs deployed by pathogens, aiding in the identification of a more complete set of substrate targets (Morisseau, 2013; Spillman et al., 2016). As we have shown, this approach can reveal unexpected active-site rearrangements and their effects on substrate selectivity. Knowledge of active-site flexibility and expansions also allowed us to improve our first generation Cif inhibitor, increasing the specificity and affinity for the next generation (Bahl et al., 2015a; Kitamura et al., 2016). Our study, along with others, highlights the possibility for inducible fit in this class of enzymes, greatly expanding the range of potential substrates, particularly among physiological signaling epoxides (Morisseau, 2013; Selvan and Anishetty, 2015; Xue et al., 2016).
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dean Madden (drm0001@dartmouth.edu).
Experimental Model and Subject Details
Strains used in Protein Purification
E. coli TOP10 strains with the relevant AmpR plasmid were streaked from frozen stocks onto LB/Amp agar plates. 10 mL LB/Amp broth starter cultures were inoculated from several colonies on the plates and grown for ~16 hours. These cultures were transferred into 1 L of terrific broth with Amp and arabinose and grown for ~72 hours. (Bahl et al., 2015a, 2016).
Method Details
Protein Purification
Recombinant carboxyl-terminal His6-tagged Cif protein, and CifE153Q and CifD129S mutant proteins encoded on the pMQ70 vector were expressed and secreted by the E. coli TOP10 strain grown in terrific broth for 72 hours (Bahl et al., 2015b, 2016; Shanks et al., 2006). Cells were pelleted out of the medium, and the supernatant was pumped over a 5 mL GE HisTrap HP column. The His-tagged protein was eluted with a 10–500 mM gradient of imidazole. The protein was concentrated with Millipore Amicon Ultra 10K MWCO centrifugal filter and dialyzed into 100 mM NaCl, 20 mM sodium phosphate, (pH 7.4 at room temperature) for biochemical assays and crystallization.
Specific-Activity and Basic Hydrolysis Assays
Specific-activity and hydrolysis assays were performed using an adapted adrenochrome reporter assay (Bahl et al., 2014; Cedrone et al., 2005). For specific-activity assays, various concentrations of Cif protein were incubated separately with 1 or 2 mM of each epoxide substrate in 2% (v/v) DMSO in 20 mM sodium phosphate, pH 7.4 (at room temperature) and 100 mM NaCl at 37°C for 10 minutes. Concentrations were adjusted to achieve linearity for each substrate reaction. Racemic mixtures used in Figures 2 and 3 were both purchased from suppliers and generated by mixing pure enantiomers to confirm the hydrolysis results reflect 1:1 mixtures. For basic hydrolysis assays, 37.1 μM Cif was incubated with 1 mM of the epoxide substrate in 2% (v/v) DMSO in 20 mM sodium phosphate, pH 7.4 and 100 mM NaCl at 37°C for 30–60 minutes. Reactions from both assay conditions were quenched with NaIO4 in 90% (v/v) acetonitrile to a final concentration equimolar to initial substrate concentrations and were then incubated at room temperature for 30 minutes. Epinephrine was added in excess to react with the residual NaIO4. Precipitated protein was removed by centrifugation. A490 values were measured in a Tecan spectrophotometer using a 96-well plate and compared to a standard curve generated with the cognate diol. If the cognate diol was not commercially available, the closest chemically related diol was used. Please refer to the Key Resource Table for a full list of epoxides and diols used in these assays. The enantiomeric excess (ee) and enantiomeric ratio (E) were calculated as described previously: E = (v1×[S2])/(v2×[S1]) (Chen et al., 1982).
Substrate selectivity analysis
A mixture of 14 EpFAs derived from linoleic acid, arachidonic acid, eicosapentaenic acid, and docosahexaenoic acid at 0.1 mM each in DMSO was prepared from purified regioisomers. Purified recombinant HsEH (0.4 μg/mL) and Cif (5 μg/mL) in 100 μL of sodium phosphate buffer (0.1 M, pH 7.4) containing 0.1 mg/mL BSA were each incubated with the mixture of EpFAs (each at a final concentration of 1 μM). After incubation at 37°C for 20 minutes (HsEH) or 120 minutes (Cif), the reactions were stopped by the addition of 100 μL of methanol containing 400 nM of 12-(3-cyclohexylureido) dodecanoic acid (CUDA) as internal standard. Incubation times were optimized to ensure that the total turnover was <5% for the preferred substrate. The amount of each diol formed was quantified by LC-MS/MS as previously described (Morisseau et al., 2014). Results are mean ± SD (n = 3).
