

Summary
Background
Inhibition of phosphatidylinositol 3-kinase (PI3K) is a promising approach to overcome resistance to endocrine therapy in breast cancer. Pictilisib is an oral inhibitor of multiple PI3K isoforms. The aim of this study is to establish if addition of pictilisib to fulvestrant can improve progression-free survival in oestrogen receptor-positive, endocrine-resistant breast cancer.
Methods
In this two-part, randomised, double-blind, placebo-controlled, phase 2 study, we recruited postmenopausal women aged 18 years or older with oestrogen receptor-positive, HER2-negative breast cancer resistant to treatment with an aromatase inhibitor in the adjuvant or metastatic setting, from 123 medical centres across 21 countries. Part 1 included patients with or without PIK3CA mutations, whereas part 2 included only patients with PIK3CA mutations. Patients were randomly allocated (1:1 in part 1 and 2:1 in part 2) via a computer-generated hierarchical randomisation algorithm to daily oral pictilisib (340 mg in part 1 and 260 mg in part 2) or placebo starting on day 15 of cycle 1, plus intramuscular fulvestrant 500 mg on day 1 and day 15 of cycle 1 and day 1 of subsequent cycles in both groups. In part 1, we stratified patients by presence or absence of PIK3CA mutation, primary or secondary aromatase inhibitor resistance, and measurable or non-measurable disease. In part 2, we stratified patients by previous aromatase inhibitor treatment for advanced or metastatic disease or relapse during or within 6 months of an aromatase inhibitor treatment in the adjuvant setting and measurable or non-measurable disease. All patients and those administering treatment and assessing outcomes were masked to treatment assignment. The primary endpoint was progression-free survival in the intention-to-treat population for both parts 1 and 2 and also separately in patients with PIK3CA-mutated tumours in part 1. Tumour assessment (physical examination and imaging scans) was investigator-assessed and done at screening and after 8 weeks, 16 weeks, 24 weeks, and 32 weeks of treatment from day 1 of cycle 1 and every 12 weeks thereafter. We assessed safety in as-treated patients who received at least one dose of study medication. This trial is registered with ClinicalTrials.gov, number NCT01437566.
Findings
In part 1, between Sept 27, 2011, and Jan 11, 2013, we randomly allocated 168 patients to the pictilisib (89 [53%]) or placebo (79 [47%]) group. In part 2, between March 18, 2013, and Jan 2, 2014, we randomly allocated 61 patients to the pictilisib (41 [67%]) or placebo (20 [33%]) group. In part 1, we found no difference in median progression-free survival between the pictilisib (6.6 months [95% CI 3.9–9.8]) and placebo (5.1 months [3.6–7.3]) group (hazard ratio [HR] 0.74 [95% CI 0.52–1.06]; p=0.096). We also found no difference when patients were analysed according to presence (pictilisib 6.5 months [95% CI 3.7–9.8] vs placebo 5.1 months [2.6–10.4]; HR 0.73 [95% CI 0.42–1.28]; p=0.268) or absence (5.8 months [3.6–11.1] vs 3.6 months [2.8–7.3]; HR 0.72 [0.42–1.23]; p=0.23) of PIK3CA mutation. In part 2, we also found no difference in progression-free survival between groups (5.4 months [95% CI 3.8–8.3] vs 10.0 months [3.6–13.0]; HR 1.07 [95% CI 0.53–2.18]; p=0.84). In part 1, grade 3 or worse adverse events occurred in 54 (61%) of 89 patients in the pictilisib group and 22 (28%) of 79 in the placebo group. 19 serious adverse events related to pictilisib treatment were reported in 14 (16%) of 89 patients. Only one (1%) of 79 patients reported treatment-related serious adverse events in the placebo group. In part 2, grade 3 or worse adverse events occurred in 15 (36%) of 42 patients in the pictilisib group and seven (37%) of 19 patients in the placebo group. Four serious adverse events related to pictilisib treatment were reported in two (5%) of 42 patients. One treatment-related serious adverse event occurred in one (5%) of 19 patients in the placebo group.
Interpretation
Although addition of pictilisib to fulvestrant did not significantly improve progression-free survival, dosing of pictilisib was limited by toxicity, potentially limiting its efficacy. For future assessment of PI3K inhibition as an approach to overcome resistance to hormonal therapy, inhibitors with greater selectivity than that of pictilisib might be needed to improve tolerability and potentially increase efficacy. No further investigation of pictilisib in this setting is ongoing.
Funding
F Hoffmann-La Roche.
Introduction
Hormone receptor-positive breast cancer is typically managed with endocrine treatment, but almost half of patients who present with oestrogen receptor-positive metastatic breast cancer do not respond to first-line endocrine treatment and nearly all those who do eventually develop resistance.1 The mechanisms of endocrine resistance remain to be fully elucidated,2 but signalling through the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT)-mTOR (PI3K-AKT-mTOR) pathway and bidirectional crosstalk between this pathway and the oestrogen receptor are emerging as important targets. In preclinical models, hyperactivation of the PI3K pathway allows breast cancer cell lines to escape hormone dependence, and this effect can be blocked by PI3K or mTOR inhibitors.3 Proteomic and transcriptional profiling of tumour samples suggest that increased PI3K signalling is also associated with lowered oestrogen receptor expression levels, which have been associated with resistance to endocrine therapy and shown to promote hormonal independence in oestrogen receptor-positive breast cancer models.3,4 Together, these data suggest that inhibitors of the PI3K-mTOR pathway could help overcome resistance to endocrine therapy.
Clinical data also provide support for a role for combined inhibition of PI3K-AKT-mTOR and oestrogen receptor signalling pathways as an approach to overcome intrinsic resistance or delay acquired resistance to endocrine therapy in oestrogen receptor-positive metastatic breast cancer. Administration of everolimus, a rapamycin analogue inhibitor of mTOR, increased the efficacy of letrozole in the neoadjuvant setting in patients with oestrogen receptor-positive breast cancer,5 and addition of everolimus to tamoxifen in a phase 2 study6 in patients with oestrogen receptor-positive metastatic breast cancer previously given an aromatase inhibitor significantly improved the clinical benefit, time to progression, and overall survival compared with single-agent tamoxifen. In a phase 3 randomised study, Breast Cancer Trials of Oral Everolimus-2 (BOLERO-2),7 addition of everolimus to exemestane more than doubled median progression-free survival in patients with oestrogen receptor-positive, HER2-negative metastatic breast cancer refractory to previous treatment with letrozole or anastrozole, although no overall survival advantage could be shown.8
Activating mutations in the PI3K catalytic subunit α (PIK3CA) are common in breast cancer and are particularly frequent in oestrogen receptor-positive, HER2-negative tumours. In multiple large population-based studies,9–12 the incidence of PIK3CA mutations in oestrogen receptor-positive, HER2-negative cancers is approximately 40%. However, the role of these mutations in mediation of downstream activation of the PI3K pathway and endocrine therapy resistance is unclear. Findings from several studies13 have shown that the presence of PIK3CA mutations is inversely associated with activation of downstream components of the pathway, such as TORC1, and associated with favourable prognostic features (low histological grade and negative lymph node status) and outcomes in patients given endocrine therapy.14,15 In the BOLERO-2 study,16 no association was found between tumour PIK3CA mutation status and benefit from everolimus, further clouding the role of these mutations in mediation of resistance to endocrine therapy. Elucidation of the role of PIK3CA mutations and PI3K signalling is further complicated by findings from studies showing that other alterations of the pathway, including loss of phosphatase and tensin homologue (PTEN), are associated with activation of downstream PI3K pathway signalling and resistance to endocrine therapy.17
Pictilisib (GDC-0941) is an orally available small-molecule inhibitor of all four class I PI3K isoforms that has shown clinically significant activity in preclinical breast cancer models18 and antitumour activity with acceptable safety in a phase 1 study19 in patients with advanced solid tumours. In this randomised phase 2 study (FERGI), we aimed to assess the combination of pictilisib plus fulvestrant (pictilisib group) versus placebo plus fulvestrant (placebo group) in patients with oestrogen receptor-positive advanced breast cancer to test the hypothesis that inhibition of PI3K can overcome endocrine therapy resistance and to explore the role of PIK3CA mutations in prediction of benefit of this therapeutic approach.
Methods
Study design and patients
In this international, multicentre, randomised, double-blind, placebo-controlled, phase 2 clinical trial, formed of two parts with separate randomisations, we recruited patients from 123 medical centres across 21 countries. For part 1, eligible patients were postmenopausal women aged 18 years or older with oestrogen receptor-positive, HER2-negative locally advanced or metastatic breast cancer appropriate for endocrine therapy (ie, fulvestrant) based on national or local treatment guidelines, relapsed during or within 6 months of aromatase inhibitor treatment in the adjuvant setting, or with progressive disease during treatment with an aromatase inhibitor in the metastatic setting. The most recent treatment before enrolment was required to be an aromatase inhibitor, with a minimum duration of 4 weeks of treatment before recurrence or progressive disease. Patients who discontinued aromatase inhibitors because of toxicity without disease progression or completion of treatment were not eligible. A separate cohort of patients was enrolled for part 2. Inclusion criteria for part 2 were the same as those for part 1, except that in part 2, all patients were required to have a PIK3CA-mutated tumour and the requirement for an aromatase inhibitor to be the last therapy was removed.
Other inclusion criteria for both parts were Eastern Cooperative Oncology Group performance status of 0 or 1, measurable disease with Response Evaluation Criteria in Solid Tumors version 1.1 or non-measurable disease with radiology scans within 28 days of day 1 of cycle 1, and adequate haematological and liver and kidney function. Key exclusion criteria included previous treatment with fulvestrant, a PI3K inhibitor, or an mTOR inhibitor for advanced or metastatic breast cancer; previous anticancer therapy or radiotherapy within 2 weeks before day 1 of cycle 1; previous treatment with more than one cytotoxic chemo therapy regimen; or recurrent disease or progressive disease on more than two previous endocrine therapies for metastatic breast cancer. We also excluded patients requiring antihyper glycaemic therapy or with clinically significant cardiac or pulmonary dysfunction, active autoimmune disease, immunocompromised status, clinically significant history of liver disease, or untreated or active CNS metastases from study participation.
The study was done in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. We obtained written informed consent from all patients before enrolment, in agreement with approved protocols from respective ethics committees at each site.
Randomisation and masking
In part 1 of the study, patients were originally randomly allocated in a 1:1:1 ratio to receive pictilisib plus fulvestrant, apitolisib (GDC-0980, a dual PI3K-mTOR inhibitor) plus fulvestrant, or placebo plus fulvestrant. Patients in the placebo group received placebo corresponding either to pictilisib (pictilisb placebo) or apotilisib (apotilisib placebo). However, after a safety review done approximately 18 weeks after the first 30 patients had been recruited in the apitolisib arm, a decision was taken to close recruitment to the apitolisib arm (and corresponding placebo arm) because of an unacceptably high proportion of grade 3 adverse events in this arm. Patients in the apitolisib arm were not eligible for crossover within the study and only the pictilisib group and placebo group are considered further in this report. Subsequent to the closing of the apitolisib arm, patients were randomly allocated 1:1 to pictilisib plus fulvestrant and placebo plus fulvestrant. As a result of this closure, some patients (who had received apotilisib placebo) were lost from the placebo arm so that a greater number of patients were randomised to the pictilisib group than to the placebo group. After completion of part 1 enrolment, we randomly allocated a seperate cohort of patients in part 2 2:1 to the pictilisib group and the placebo group.
Patients enrolled by investigators at participating centres were allocated to the study arms by Bracket Randomization and Trial Supply Management (San Francisco, CA, USA) using the IXRS voice and web response system (Almac Group, Craigavon, UK) via a computer-generated hierarchical randomisation algorithm with predefined stratification variables. Randomisation codes were generated through an algorithm that adjusted the probability that a patient would receive a given treatment according to the characteristics of the patient and previously randomly allocated patients. All patients and those administering treatment and assessing outcomes were masked to treatment group assignment. In part 1 of the study, we stratified patients according to presence or absence of PI3K mutation, measurable disease, and primary or secondary resistance (ie, after an initial response) to previous aromatase inhibitor therapy. In part 2, we stratified patients according to measurable disease and previous treatment with an aromatase inhibitor for advanced or metastatic disease or relapse during or within 6 months of treatment with an aromatase inhibitor in the adjuvant setting.
Procedures
In part 1, patients received 28 day cycles of 500 mg intramuscular fulvestrant on day 1 and day 15 of cycle 1 and day 1 of subsequent cycles, with daily 340 mg oral pictilisib or placebo starting on day 15 of cycle 1. Before recruitment was halted to the apitolisib arm, patients in this arm received 30 mg daily of oral apitolisib. In part 2, fulvestrant 500 mg was administered on day 1 and day 15 of cycle 1 and day 1 of subsequent 28 day cycles, with pictilisib at a reduced dose of 260 mg or placebo daily starting on day 1 of cycle 1. The protocol was amended on Oct 25, 2012, to reduce the pictilisib dose in part 2 to 260 mg because of a high proportion of discontinuations in part 1. Patients in both parts of the study received treatment until disease progression, intolerable toxicity, elective withdrawal from the study, or study completion or termination. Patients who had progressive disease while receiving placebo treatment and continued to meet the eligibility criteria had the opportunity to receive crossover therapy with open-label pictilisib as a single agent or with fulvestrant.
PIK3CA mutation was detected with quantitative real-time PCR for activating missense mutations Cys420Arg; Glu542Lys; Glu545Ala, Glu545Gly, or Glu545Lys; and His1047Leu, His1047Arg, or His1047Tyr at a central laboratory. Pictilisib could be temporarily suspended for up to 28 days or a fulvestrant dose could be missed if a patient had toxicity related to treatment. Patients who missed more than 28 consecutive days of treatment or two consecutive fulvestrant treatments because of adverse events discontinued that treatment, but could continue on single-agent fulvestrant or pictilisib at the discretion of the investigator. Also at the discretion of the investigator, the dose of pictilisib could be reduced stepwise from 340 mg to 260 mg, then to 200 mg in part 1, and from 260 mg to 200 mg, then to 140 mg in part 2. If patients on the lowest dose had an indication for further dose reduction, pictilisib was discontinued.