Enantioselective hydrolysis of 19,20-EDP
Purified recombinant Cif (100 μg/mL) in 2 mL of sodium phosphate buffer (20mM, pH 7.0) containing 50 mM NaCl and 0.1 mg/mL BSA was incubated with 20 μL of 5 mM rac-19,20-EDP in DMSO ([S]final = 50 μM) at 37°C. At different time points, 100 μL of the mixture was taken and added to 100 μL of cold methanol containing 400 nM of CUDA as internal standard. The fixed samples were kept at −20°C until analysis. Both enantiomers of 19,20-EDP were separated on a chiral column (Phenomenex Lux Cellulose-3, 4.6 × 250 mm, 5 μm particle size) under isocratic conditions (60% [v/v] methanol in 5 mM ammonium bicarbonate, pH 8.6), and quantified by LC-MS/MS as described (Morisseau et al., 2010). The diol product (19,20-DiHDP) was also quantified by analytical mass spectrometry (Morisseau et al., 2010). Results are mean ± SD (n = 3). Enantiomeric ee and E values were calculated as described above.
Protein Crystallization
CifE153Q protein was dialyzed into buffer containing 100 mM NaCl, 20 mM sodium phosphate (pH 7.4 at room temperature), 2% (v/v) DMSO, and 20 mM (R-EpO, rac-EpO, rac-SOx) or 2 mM (all others) epoxide substrate over 72 hours. For EpFA substrates, 50 μL of 5 mg/mL CifE153Q protein in buffer with 2% (v/v) DMSO was added to neat EpFA and incubated at 4°C overnight. Crystals were obtained by vapor diffusion against 400 μL of reservoir solution in a 4 μL, 1:1 ratio hanging drop set up at 291 K (Bahl et al., 2010). Reservoir solution contained 12–16% (w/v) PEG 8000, 125 mM CaCl2, 100 mM sodium acetate (pH 5 at room temperature), and 2% (v/v) DMSO. Except for EpFA substrates, 2 or 20 mM of the relevant epoxide substrate was included in the reservoir solution. Crystals were soaked in cryoprotectant containing reservoir solution supplemented with 20% (v/v) glycerol and flash cooled in liquid nitrogen.
Data Collection
Oscillation data for crystals were collected at: Beamline X6A of the National Synchrotron Light Source at Brookhaven National Laboratory, Beamline 5.0.3 at the Advanced Light Source of the Lawrence Berkley National Laboratory, or Beamlines 14-1 or 9-3 at Stanford Synchrotron Radiation Lightsource at the SLAC National Accelerator Laboratory (Please refer to data tables for PDB IDs and beamline information). All data sets were indexed, integrated, and scaled with the XDS package (Kabsch, 1993). The Rfree test set was selected in thin shells using the Phenix Reflection File Editor module (Adams et al., 2010).
Structure Determination and Refinement
Molecular replacement using Chain A of the wild-type Cif protein (PDB ID: 3KD2), and iterative rounds of refinement were conducted using Phenix (Adams et al., 2010). Manual rounds of model adjustment were completed using WinCoot between automated refinement steps (Emsley et al., 2010). Hydroxyalkyl-enzyme intermediates and bound ligands were added after two or three rounds of refinement. Images were prepared using Marvin Sketch (v16.12.12), PyMOL (v1.8.4), and Adobe Illustrator (vCS6). As previously reported for the structures of ligand-free WT Cif (Bahl et al., 2010) and CifE153Q adducts (Bahl et al., 2016), the asymmetric unit consists of two Cif homodimers (chains A and B and chains C and D). The homodimers (AB vs. CD) superimpose with RMSD values comparable to the maximum-likelihood coordinate error, with slightly larger differences observed between the monomers in each dimer (e.g., A vs. B).
Synthesis of (R)-2-Butyloxirane
(R)-1-chlorohexan-2-ol
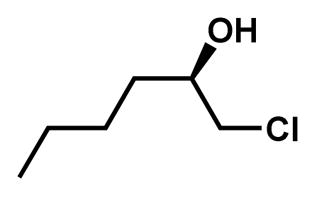
The title compound was synthesized by the method described previously (Moriarty et al., 2004). To a solution of (R)-(−)-epichlorohydrin (5.08 mL, 64.9 mmol) in 10 mL THF was added copper iodide (1.2 g, 6.3 mmol) and cooled to 0°C. To this solution was slowly added 2M propylmagnesium chloride solution in diethyl ether (34 mL, 68 mmol) and stirred overnight at RT. The reaction mixture was cooled to 0°C and to this was slowly added 10 mL of 15% aqueous solution of NH4OH saturated with NH4Cl. Solid was removed by filtration over celite and washed with ethyl acetate. Filtrate was washed twice with 15% aqueous NH4OH saturated with NH4Cl and brine, dried (MgSO4), filtered and concentrated in vacuo. The target compound was purified by column chromatography (hexane: ethyl acetate=10: 1) to afford 6.40 g (46.9 μmol, 72%) of (R)-1-chlorohexan-2-ol as a clear yellowish oil. 1H NMR (600 MHz, CDCl3) δ 3.81 (m, 1H), 3.64 (dd, J = 11.0, 3.3 Hz, 1H), 3.48 (dd, J = 11.1, 7.1 Hz, 1H), 2.15 (d, J = 4.6 Hz, 1H), 1.59 – 1.53 (m, 2H), 1.45 (m, 2H), 1.35 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H).