Progression was assessed by the local investigator on the basis of physical examination and imaging scans using the Response Evaluation Criteria in Solid Tumors version 1.1. Tumour assessment was done at screening and after 8 weeks, 16 weeks, 24 weeks, and 32 weeks of treatment from day 1 of cycle 1 and every 12 weeks thereafter.
Outcomes
The primary endpoint was progression-free survival, defined as the time from the date of the first dose of fulvestrant to the first occurrence of disease progression or death from any cause. Secondary endpoints were objective response (defined as the proportion of patients achieving a complete or partial response), clinical benefit (defined as the proportion of patients who have a complete response, a partial response, or stable disease lasting for at least 6 months), duration of response, the prognostic effect on progression-free survival of PIK3CA mutations, pharma cokinetics of pictilisib in combination with fulvestrant, and the prevalence of PIK3CA mutation and PTEN loss in tumour samples from patients with oestrogen receptor-positive advanced breast cancer.
We graded safety according to the National Cancer Institute Common Toxicity Criteria for Adverse Events version 4.0. We assessed patients for safety every 2 weeks during the first two cycles of treatment and before administration of fulvestrant at the beginning of subsequent 28 day cycles.
Statistical analysis
The trial was hypothesis generating and not powered to detect minimal clinically meaningful differences between treatment groups at a significant (type I) error level of 5%. The trial was designed to obtain meaningful estimates of the hazard ratio (HR). At a targeted HR of 0.40 in patients with PIK3CA-mutant tumours in part 1, a two-sided upper 90% CI of 0.22–0.73 at 35 events would be considered meaningful. Based on the assumption that 30% of randomly allocated patients would be unassessable for efficacy and that 35% of patients would have a PIK3CA mutation, approximately 160 patients would be required. Similarly, in part 2, at a targeted HR of 0.57 in patients with PIK3CA-mutant tumours, a two-sided upper 90% CI of 0.33–0.99 at 35 events would be considered meaningful and approximately 60 patients would be required in a 2:1 randomisation.
We assessed efficacy in the intention-to-treat (ITT) population in both parts of the trial and separately in patients with PIK3CA-mutated tumours in part 1. We assessed progression-free survival with a log-rank test. We assessed safety according to treatment received in all patients who received at least one dose of study medication. We censored data for randomly allocated patients who had not had an event of progressive disease or death at the date of their last tumour assessment. The HR estimate of progression-free survival and the 95% CI comparing the pictilisib group with the placebo group were provided by the Cox model for the ITT population in both parts, as well as for the PIK3CA-mutated subgroup in part 1. Additional post-hoc analyses included HR estimates of progression-free survival by the Cox model, adjusted by prespecified stratification factors. We parsed Kaplan-Meier curves by treatment for the ITT population in both parts and by PIK3CA mutation status versus treatment in part 1. We assessed objective response and clinical benefit with χ2 tests and generated 95% CIs with the Clopper-Pearson method. We assessed duration of response with log-rank tests and used Cox models to generate the HRs and 95% CIs. We used SAS version 9.2 for statistical analyses.
Role of the funding source
The funder of the study was involved in study design and data interpretation. Employees of the funder (SG, MDe, ML, GL, JQ, and JH) collected, managed, and analysed data, had access to the raw data, and were involved in writing of the report. The funder funded third-party writing assistance. The corresponding author had full access to all of the data in the study and had final responsibility to submit for publication.
Results
In part 1, between Sept 27, 2011, and Jan 11, 2013, we randomly allocated 168 patients to the pictilisib group (89 [53%] patients) or placebo group (79 [47%] patients). All patients were assessable for efficacy and safety. In part 2, between March 18, 2013, and Jan 2, 2014, we randomly allocated 61 patients to the pictilisib group (41 [67%]) or placebo group (20 [33%]; figure 1). All patients were assessable for efficacy in the two treatment groups as randomly allocated. One patient randomly allocated to the placebo group in part 2 received one cycle of pictilisib in error and was included in the pictilisib group for safety analysis. The number of events required for final analysis of the primary endpoints had been reached or exceeded in both parts by the time of data cutoff (part 1: March 7, 2014; part 2: Sept 12, 2014).
Figure 1.
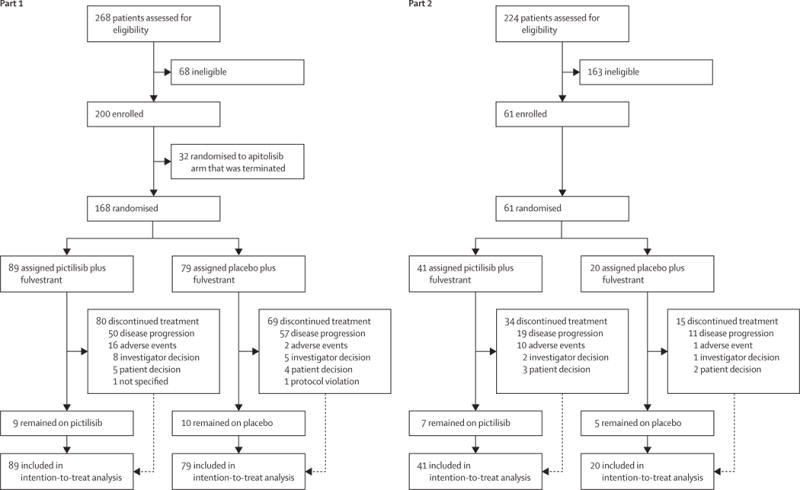
Trial profile
Baseline characteristics are shown in table 1. In part 1, 70 (41%) of 170 patients had a PIK3CA-mutated tumour (pictilisib 38 [43%] vs placebo 32 [41%]). In part 2, one (2%) of 41 patients in the pictilisib group with a mutated tumour according to testing at the local laboratory was subsequently found not to have a PIK3CA mutation after analysis at the central laboratory. Of the 229 tumour samples that were used for biomarker analysis, 115 (50%) were collected at the time that the patient was diagnosed with primary breast cancer, 107 (47%) were collected at diagnosis of locally advanced or metastatic disease, and seven (3%) were unknown. We enrolled a higher proportion of patients in part 2 than in part 1 with three or more metastatic sites and who had three or more previous therapies in the metastatic setting, reflecting the fact that patients in part 2 had more advanced disease than did those in part 1.
Table 1.