(R)-2-butyloxirane
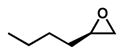
The title compound was synthesized from (R)-1-chlorohexan-2-ol by the method described previously (Van Zyl et al., 1953). To 3.0 g (21.9 mmol) of (R)-1-chlorohexan-2-ol was added NaOH (1 g, 25 mmol) in 5.8 mL of water, and stirred vigorously for 4 hours. The stirring was stopped and the oily upper layer was separated. The aqueous layer was extracted with pentane. The organic layer was combined and distilled to give 1.65 g (16.5 mmol, 75% yield) of title compound as a colorless oil. The percent enantiomeric excess (ee) was determined using 1H NMR spectroscopy using Eu[(+)-3- (trifluoromethylhydroxymethylene)-(+)-camphorate]3 in C6D6 at ambient temperature as described previously and found to be approximately 95% (Hirahata et al., 2008). The purity of the title compound was determined using a gas chromatograph (Model 6980; Hewlett Packard) equipped with an MSD 5973 mass spectrometer (Agilent Technologies). Data acquisition and processing were carried out with the Hewlett Packard UX ChemStation data system. GC separation was performed on a J&W DB-1 column (15 m 0.25 mm i.d.; 0.25 μm film thickness) in splitless mode with helium as the carrier gas. Optical rotation was measured on an Autopol IV polarimeter (Rudolph Research Analytical). bp 35–45°C (20 mm.). 1H NMR (600 MHz, CDCl3) δ 2.91 (ddt, J = 5.8, 2.8, 1.5 Hz, 1H), 2.75 (dd, J = 5.0, 4.0 Hz, 1H), 2.46 (dd, J = 5.1, 2.8 Hz, 1H), 1.55 – 1.52 (m, 2H), 1.44 (dt, J = 15.1, 7.0 Hz, 2H), 1.41 – 1.37 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 52.5, 47.2, 32.3, 28.2, 22.6, 14.1. (c 1.0, CHCl3). Purity (GC-MS): 97%.
Quantification and Statistical Analysis
All significance tests performed within this manuscript are Welch’s t-tests against the substrate-only condition, unless otherwise specified. Statistical analyses were carried out using R (v3.3.1). The number of independent experiments is indicated by “n” and all measures are mean ± SD, unless otherwise specified. For all biochemical experiments, n ≥ 3. Significance is defined as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Structural alignments and comparisons were carried out by aligning all main-chain atoms within 10 Å of the Cα atom of Asp129 (Figure S1) of either the A or C chain within the respective asymmetric units, except for Figure 6D, for which chain B is shown representing an alternative conformation. His177 and Tyr239 mFo-DFc omit maps (Figure S3) were generated by removal of these residues from all chains followed by simulated annealing in the Phenix Refine module without averaging (Adams et al., 2010). Hydroxyalkyl-enzyme intermediate mFo-DFc density is derived from maps arising from the round of refinement before the ligand was placed, without averaging, for all structures except 5TNK and 5TNS (Figures S4 and S7). For 5TNK and 5TNS, the mFo-DFc map is generated by ligand removal and simulated annealing after the last round of refinement without averaging. Hydroxyalkyl-enzyme intermediate 2mFo-DFc density is derived from the 2mFo-DFc map after the last round of refinement. The identity of the chains displayed within Figures 6, S4, and S7 can be found in their respective legends.
Data and Software Availability
The structures determined by X-ray crystallography have been deposited in the wwPDB under PDB ID codes 5TNL, 5TNN, 5TNM, 5TNK, 5TNI, 5TNQ, 5TNP, 5TND, 5TNJ, 5TNE, 5TNS, 5TNR, 5TNF, 5TNG, and 5TNH.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| E. coli TOP10 Bacterial Strain | Gift from Dr. George O’Toole | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| (±)-epibromohydrin | Sigma Aldrich | Cat#E1012 |
| 1,2-cyclohexane oxide | Sigma Aldrich | Cat#C102504 |
| 4-(±)-vinyl-1-cyclohexene 1,2-(±)-epoxide | Sigma Aldrich | Cat#152544 |
| 1,2-(±)-epoxyhexane | Sigma Aldrich | Cat#377171 |
| 1,2-(±)-epoxyoctane | Sigma Aldrich | Cat#260258 |
| (R)-epoxyoctane | Sigma Aldrich | Cat#45366 |
| cis-stilbene oxide | Sigma Aldrich | Cat#308323 |
| 1,2-(±)-styrene oxide | Sigma Aldrich | Cat#W513504 |
| (R)-styrene oxide | Sigma Aldrich | Cat#540099 |
| (S)-styrene oxide | Sigma Aldrich | Cat#540102 |
| (+)-limonene 1,2-epoxide | Sigma Aldrich | Cat#218324 |
| (−)-limonene 1,2-epoxide | Sigma Aldrich | Cat#218332 |
| fosfomycin sodium | Sigma Aldrich | Cat#34089 |
| (R)-1-(4-nitrophenyl)-oxirane | Sigma Aldrich | Cat#69167 |
| (S)-1-(4-nitrophenyl)-oxirane | Sigma Aldrich | Cat#75914 |
| (±)-propylene oxide | Sigma Aldrich | Cat#56671 |
| 3-bromo-1,2-propanediol | Sigma Aldrich | Cat#226130 |
| trans-1,2-cyclohexane diol | Sigma Aldrich | Cat#141712 |
| 1-phenyl-1,2-ethanediol | Sigma Aldrich | Cat#P24055 |
| 1,2-hexanediol | Sigma Aldrich | Cat#213691 |
| 1,2-octanediol | Sigma Aldrich | Cat#213705 |
| (±)-1-(4-nitrophenyl)-ethane-1,2-diol | Sigma Aldrich | Cat#R416576 |
| (±)-trans-oxirane-2,3-dicarboxylic acid | TCI America | Cat#E0350 |
| cis-oxirane-2,3-dicarboxylic acid | TCI America | Cat#E0449 |
| (S)-1,2-epoxyhexane | Maybridge Chemical | Cat#MAY00281CE |
| (S)-1,2-epoxyoctane | Enamine | Cat#BBV-39745467 |
| (R)-1,2-epoxyhexane | This study | N/A |
| (±)19,20-dihydroxy-4Z,7Z,10Z,13Z,16Z-docosapentaenoic acid | Cayman Chemical | Cat#10007001 |
| (±)19,20-dihydroxy-4Z,7Z,10Z,13Z,16Z-docosapentaenoic acid | This study (Morisseau et al., 2010) | N/A |
| (±)17,18-dihydroxy-5Z,8Z,11Z,14Z-eicosatetraenoic acid | This Study (Morisseau et al., 2010) | N/A |
| (±)14,15-dihydroxy-5Z,8Z,11Z,17Z-eicosatetraenoic acid | Cayman Chemical | Cat#10006998 |
| (±)19,20-epoxy-4Z,7Z,10Z,13Z,16Z-docosapentaenoic acid | Cayman Chemical | Cat#10175 |
| (±)16,17-epoxy-4Z,7Z,10Z,13Z,19Z-docosapentaenoic acid | Cayman Chemical | Cat#10174 |
| (±)-(4Z,7Z,10Z)-12-[3-(2Z,5Z)-2,5-octadien-1-yl-2-oxiranyl]-4,7,10-dodecatrienoic acid | Cayman Chemical | CAT#10464; CAS ID: 895127-64-7 |
| (±)17,18-epoxy-5Z,8Z,11Z,14Z-eicosatetraenoic acid | This Study (Morisseau et al., 2010) | N/A |
| (±)14,15-epoxy-5Z,8Z,11Z,17Z-eicosatetraenoic acid | Cayman Chemical | Cat#10173; CAS ID: 131339-24-7 |
| (±)11,12-epoxy-5Z,8Z,14Z,17Z- eicosatetraenoic acid | Cayman Chemical | Cat#10462; CAS ID: 504435-15-8 |
| (±)13-(3-pentyl-2-oxiranyl)-8Z,11Z-tridecadienoic acid | Cayman Chemical | Cat#10007527; CAS ID: 351533-80-7 |
| Epoxy-Fatty Acids, Mixture of 14 | This study (Morisseau et al., 2010) | N/A |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Crystal Structure: CifE153Q-(S)-2-Butyloxirane | This study | PDB ID: 5TNL |
| Crystal Structure: CifE153Q-(S)-2-Hexyloxirane | This study | PDB ID: 5TNN |
| Crystal Structure: CifE153Q-(R)-2-Hexyloxirane | This study | PDB ID: 5TNM |
| Crystal Structure: CifE153Q-rac-2-Hexyloxirane | This study | PDB ID: 5TNK |
| Crystal Structure: CifE153Q-(S)-2-phenyloxirane | This study | PDB ID: 5TNI |
| Crystal Structure: CifE153Q-(R)-2-phenyloxirane | This study | PDB ID: 5TNQ |
| Crystal Structure: CifE153Q-rac-2-phenyloxirane | This study | PDB ID: 5TNP |
| Crystal Structure: CifE153Q-7-oxabicyclo[4.1.0]heptane | This study | PDB ID: 5TND |
| Crystal Structure: CifE153Q-3-Vinyl-7-oxabicyclo[4.1.0]heptane | This study | PDB ID: 5TNJ |
| Crystal Structure: CifE153Q-(2R,3S)-2,3-diphenyloxirane | This study | PDB ID: 5TNE |