Baseline characteristics
| Part 1
|
Part 2
|
|||
|---|---|---|---|---|
| Pictilisib plus fulvestrant (n=89) | Placebo plus fulvestrant (n=79) | Pictilisib plus fulvestrant (n=41) | Placebo plus fulvestrant (n=20) | |
| Age (years) | 60 (36–90) | 63 (40–82) | 58 (32–79) | 63 (45–84) |
|
| ||||
| Aged ≥65 years | 29 (33%) | 29 (37%) | 13 (32%) | 9 (45%) |
|
| ||||
| Race | ||||
| White | 78 (88%) | 68 (86%) | 31 (76%) | 18 (90%) |
| Asian | 5 (6%) | 8 (10%) | 8 (20%) | 1 (5%) |
| Black | 2 (2%) | 1 (1%) | 0 | 1 (5%) |
| Other | 4 (4%) | 1 (1%) | 2 (5%) | 0 |
| Multiple | 0 | 1 (1%) | 0 | 0 |
|
| ||||
| Measurable disease | 51 (57%) | 43 (54%) | 29 (71%) | 13 (65%) |
|
| ||||
| Aromatase inhibitor resistance | ||||
| Primary | 33 (37%) | 33 (42%) | 11 (27%) | 11 (55%) |
| Secondary | 54 (61%) | 45 (57%) | 28 (68%) | 9 (45%) |
| Unknown | 2 (2%) | 1 (1%) | 2 (5%) | 0 |
|
| ||||
| Progesterone receptor status* | ||||
| Positive | 58 (65%) | 58 (73%) | 27 (66%) | 12 (60%) |
| Negative | 21 (24%) | 14 (18%) | 7 (17%) | 6 (30%) |
| Unknown | 10 (11%) | 7 (9%) | 7 (17%) | 2 (10%) |
|
| ||||
| Visceral disease | 51 (57%) | 42 (53%) | 21 (51%) | 10 (50%) |
|
| ||||
| Bone-only disease | 19 (21%) | 17 (22%) | 7 (17%) | 5 (25%) |
| ≥3 metastatic sites | 24 (27%) | 31 (39%) | 16 (39%) | 9 (45%) |
|
| ||||
| Number of previous therapies in the metastatic setting | ||||
| 0 | 24 (27%) | 20 (25%) | 5 (12%) | 2 (10%) |
| 1 | 33 (37%) | 36 (46%) | 11 (27%) | 7 (35%) |
| 2 | 23 (26%) | 15 (19%) | 8 (20%) | 5 (25%) |
| ≥3 | 9 (10%) | 8 (10%) | 17 (41%) | 6 (30%) |
|
| ||||
| PIK3CA mutation | ||||
| PIK3CA mutation positive | 38 (43%) | 32 (41%)† | 40 (98%)‡ | 20 (100%) |
| Helical domain | 18 (20%) | 11 (14%) | 20 (49%) | 7 (35%) |
| Kinase domain | 20 (22%) | 22 (28%) | 20 (49%) | 13 (65%) |
Data are median (range) or n (%). PIK3CA=phosphatidylinositol 3-kinase catalytic subunit α.
Based on central assessment; positivity was defined as 10% or more of cells positive.
One patient had a mutation in each domain.
One patient was found to have no PIK3CA mutation after enrolment.
In part 1, after a median follow-up of 17.5 months (IQR 15.4–19.4), 61 (69%) of 89 patients in the pictilisib group and 59 (75%) of 79 in the placebo group had progressed. Median progression-free survival was 6.6 months (95% CI 3.9–9.8) in the pictilisib group and 5.1 months (3.6–7.3) in the placebo group (HR 0.74 [95% CI 0.52–1.06]; p=0.096; figure 2A). All contributing progression-free survival events were due to disease progression, with no deaths reported in either group in the absence of documented progression. When patients were analysed according to PIK3CA mutation, median progression-free survival was 6.5 months (95% CI 3.7–9.8) in the pictilisib group versus 5.1 months (2.6–10.4) in the placebo group in patients with a PIK3CA-mutated tumour (HR 0.73 [95% CI 0.42–1.28]; p=0.268; figure 2B) and 5.8 months (3.6–11.1) versus 3.6 months (2.8–7.3) in those in which the mutation was not detected (HR 0.72 [95% CI 0.42–1.23]; p=0.23). Further post-hoc subgroup analyses according to baseline characteristics were consistent with findings in the overall population (figure 3), with the exception of patients whose tumours were positive for progesterone receptor. Exploratory subset analyses in patients with progesterone receptor-positive tumours treated with pictilisib had a median progression-free survival of 7.4 months (95% CI 5.6–12.8) versus 3.7 months (2.8–5.4) for those treated with placebo (HR 0.44 [95% CI 0.28–0.69]; p=0.0002; appendix p 9).
Figure 2. Progression-free survival in part 1.

(A) Intention-to-treat population. (B) Patients with phosphatidylinositol 3-kinase catalytic subunit α mutation. Crosses denote censored patients. HR=hazard ratio. FLV=fulvestrant.
Figure 3. Forest plot of hazard ratios for progression-free survival in part 1 according to patient characteristics at baseline.

ECOG=Eastern Cooperative Oncology Group. FLV=fulvestrant. NE=not estimable. PIK3CA=phosphatidylinositol 3-kinase catalytic subunit α. PFS=progression-free survival. PR=progesterone receptor.
*Data not available for one patient in the placebo group. †Based on central assessment; positivity was defined as 10% or more of cells positive.
Seven (7.9% [95% CI 3.2–15.5]) of 89 patients achieved an objective response in the pictilisib group compared with five (6.3% [2.1–14.2]) of 79 in the placebo group (p=0.70), whereas the proportion of patients who achieved clinical benefit was 22 (24.7%) of 89 (95% CI 16.2–35.0) patients in the pictilisib group versus 14 (17.7%) of 79 (10.0–27.9) in the placebo group (p=0.27). We also found no significant difference in median duration of response (9.43 months [4.1 to not estimable] vs 6.5 months [3.7 months to not estimable]; p=0.65). In patients with a PIK3CA mutation, six (16%) of 38 patients achieved an objective response in the pictilisib group and one (3%) of 32 achieved an objective response in the placebo group (p=0.73).
In part 2, after a median follow-up of 12.9 months (IQR 12.0–13.7), 23 (56%) of 41 patients had progressed in the pictilisib group, whereas 12 (60%) of 20 in the placebo group had a progression event, including one (5%) who died without progression. Median progression-free survival was 5.4 months (95% CI 3.8–8.3) in the pictilisib group and 10.0 months (3.6–13.0) in the placebo group (HR 1.07 [95% CI 0.53–2.18]; p=0.84). Three (7.3% [95% CI 1.5–19.9]) patients in the pictilisib group and one (5.0% [0.1–24.9]) in the placebo group had an objective response (p=0.73), whereas the proportion of patients who achieved a clinical response was eight (19.5% [95% CI 8.8–34.9) of 41 in the pictilisib group and seven (35.0% [15.4–59.2]) of 20 in the placebo group (p=0.19). Median duration of response was not reached in either group.
Secondary endpoints of the prognostic effect of PIK3CA mutation, pharmacokinetics of pictilisib with fulvestrant, and prevalence of PIK3CA mutation and PTEN loss have not been investigated further at this stage and are not included in this report because of the absence of benefit for pictilisib shown by the primary endpoint.
In part 1, observed adverse events were consistent with those previously described for single-agent pictilisib in phase 1a studies19,20 and for fulvestrant21 and were mostly of grade 1 or 2 severity (appendix p 6). Gastrointestinal disorders were the most common adverse event in both groups and skin or subcutaneous disorders were more frequent in the pictilisib group than in the placebo group. We typically observed the onset of gastrointestinal and dermatological toxicities during the first few treatment cycles. Hyperglycaemia (16 [18%] of 89 patients vs six [8%] of 79 patients) and pneumonitis (7 [8%] of 89 vs 1 [1%] of 79), treatment-related adverse events that have been associated with agents targeting the PI3K-AKT-mTOR pathway, were seen more frequently in the pictilisib group than in the placebo group.