| Crystal Structure: CifD129S + 7-oxabicyclo[4.1.0]heptane | This study | PDB ID: 5TNS |
| Crystal Structure: CifE153Q-rac-cis-16,17-EDP | This study | PDB ID: 5TNR |
| Crystal Structure: CifE153Q-rac-cis-19,20-EDP | This study | PDB ID: 5TNF |
| Crystal Structure: CifE153Q-rac-cis-14,15-EEQ | This study | PDB ID: 5TNG |
| Crystal Structure: CifE153Q-rac-cis-17,18-EEQ | This study | PDB ID: 5TNH |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| pDPM73: C-terminal His6-tagged Cif | Gift from Dr. George O’Toole (MacEachran et al., 2007) | N/A |
| pMQ70::CifE153Q: C-terminal His6-tagged CifE153Q | This lab (Bahl et al., 2016) | N/A |
| pMQ70::CifD129S: C-terminal His6-tagged CifD129S | This lab (Bahl et al., 2015) | N/A |
| Software and Algorithms | ||
| Other | ||
Supplementary Material
Highlights.
Adducts probe the active-site geometry of an epoxide-hydrolase virulence factor
Localized conformational flexibility enables Cif to hydrolyze epoxy-fatty acids
Substrate competition can modulate Cif specific activity for targets in vivo
Cif allows P. aeruginosa to perturb important host regulatory signals
Acknowledgments
We would like to thank Dr. Vivian Stojanoff, Dr. Jean Jakonic, and Edwin Lazo at Beamline X6A of NSLS at BNL, Dr. Clyde Smith and the staff of Beamlines 14-1 and 9-3 of SSRL at SLAC, and Drs. Diane Bryant and Corie Ralston at Beamline 5.0.3 of ALS for help with data collection. We would also like to thank Drs. Scott Gerber, George O’Toole, and Peter Jacobi for helpful discussions.
This work was supported by the NIH (R01AI091699, P20GM113132, P30GM106394, U24DK097154, T32GM008704, R01ES002710, P42ES004699 and K99ES024806), the Cystic Fibrosis Foundation (STANTO19R0), and the Munck-Pfefferkorn Fund. S.K. was financially supported by Japan Student Services Organization. Beamline access was supported in part by the NIH (GM-0080, P41-GM111244, P41-GM103393) and the DOE (DE-SC0012704, DE-AC02-76SF00515).
D.R.M., C.D.B., C.M., and B.D.H are the co-inventors of Cif inhibitor intellectual property held by Dartmouth College. B.D.H is a founder and CEO of EicOsis, a company that develops sEH inhibitors for treatment of neuropathic pain.
Footnotes
Supplemental Information includes 2 Tables and 7 Figures and can be found in CifSubstrateSnapshots_SupplementalInfo.pdf.
Author Contributions
K.L.H., C.D.B., B.D.H, C.M., and D.R.M. conceptualized the study and designed experiments; K.L.H., C.D.B., and C.M. performed pilot studies; K.L.H., S.K., K.S.S.L., and C.M. conducted the experiments; K.L.H., K.S.S.L., C.M., and D.R.M analyzed the data; K.L.H., C.M., and D.R.M. wrote the manuscript. All authors edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arand M, Cronin A, Adamska M, Oesch F. Epoxide hydrolases: Structure, function, mechanism, and assay. Methods Enzymol. 2005;400:569–588. doi: 10.1016/S0076-6879(05)00032-7. [DOI] [PubMed] [Google Scholar]
- Bahl CD, Madden DR. Pseudomonas aeruginosa Cif defines a distinct class of α/β epoxide hydrolases utilizing a His/Tyr ring-opening pair. Protein Pept Lett. 2012;19:186–193. doi: 10.2174/092986612799080392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl CD, Morisseau C, Bomberger JM, Stanton BA, Hammock BD, O’Toole GA, Madden DR. Crystal structure of the cystic fibrosis transmembrane conductance regulator inhibitory factor Cif reveals novel active-site features of an epoxide hydrolase virulence factor. J Bacteriol. 2010a;192:1785–1795. doi: 10.1128/JB.01348-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl CD, MacEachran DP, O’Toole Ga, Madden DR. Purification, crystallization and preliminary X-ray diffraction analysis of Cif, a virulence factor secreted by Pseudomonas aeruginosa. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010b;66:26–28. doi: 10.1107/S1744309109047599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl CD, Hvorecny KL, Bridges AA, Ballok AE, Bomberger JM, Cady KC, O’Toole GA, Madden DR. Signature motifs identify an Acinetobacter cif virulence factor with epoxide hydrolase activity. J Biol Chem. 2014;289:7460–7469. doi: 10.1074/jbc.M113.518092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl CD, Hvorecny KL, Bomberger JM, Stanton BA, Hammock BD, Morisseau C, Madden DR. Inhibiting an epoxide hydrolase virulence factor from Pseudomonas aeruginosa protects CFTR. Angew Chemie Int Ed. 2015a;54:9881–9885. doi: 10.1002/anie.201503983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl CD, Hvorecny KL, Bomberger JM, Stanton BA, Hammock BD, Morisseau C, Madden DR. Inhibiting an Epoxide Hydrolase Virulence Factor from Pseudomonas aeruginosa Protects CFTR. Angew Chemie - Int Ed. 2015b;54:9881–9885. doi: 10.1002/anie.201503983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl CD, Hvorecny KL, Morisseau C, Gerber SA, Madden DR. Visualizing the mechanism of epoxide hydrolysis by the bacterial virulence enzyme Cif. Biochemistry. 2016;55:788–797. doi: 10.1021/acs.biochem.5b01229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballok AE, Bahl CD, Dolben EL, Lindsay AK, St Laurent JD, Hogan DA, Madden DR, O’Toole GA. Epoxide-mediated CifR repression of cif gene expression utilizes two binding sites in Pseudomonas aeruginosa. J Bacteriol. 2012;194:5315–5324. doi: 10.1128/JB.00984-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth S, Fischer M, Schmid RD, Pleiss J. Sequence and structure of epoxide hydrolases: A systematic analysis. Proteins Struct Funct Genet. 2004;55:846–855. doi: 10.1002/prot.20013. [DOI] [PubMed] [Google Scholar]
- Bauer P, Carlsson ÅJ, Amrein BA, Dobritzsch D, Widersten M, Kamerlin SCL. Conformational diversity and enantioconvergence in potato epoxide hydrolase 1. Org Biomol Chem. 2016;14:5639–5651. doi: 10.1039/c6ob00060f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci G, Chiappe C, Cordoni A, Ingrosso G. Enantioconvergent transformation of racemic cis-dialkyl substituted epoxides to (R,R) threo diols by microsomal epoxide hydrolase catalysed hydrolysis. Tetrahedron Lett. 1996;37:9089–9092. [Google Scholar]
- Bishop-Bailey D, Thomson S, Askari A, Faulkner A, Wheeler-Jones C. Lipid-metabolizing CYPs in the regulation and dysregulation of metabolism. Annu Rev Nutr. 2014;34:261–279. doi: 10.1146/annurev-nutr-071813-105747. [DOI] [PubMed] [Google Scholar]
- Bomberger JM, Ye S, MacEachran DP, Koeppen K, Barnaby RL, O'Toole GA, Stanton BA. A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson ÅJ, Bauer P, Ma H, Widersten M. Obtaining optical purity for product diols in enzyme-catalyzed epoxide hydrolysis: Contributions from changes in both enantio- and regioselectivity. Biochemistry. 2012;51:7627–7637. doi: 10.1021/bi3007725. [DOI] [PubMed] [Google Scholar]
- Carlsson ÅJ, Bauer P, Dobritzsch D, Nilsson M, Kamerlin SCL, Widersten M. Laboratory-evolved enzymes provide snapshots of the development of enantioconvergence in enzyme-catalyzed epoxide hydrolysis. Chem Bio Chem. 2016;17:1693–1697. doi: 10.1002/cbic.201600330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedrone F, Bhatnagar T, Baratti JC. Colorimetric assays for quantitative analysis and screening of epoxide hydrolase activity. Biotechnol Lett. 2005;27:1921–1927. doi: 10.1007/s10529-005-3904-1. [DOI] [PubMed] [Google Scholar]
- Chen C, Fujimoto Y, Girdaukas G, Sih CJ. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J Am Chem Soc. 1982;104:7294–7299. [Google Scholar]
- Deline M, Keller J, Rothe M, Schunck WH, Menzel R, Watts JL. Epoxides derived from dietary dihomo-gamma-linolenic acid induce germ cell death in C. elegans. Sci Rep. 2015;5:15417. doi: 10.1038/srep15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfström LT, Widersten M. Catalysis of potato epoxide hydrolase, StEH1. Biochem J. 2005;390:633–640. doi: 10.1042/BJ20050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flitter BA, Hvorecny KL, Ono E, Eddens T, Yang J, Kwak DH, Bahl CD, Hampton TH, Morisseau C, Hammock BD, et al. Pseudomonas aeruginosa sabotages the generation of host proresolving lipid mediators. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1610242114. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Ga, Morisseau C, Hammock BD, Christianson DW. Human soluble epoxide hydrolase: structural basis of inhibition by 4-(3-cyclohexylureido)-carboxylic acids. Protein Sci. 2006;15:58–64. doi: 10.1110/ps.051720206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahata W, Thomas RM, Lobkovsky EB, Coates GW. Enantioselective polymerization of epoxides: A highly active and selective catalyst for the preparation of stereoregular polyethers and enantiopure epoxides. J Am Chem Soc. 2008;130:17658–17659. doi: 10.1021/ja807709j. [DOI] [PubMed] [Google Scholar]