Grade 3 or worse adverse events in three or more patients are shown in table 2. In part 1, 46 serious adverse events were reported in 28 (31%) of 89 patients in the pictilisib group (table 3). Grade 1–2: vomiting, pneumonitis, bronchopneumonia, abdominal pain, meningioma, and pleural effusion (one [1%] patient each); grade 3: rash (three [3%] patients), colitis, abdominal pain, epilepsy, and femur fracture (two [2%] patients each), vomiting, diarrhoea, haemorrhagic diarrhoea, nausea, hypokalaemia, agitation, dysphagia, fall, headache, hyperglycaemia, hypertension, hypophagia, infectious colitis, joint dislocation, oral candidiasis, osteoarthritis, paratyphoid fever, pneumonia, pneumonitis, pulmonary embolism, maculopapular rash, and generalised rash (one [1%] patient each); grade 4: asthenia, cardiac tamponade, hyperglycaemia, hyponatraemia, and neutropenia (one [1%] patient each). Two (2%) patients had fatal serious adverse events of breast cancer progression. 19 serious adverse events in 14 (16%) patients were considered related to treatment. In the placebo group, 17 serious adverse events were reported in ten (13%) of 79 patients. Grade 1–2: dyspnoea, eye pain, fatigue, palpitations, and ascites (one [1%] patient each); grade 3: breast cancer progression, dyspnoea, bone pain, pancreatitis, pulmonary embolism, sinusitis, syncope, viral infection, and vomiting (1 [1%] patient each); grade 4: mitral valve disease and small intestinal obstruction (one [1%] patient each). One (1%) patient had a fatal serious adverse event of breast cancer progression. Four serious adverse events (all in the same patient) were considered to be related to treatment. Adverse events leading to discontinuation of pictilisib in more than one patient in part 1 were rash (four [4%]), pneumonitis (three [3%]), diarrhoea (three [3%]), abdominal pain (two [2%]), stomatitis (two [2%]), and elevation of alanine or aspartate aminotransferase concentration (three [3%]). No adverse events causing discontinuation of placebo in more than one patient occurred.
Table 2.
Grade 3 or worse adverse events in three or more patients
| Pictilisib plus fulvestrant (n=89; part 2 n=42)
|
Placebo plus fulvestrant (part 1 n=79; part 2 n=19)
|
|||||
|---|---|---|---|---|---|---|
| Grade 3 | Grade 4 | Grade 5 | Grade 3 | Grade 4 | Grade 5 | |
| Part 1 | ||||||
|
| ||||||
| Maculopapular rash | 8 (9%) | 0 | 0 | 0 | 0 | 0 |
| Diarrhoea | 6 (7%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Fatigue | 7 (8%) | 0 | 0 | 0 | 0 | 0 |
| ALT concentration increased | 4 (4%) | 1 (1%) | 0 | 1 (1%) | 0 | 0 |
| Rash | 6 (7%) | 0 | 0 | 0 | 0 | 0 |
| AST concentration increased | 3 (3%) | 1 (1%) | 0 | 2 (3%) | 0 | 0 |
| Hyperglycaemia | 3 (3%) | 1 (1%) | 0 | 0 | 0 | 0 |
| Anaemia | 1 (1%) | 0 | 0 | 2 (3%) | 1 (1%) | 0 |
| Hypertension | 4 (4%) | 0 | 0 | 0 | 0 | 0 |
| Hypokalaemia | 2 (2%) | 1 (1%) | 0 | 1 (1%) | 0 | 0 |
| Vomiting | 3 (3%) | 0 | 0 | 1 (1%) | 0 | 0 |
| Colitis | 3 (3%) | 0 | 0 | 0 | 0 | 0 |
| Generalised rash | 3 (3%) | 0 | 0 | 0 | 0 | 0 |
| Nausea | 3 (3%) | 0 | 0 | 0 | 0 | 0 |
| Neutropenia | 2 (2%) | 1 (1%) | 0 | 0 | 0 | 0 |
|
| ||||||
| Part 2 | ||||||
|
| ||||||
| Diarrhoea | 4 (10%) | 0 | 0 | 0 | 0 | 0 |
| Rash | 3 (7%) | 0 | 0 | 0 | 0 | 0 |
Data are n (%). AE=adverse event. ALT=alanine aminotransferase. AST=aspartate aminotransferase.
Table 3.
Summary of adverse
| Part 1
|
Part 2
|
|||
|---|---|---|---|---|
| Pictilisib plus fulvestrant (n=89) | Placebo plus fulvestrant (n=79) | Pictilisib plus fulvestrant (n=42) | Placebo plus fulvestrant (n=19) | |
| Grade ≥3 AEs | 54 (61%) | 22 (28%) | 15 (36%) | 7 (37%) |
|
| ||||
| AEs leading to discontinuation | 20 (22%) | 4 (5%) | 10 (24%) | 1 (5%) |
|
| ||||
| AEs leading to dose reduction | 21 (24%) | 1 (1%) | 7 (17%) | 2 (11%) |
|
| ||||
| Serious AEs* | ||||
| All | 28 (31%) | 10 (13%) | 4 (10%) | 3 (16%) |
| Grade 1–2 | 5 (6%) | 3 (4%) | 0 (0%) | 1 (5%) |
| Grade 3 | 22 (25%) | 8 (10%) | 4 (10%) | 2 (11%) |
| Grade 4 | 4 (4%) | 2 (3%) | 0 (0%) | 0 (0%) |
| Grade 5 | 1 (1%) | 1 (1%) | 0 (0%) | 0 (0%) |
AE=adverse event.
Patients with separate serious AEs of different grades can be recorded more than once.
Median exposure to pictilisib in part 1 was 2.9 months (IQR 1.5–6.5) and to placebo was 3.9 months (1.5–10.8). Many patients did not receive the full dose of 340 mg pictilisib until progression, with 40 (45%) of 89 patients having at least one pictilisib dose modification to 260 mg or 200 mg for adverse events (21 [24%] of 89; most commonly for diarrhoea: nine [10%] of 89) or discontinuing pictilisib for reasons other than a disease progression event (30 [34%] of 89), or both (tables 3, 4). In the placebo group, one (1%) of 79 patients had a placebo dose reduction for arthralgia and 12 (15%) of 79 discontinued because of non-progressive disease events (table 3).
Table 4.
Reasons for discontinuation
| Part 1
|
Part 2
|
|||
|---|---|---|---|---|
| Pictilisib plus fulvestrant (n=89) | Placebo plus fulvestrant (n=79) | Pictilisib plus fulvestrant (n=41) | Placebo plus fulvestrant (n=20) | |
| Discontinued | 80 (90%) | 69 (87%) | 34 (83%) | 15 (75%) |
| Disease progression | 50 (56%) | 57 (72%) | 19 (46%) | 11 (55%) |
| Non-disease progression events | 30 (34%) | 12 (15%) | 15 (37%) | 4 (20%) |
| AE | 16 (18%) | 2 (3%) | 10 (24%) | 1 (5%) |
| Investigator decision | 8 (9%) | 5 (6%) | 2 (5%) | 1 (5%) |
| Patient decision | 5 (6%) | 4 (5%) | 3 (7%) | 2 (10%) |
| Protocol violation | 0 | 1 (1%) | 0 | 0 |
| Not specified | 1 (1%) | 0 | 0 | 0 |
Where the reason for discontinuation was reported in this table as an investigator or patient decision, an AE might still have been reported as leading to discontinuation in table 3. AE=adverse event.