- Horsman GP, Lechner A, Ohnishi Y, Moore BS, Shen B. Predictive model for epoxide hydrolase-generated stereochemistry in the biosynthesis of nine-membered enediyne antitumor antibiotics. Biochemistry. 2013;52:5217–5224. doi: 10.1021/bi400572a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig J, Hammock B. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- Kitamura S, Hvorecny KL, Niu J, Hammock BD, Madden DR, Morisseau C. Rational design of potent and selective inhibitors of an epoxide hydrolase virulence factor from Pseudomonas aeruginosa. J Med Chem. 2016;59:4790–4799. doi: 10.1021/acs.jmedchem.6b00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaar AL, Yang L, Boardley RL, Goyal NS, Robertson J, Baldwin SJ, Newby DE, Wilkinson IB, Tal-Singer R, Mayer RJ, et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol. 2015;81:971–979. doi: 10.1111/bcp.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg D, De La Revenga MF, Widersten M. Temperature and pH dependence of Enzyme-catalyzed hydrolysis of trans-methylstyrene oxide. A unifying kinetic model for observed hysteresis, Cooperativity, and regioselectivity. Biochemistry. 2010;49:2297–2304. doi: 10.1021/bi902157b. [DOI] [PubMed] [Google Scholar]
- van Loo B, Kingma J, Arand M, Wubbolts MG, Janssen DB. Diversity and biocatalytic potential of epoxide hydrolases identified by genome analysis. Appl Environ Microbiol. 2006;72:2905–2917. doi: 10.1128/AEM.72.4.2905-2917.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEachran DP, Ye S, Bomberger JM, Hogan DA, Swiatecka-Urban A, Stanton BA, O’Toole GA. The Pseudomonas aeruginosa secreted protein PA2934 decreases apical membrane expression of the cystic fibrosis transmembrane conductance regulator. Infect Immun. 2007;75:3902–3912. doi: 10.1128/IAI.00338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monterde MI, Lombard M, Archelas A, Cronin A, Arand M, Furstoss R. Enzymatic transformations. Part 58: Enantioconvergent biohydrolysis of styrene oxide derivatives catalysed by the Solanum tuberosum epoxide hydrolase. Tetrahedron: Asymmetry. 2004;15:2801–2805. [Google Scholar]
- Moriarty RM, Rani N, Enache LA, Rao MS, Batra H, Guo L, Penmasta RA, Staszewski JP, Tuladhar SM, Prakash O, et al. The intramolecular asymmetric pauson-khand cyclization as a novel and general stereoselective route to benzindene prostacyclins: synthesis of UT-15 (Treprostinil) J Org Chem. 2004;69:1890–1902. doi: 10.1021/jo0347720. [DOI] [PubMed] [Google Scholar]
- Morisseau C. Role of epoxide hydrolases in lipid metabolism. Biochimie. 2013;95:91–95. doi: 10.1016/j.biochi.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Hammock BD. Epoxide Hydrolases: Mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Inceoglu B, Schmelzer K, Tsai H, Jinks SL, Hegedus CM, Hammock BD. Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. 2010;51:3481–3490. doi: 10.1194/jlr.M006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Wecksler AT, Deng C, Dong H, Yang J, Lee KSS, Kodani SD, Hammock BD. Effect of soluble epoxide hydrolase polymorphism on substrate and inhibitor selectivity and dimer formation. J Lipid Res. 2014;55:1131–1138. doi: 10.1194/jlr.M049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hagan D, Schmidberger JW. Enzymes that catalyse SN2 reaction mechanisms. Nat Prod Rep. 2010;27:900–918. doi: 10.1039/b919371p. [DOI] [PubMed] [Google Scholar]
- Oesch F, Schladt L, Hartmann R, Timms C, Wörner W. Rat cytosolic epoxide hydrolase. Adv Exp Med Biol. 1986;197:195–201. doi: 10.1007/978-1-4684-5134-4_16. [DOI] [PubMed] [Google Scholar]
- Reetz MT, Bocola M, Wang LW, Sanchis J, Cronin A, Arand M, Zou J, Archelas A, Bottalla AL, Naworyta A, et al. Directed evolution of an enantioselective epoxide hydrolase: Uncovering the source of enantioselectivity at each evolutionary stage. J Am Chem Soc. 2009;131:7334–7343. doi: 10.1021/ja809673d. [DOI] [PubMed] [Google Scholar]
- Ribeiro BD, Coelho MAZ, De Castro AM. Principles of Green Chemistry and White Biotechnology. In: Ribeiro BD, Coelho MAZ, editors. White Biotechnology for Sustainable Chemistry. The Royal Society of Chemistry; 2015. pp. 1–8. [Google Scholar]
- Ringholz FC, Buchanan PJ, Clarke DT, Millar RG, McDermott M, Linnane B, Harvey BJ, McNally P, Urbach V. Reduced 15-lipoxygenase 2 and lipoxin A4/leukotriene B4 ratio in children with cystic fibrosis. Eur Respir J. 2014;44:394–404. doi: 10.1183/09031936.00106013. [DOI] [PubMed] [Google Scholar]
- Schäuble S, Stavrum AK, Puntervoll P, Schuster S, Heiland I. Effect of substrate competition in kinetic models of metabolic networks. FEBS Lett. 2013;587:2818–2824. doi: 10.1016/j.febslet.2013.06.025. [DOI] [PubMed] [Google Scholar]
- Schiøtt B, Bruice TC. Reaction mechanism of soluble epoxide hydrolase: insights from molecular dynamics simulations. J Am Chem Soc. 2002;124:14558–14570. doi: 10.1021/ja021021r. [DOI] [PubMed] [Google Scholar]
- Selvan A, Anishetty S. Cavities create a potential back door in epoxide hydrolase Rv1938 from Mycobacterium tuberculosis - A molecular dynamics simulation study. Comput Biol Chem. 2015;58:222–230. doi: 10.1016/j.compbiolchem.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks RMQ, Caiazza NC, Hinsa SM, Toutain CM, O’Toole GA. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl Environ Microbiol. 2006;72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillman NJ, Dalmia VK, Goldberg E. Exported epoxide hydrolases modulate erythrocyte vasoactive lipids during Plasmodium falciparum infection. MBio. 2016;7:1–13. doi: 10.1128/mBio.01538-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerer S, Hanano A, Utsumi S, Arand M, Schuber F, Blée E. Stereochemical features of the hydrolysis of 9,10-epoxystearic acid catalysed by plant and mammalian epoxide hydrolases. Biochem J. 2002;366:471–480. doi: 10.1042/BJ20011778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatecka-Urban A. Pseudomonas aeruginosa inhibits endocytic recycling of CFTR in polarized human airway epithelial cells. AJP Cell Physiol. 2005;290:C862–C872. doi: 10.1152/ajpcell.00108.2005. [DOI] [PubMed] [Google Scholar]
- Xue Y, Olsson T, Johansson CA, Öster L, Beisel HG, Rohman M, Karis D, Bäckström S. Fragment Screening of Soluble Epoxide Hydrolase for Lead Generation - Structure-Based Hit Evaluation and Chemistry Exploration. Chem Med Chem. 2016;11:497–508. doi: 10.1002/cmdc.201500575. [DOI] [PubMed] [Google Scholar]
- Yandek LE, Lin HC, Harris ME. Alternative substrate kinetics of escherichia coli ribonuclease P: Determination of relative rate constants by internal competition. J Biol Chem. 2013;288:8342–8354. doi: 10.1074/jbc.M112.435420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldin DC, Wei S, Falck JR, Hammock BD, Snapper JR, Capdevila JH. Metabolism of Epoxyeicosatrienoic Acids by Cytosolic Epoxide Hydrolase: Substrate Structural Determinants of Asymmetric Catalysis. Arch Biochem Biophys. 1995;316:443–451. doi: 10.1006/abbi.1995.1059. [DOI] [PubMed] [Google Scholar]
- Zhao L, Han B, Huang Z, Miller M, Huang H, Malashock DS, Zhu Z, Milan A, Robertson DE, Weiner DP, et al. Epoxide hydrolase-catalyzed enantioselective synthesis of chiral 1,2-diols via desymmetrization of meso-epoxides. J Am Chem Soc. 2004;126:11156–11157. doi: 10.1021/ja0466210. [DOI] [PubMed] [Google Scholar]
- Van Zyl G, Zuidema GD, Zack JF, Kromann B. The course of ring opening of glycidyl ethers with nucleophilic reagents. J Am Chem Soc. 1953;75:5002–5006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.