The safety profile observed in part 2 was similar to that seen in part 1, although the pictilisib dose was reduced to 260 mg in part 2 after a protocol amendment due to the high proportion of discon tinuations in part 1. Most adverse events were mild to moderate (table 3, appendix p 8). The most common adverse events leading to discontinuation of pictilisib were diarrhoea (three [7%]) and rash (four [10%]). One (5%) patient discontinued placebo because of grade 3 haemolytic anaemia, with grade 1 dyspepsia and elevations in liver enzymes. Seven (17%) patients in the pictilisib group had dose reductions for adverse events, namely grade 2 maculopapular rash (two [5%]) and grade 3 rash, grade 1 pruritus, grade 3 diarrhoea, grade 2 stomatitis, and increased grade 2 aspartate amino transferase concentration (one [2%] each). Reasons for dose reductions for adverse events in the placebo group were grade 1 anaemia and grade 2 myalgia. In the pictilisib group, six serious adverse events were reported in four (10%) patients. Grade 3: pleural effusion, colitis, bone pain, diarrhoea, nausea, and vomiting (one [2%] patient each). In the placebo group, three serious adverse events were reported in three (15%) patients. Grade 3: pleural effusion, haemolytic anaemia, and asthenia (one [5%] patient each). No fatal treatment-related adverse events occurred. Two (5%) patients in the pictilisib group had treatment-related adverse events (colitis in one [2%] patient and diarrhoea, nausea, and vomiting in another). One treatment-related serious adverse event occurred in one (5%) of 19 patients in the placebo group (haemolytic anaemia). Median exposure to pictilisib was 4.2 months (IQR 3.0–8.0) compared with 8.6 months (2.0–12.1) for placebo. Details of adverse events in part 2 are shown in the appendix (p 8).
Discussion
In our study, addition of pictilisib to fulvestrant was not associated with improved progression-free survival for patients with endocrine-resistant advanced breast cancer, regardless of the presence or absence of PIK3CA mutation. Many patients could not receive the full planned dose of pictilisib due to the toxicity of this agent. An unmet medical need exists for treatments to overcome endocrine resistance in patients with hormone receptor-positive breast cancer. Increased PI3K signalling has been implicated in endocrine resistance,3,4 and inhibition of the PI3K-AKT-mTOR pathway by the mTOR inhibitor everolimus has shown promising activity in patients who are refractory to letrozole or anastrozole.7 Our study is, to our knowledge, the first report of a randomised clinical study assessing a pan-PI3K inhibitor in patients with metastatic breast cancer. Safety was consistent with that previously observed in phase 1.19 Gastrointestinal and skin disorders were the most significant toxicities in both parts of the study.
The presence or absence of PIK3CA mutation in patients’ tumours was not associated with pictilisib benefit as measured by progression-free survival or objective response. A confounding factor in interpretation of these data is that exposure was limited by tolerability and thus many patients might not have achieved sustained pharmacological pathway inhibition. The predictive value of PIK3CA mutations in this setting therefore remains unclear. However, consistent with the results from this study, other investigators have found little or no association between PIK3CA mutation status and the efficacy of other PI3K pathway inhibitors such as everolimus in combination with exemestane,16 buparlisib (BKM120) in patients with hormone receptor-positive metastatic disease,22 or pictilisib in the presurgical setting.23 The inability of PIK3CA mutation status to predict benefit of these PI3K pathway inhibitors could reflect the absence of observed association between PIK3CA mutation status and PI3K pathway activation status in hormone receptor-positive breast cancer. In HER2-positive cancers, an association has been shown between PIK3CA mutation status and the efficacy of everolimus with trastuzumab and chemotherapy in a retrospective combined analysis of BOLERO-1 and BOLERO-3.24 Thus, disease context, therapeutic com binations, and inhibitor selectivity could all play an important role in establishing if PIK3CA mutations have predictive value, and all of these factors should be assessed carefully in future diagnostic-focused studies.
This study was designed to be hypothesis generating; conclusions regarding the efficacy of pictilisib are limited by the modest sample size. However, the absence of pictilisib benefit is different from what might have been predicted on the basis of observations made in the presurgical setting,23 which showed that the combination of pictilisib and anastrozole had a significantly greater anti proliferative effect in tumours than did anastrozole alone. The absence of efficacy in our study might have been due to an inability to maintain pictilisib exposures that adequately inhibit the PI3K pathway for sustained periods. We selected the 340 mg dose used in part 1 of the study on the basis of the single-agent maximum tolerated dose, as well as pharmacokinetic and pharmacodynamic data from a phase 1 study of pictilisib in solid tumours.19 A pictilisib dose of 450 mg as a single agent was administered in phase 1, which had more consistent pharmacodynamic modulation than did the 340 mg dose, on the basis of predose and postdose tumour biopsies of downstream PI3K markers, but this dose was not well tolerated enough to move forward as a phase 2 dose.19 In this study, 45% of patients in part 1 did not receive the full 340 mg dose of pictilisib because of tolerability issues, particularly rash, and either received a reduced dose or discontinued for reasons other than tumour progression. The inability to consistently achieve pictilisib exposures required for robust pharmacodynamic modulation suggests that, in many patients, only partial target blockade was achieved, which could explain the absence of significant improvement in progression-free survival observed.
Given these observations and the absence of benefit for pictilisib according to the primary endpoint in either part of the study, pictilisib does not appear to have a sufficient therapeutic index to support further investigation in this setting. Nevertheless, targeting of the PI3K-AKT-mTOR pathway overall remains a promising approach to extending endocrine sensitivity for oestrogen receptor-driven tumours. Identification of oestrogen receptor 1-activating mutations has shown that oestrogen receptor-positive tumours might remain highly dependent on oestrogen receptor signalling even after multiple rounds of treatment.25 Together with the very high prevalence of PI3K pathway alterations in oestrogen receptor-positive tumours, further dissection of mechanisms of endocrine resistance will be essential to develop the next generation of combination therapies. Defined subsets within oestrogen receptor-positive breast cancer might be needed to maximise the benefit to risk ratio for these patients.
Exploratory subset analyses in patients with progesterone receptor-positive tumours showed an improvement in progression-free survival with addition of pictilisib. These data generate hypotheses regarding tumours with evidence of active oestrogen receptor signalling (as shown by transcriptional progesterone receptor expression) that might be particularly sensitive to the co-dependencies of oestrogen receptor and PI3K pathways. Patients with progesterone receptor-negative tumours tend to develop endocrine resistance quickly and have different oestrogen receptor-driven transcriptional outputs, which probably reflect very different mechanisms of endocrine resistance.26 Given the exploratory nature of these analyses, further studies are necessary to determine the extent of the role of PI3K signalling in endocrine resistance, specifically in progesterone receptor-positive tumours.
Alternative strategies to specifically target the α subunit of PI3K or the mutated kinase, which could have a wider therapeutic index than do pan-PI3K inhibitors, are under development. These strategies include alpelisib, which is equipotent against both the wild-type and mutated PI3Kα subunit and showed some single-agent activity in patients with PIK3CA-mutated breast cancer,27 and taselisib, which displays greater selectivity for mutant than for wild-type PI3Kα28 and might afford a better therapeutic index. Taselisib has also shown increased clinical activity in PIK3CA-mutant tumours, both as a single agent and in combination with fulvestrant or letrozole in patients with hormone receptor-positive advanced breast cancer.29,30 A phase 3 randomised trial is underway of taselisib in combination with fulvestrant in patients with advanced or metastatic breast cancer recurrent after treatment with an aromatase inhibitor and enriched for patients with PIK3CA-mutant tumours (NCT02340221).
Research in context.
Evidence before this study
To put this trial into the context of other studies of endocrine-resistant metastatic breast cancer, we searched PubMed and abstracts from the American Society of Clinical Oncology annual meeting, San Antonio Breast Cancer Symposium, European Society for Medical Oncology biennial meeting, and European Cancer Congress biennial meeting using the search terms “metastatic breast cancer”, “endocrine-resistant”, “PI3 kinase”, and “mTOR”, including full names and abbreviations, selecting relevant publications from Jan 1, 2011, to Aug 31, 2015, for full manuscripts andJan 1, 2006, and Aug 31, 2015, for abstracts, in English. The search revealed phase 1 and 2 data showing activity of phosphatidylinositol 3-kinase (PI3K)-protein kinase B-mTOR inhibitors in breast cancer in various settings and phase 3 data for the mTOR inhibitor everolimus in endocrine-resistant metastatic breast cancer.
Added value of this study
This study of pictilisib is, to our knowledge, the first randomised clinical study assessing a PI3K inhibitor in patients with metastatic breast cancer. Addition of pictilisib to fulvestrant was associated with a progression-free survival hazard ratio of 0.74 (95% CI 0.52–1.06) compared with placebo and fulvestrant, which was not significant (p=0.096). PIK3CA mutation status had no effect on pictilisib benefit. Pictilisib dosing was limited by substantial toxicity, with 45% of patients requiring a dose reduction or discontinuation.
Implications of all the available evidence
The mTOR inhibitor everolimus has shown promising activity in combination with exemestane in patients with metastatic breast cancer who are refractory to letrozole or anastrozole, whereas pictilisib and other pan-PI3K inhibitors have shown some activity in phase 1 and 2 studies. Although efficacy of pictilisib was potentially limited by the inability to administer a mechanistically optimal dose, this study provides evidence that will help guide future research, suggesting that an agent with higher selectivity for specific PI3K isoforms or mutants than that used in this study might improve tolerability and afford a more robust therapeutic index.
Acknowledgments
We thank the patients, their families, the nurses, and the investigators who participated in this study. Third-party writing assistance for this manuscript was provided by John Carron.
IEK reports grant support from Genentech during the conduct of the study and personal fees from Amgen. IAM reports research support from Novartis. MDi reports grants from Genentech/Roche during the conduct of the study and personal fees from Genentech/Roche and Novartis, grants from Eli Lilly, and grants and personal fees from Pfizer and AstraZeneca, outside the submitted work. SJ reports personal fees from Roche/Genentech, Eli Lilly, AstraZeneca, Novartis, and GlaxoSmithKline, and research funding from Pfizer. DAY reports personal fees from Genentech/Roche outside the submitted work. BM reports personal fees and travel support from Roche, Novartis, and Merck, and personal fees from AstraZeneca, Pfizer, Bristol-Myers Squibb, Astellas, Merck Sharp & Dohme, Janssen, Bayer, and Amgen, outside the submitted work. SCL reports personal fees and travel support from Roche and AstraZeneca, personal fees from Novartis, and research support from Elsai and GlaxoSmithKline, outside the submitted work. RdB reports personal fees from Roche Australia outside the submitted work. EAP reports personal fees from Genentech outside the submitted work and became an employee of Genentech after the submitted work was completed. MP reports personal fees from Genentech outside the submitted work. ME reports personal fees and non-financial support from Bioclassifier outside the submitted work, has a patent pending for PAM50, and has interest in Prosigna through patents and ownership of commercialisation rights. EW reports grants from Genentech and Novartis outside the submitted work. SG, MDe, ML, GL, JQ, and JH are employees of Genentech/Roche. MDe has a pending patent (pictilisib treatment in breast cancer; 62/001,764).
Footnotes
Contributors
IEK, PS, MP, ME, GL, SG, ML, JH, and MDe conceived and designed the study. AF-T, IAM, DAY, SJ, BM, SCL, RdB, SJ, PS, SM, SV, and VG collected data. IEK, IAM, VG, SJ, BM, AF-T, RdB, EAP, MP, ME, EW, MDe, ML, GL, and JQ analysed data. IEK, IAM, VG, SJ, BM, AF-T, RdB, EAP, MP, ME, EW, MDe, ML, GL, and JH interpreted data. IEK, IAM, VG, MDi, SJ, DAY, BM, AF-T, SCL, RdB, EAP, ME, EW, SG, MDe, ML, GL, JQ, and PS wrote the manuscript and IAM, VG, DAY, AF-T, SCL, RdB, KP, MP, EW, GL, JH, SM, SV, and PS reviewed the manuscript. All authors approved the final version of the submitted report.
Declaration of interests
All other authors declare no competing interests.
Contributor Information
Ian E Krop, Dana-Farber Cancer Institute, Boston, MA, USA.
Ingrid A Mayer, Vanderbilt-Ingram Cancer Center, Nashville, TN, USA.
Vinod Ganju, Peninsula Oncology Centre, Melbourne, VIC, Australia.
Maura Dickler, Memorial Sloan Kettering Cancer Center, New York, NY, USA.
Stephen Johnston, The Royal Marsden Hospital, London, UK.
Serafin Morales, Hospital Arnau de Vilanova, Lleida, Spain.
Denise A Yardley, Sarah Cannon Research Institute, and Tennessee Oncology, Nashville, TN, USA.
Prof. Bohuslav Melichar, Palacky University Medical School and Teaching Hospital, Olomouc, Czech Republic.
Andres Forero-Torres, University of Alabama, Birmingham, AL, USA.
Soo Chin Lee, National University Hospital, Singapore.
Richard de Boer, Royal Melbourne Hospital, Parkville, VIC, Australia.
Katarina Petrakova, Masaryk Memorial Cancer Institute, Brno, Czech Republic.
Susanne Vallentin, Herlev University Hospital, Copenhagen, Denmark.
Edith A Perez, Mayo Clinic, Jacksonville, FL, USA.
Prof. Martine Piccart, Institut Jules Bordet, Université Libre de Bruxelles, Brussels, Belgium.
Prof. Matthew Ellis, Washington University School of Medicine, St Louis, MO, USA.
Prof. Eric Winer, Dana-Farber Cancer Institute, Boston, MA, USA.
Steven Gendreau, Genentech, South San Francisco, CA, USA.
Mika Derynck, Genentech, South San Francisco, CA, USA.
Mark Lackner, Genentech, South San Francisco, CA, USA.
Gallia Levy, Genentech, South San Francisco, CA, USA.
Jiaheng Qiu, Genentech, South San Francisco, CA, USA.
Jing He, Genentech, South San Francisco, CA, USA.
Prof. Peter Schmid, Barts Cancer Institute, Queen Mary University of London, London, UK.
References
- 1.Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004;11:643–58. doi: 10.1677/erc.1.00776. [DOI] [PubMed] [Google Scholar]
- 2.Johnston SR. New strategies in estrogen receptor-positive breast cancer. Clin Cancer Res. 2010;16:1979–87. doi: 10.1158/1078-0432.CCR-09-1823. [DOI] [PubMed] [Google Scholar]
- 3.Miller TW, Hennessy BT, González-Angulo AM, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120:2406–13. doi: 10.1172/JCI41680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shou J, Massarweh S, Osborne CK, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–35. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 5.Baselga J, Semiglazov V, van Dam P, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009;27:2630–37. doi: 10.1200/JCO.2008.18.8391. [DOI] [PubMed] [Google Scholar]
- 6.Bachelot T, Bourgier C, Cropet C, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30:2718–24. doi: 10.1200/JCO.2011.39.0708. [DOI] [PubMed] [Google Scholar]
- 7.Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–29. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piccart M, Hortobagyi GN, Campone M, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2. Ann Oncol. 2014;25:2357–62. doi: 10.1093/annonc/mdu456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maruyama N, Miyoshi Y, Taguchi T, Tamaki Y, Monden M, Noguchi S. Clinicopathologic analysis of breast cancers with PIK3CA mutations in Japanese women. Clin Cancer Res. 2007;13:408–14. doi: 10.1158/1078-0432.CCR-06-0267. [DOI] [PubMed] [Google Scholar]
- 10.Kalinsky K, Jacks LM, Heguy A, et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res. 2009;15:5049–59. doi: 10.1158/1078-0432.CCR-09-0632. [DOI] [PubMed] [Google Scholar]
- 11.Miller TW, Pérez-Torres M, Narasanna A, et al. Loss of phosphatase and tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69:4192–201. doi: 10.1158/0008-5472.CAN-09-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abramson VG, Cooper Lloyd M, Ballinger T, et al. Characterization of breast cancers with PI3K mutations in an academic practice setting using SNaPshot profiling. Breast Cancer Res Treat. 2014;145:389–99. doi: 10.1007/s10549-014-2945-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loi S, Haibe-Kains B, Majjaj S, et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc Natl Acad Sci USA. 2010;107:10208–13. doi: 10.1073/pnas.0907011107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loi S, Michiels S, Baselga J, et al. PIK3CA genotype and a PIK3CA mutation-related gene signature and response to everolimus and letrozole in estrogen receptor positive breast cancer. PLoS One. 2013;8:e53292. doi: 10.1371/journal.pone.0053292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabine VS, Crozier C, Brookes CL, et al. Mutational analysis of PI3K/AKT signaling pathway in tamoxifen exemestane adjuvant multinational pathology study. J Clin Oncol. 2014;32:2952–58. doi: 10.1200/JCO.2013.53.8272. [DOI] [PubMed] [Google Scholar]
- 16.Hortobagyi GN, Piccart-Gebhart MJ, Rugo HS, et al. Correlation of molecular alterations with efficacy of everolimus in hormone receptor-positive, HER2-negative advanced breast cancer: results from BOLERO-2. J Clin Oncol. 2013;31(suppl) abstract LBA509. [Google Scholar]
- 17.Fu X, Creighton C, Biswal NC, et al. Overcoming endocrine resistance due to reduced PTEN levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase B, or mitogen-activated protein kinase kinase. Breast Cancer Res. 2014;16:430. doi: 10.1186/s13058-014-0430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Brien C, Wallin JJ, Sampath D, et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3′ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010;16:3670–83. doi: 10.1158/1078-0432.CCR-09-2828. [DOI] [PubMed] [Google Scholar]
- 19.Sarker D, Ang JE, Baird R, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:77–86. doi: 10.1158/1078-0432.CCR-14-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Hoff DD, LoRusso P, Demetri GD, et al. A phase I dose-escalation study to evaluate GDC-0941, a pan-PI3K inhibitor, administered QD or BID in patients with advanced or metastatic solid tumors. J Clin Oncol. 2011;29(suppl) abstract 3052. [Google Scholar]
- 21.Robertson JF, Osborne CK, Howell A, et al. Fulvestrant versus anastrozole for the treatment of advanced breast carcinoma in postmenopausal women: a prospective combined analysis of two multicenter trials. Cancer. 2003;98:229–38. doi: 10.1002/cncr.11468. [DOI] [PubMed] [Google Scholar]
- 22.Mayer IA, Abramson VG, Balko JM, et al. SU2C phase Ib study of pan-PI3K inhibitor BKM120 with letrozole in ER+/HER2- metastatic breast cancer (MBC) J Clin Oncol. 2012;30(suppl) abstract 510. [Google Scholar]
- 23.Schmid P, Pinder SE, Wheatley D, et al. Preoperative window of opportunity study of the PI3K inhibitor pictilisib (GDC-0941) plus anastrozole vs anastrozole alone in patients with ER+, HER2-negative operable breast cancer (OPPORTUNE study) Cancer Res. 2015;75 abstract S2-03. [Google Scholar]
- 24.Slamon DJ, Hurvitz SA, Chen D, et al. Predictive biomarkers of everolimus efficacy in HER2+ advanced breast cancer: combined exploratory analysis from BOLERO-1 and BOLERO-3. J Clin Oncol. 2015;33(suppl) abstract 512. [Google Scholar]
- 25.Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross-Innes CS, Stark R, Teschendorff AE, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–93. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juric D, Argiles G, Burris HA, et al. Phase I study of BYL719, an alpha-specific PI3K inhibitor, in patients with PIK3CA mutant advanced solid tumors: preliminary efficacy and safety in patients with PIK3CA mutant ER-positive (ER+) metastatic breast cancer (MBC) Cancer Res. 2012;72 abstract 610-07. [Google Scholar]
- 28.Olivero AG, Heffron TP, Baumgardner M, et al. Discovery of GDC-0032: a beta-sparing PI3K inhibitor active against PIK3CA mutant tumors. Cancer Res. 2013;73 abstract DDT02-01. [Google Scholar]
- 29.Saura C, Sachdev J, Patel MR, et al. Ph1b study of the PI3K inhibitor taselisib (GDC-0032) in combination with letrozole in patients with hormone receptor-positive advanced breast cancer. Cancer Res. 2015;75 abstract D5-2. [Google Scholar]
- 30.Juric D, Krop I, Ramanathan RK, et al. GDC-0032, a beta isoform-sparing PI3K inhibitor: results of a first-in-human phase Ia dose escalation study. Cancer Res. 2013;73 abstract LB-64. [Google Scholar]