

Abstract
Enzymes catalyze chemical transformations with outstanding stereo- and regio-specificities, but many enzymes are limited by their long reaction times. Here, we describe a general method to accelerate enzymes using pressure waves contained within thin films. Each enzyme responds best to specific frequencies of pressure waves, and we report acceleration landscapes for each protein. A vortex fluidic device introduces pressure waves that drive increased rate constants (kcat) and enzymatic efficiency (kcat/Km). Four enzymes displayed an average seven-fold acceleration with deoxyribose-5-phosphate aldolase (DERA) achieving an average 15-fold enhancement through this approach. In solving a common problem in enzyme catalysis, we have uncovered a powerful, generalizable tool for enzyme acceleration. This research provides new insights into previously uncontrolled factors affecting enzyme function.
Keywords: Biocatalysis, Enzyme acceleration, Aldolase, Hydrolases, Vortex Fluidics
Graphical abstract
Enzymes – They’re Picking Up Good Vibrations: A simple, generalizable approach to accelerate enzymes through the use of pressure waves in thin films has been developed. Each enzyme responds best to specific vibrations uncovering a previously unappreciated aspect of biocatalysis.
Enzymes make life possible by catalyzing diverse and challenging chemical transformations with exquisite specificity. Applications in both industry[1] and academia[2] rely on the selectivity and power of enzymes to catalyze otherwise challenging transformations. Biocatalysts offer remarkable rate accelerations compared to uncatalyzed reactions, with typical rate accelerations (kcat/kuncat) of 105- to 1015-fold faster.[3] Though some enzymes are diffusion-limited,[4] the catalytic rates of enzymes are often more typically limited by their catalytic efficiency (kcat/Km); additionally, molecular crowding, along with product and substrate inhibition, can reduce enzyme efficiency.[5] Though some enzymes catalyse transformations with rapid rates (e.g., laccases, fumarases and alcohol dehydrogenases),[6] other enzymes operate at only modest reaction rates, requiring long reaction times and carefully optimized conditions; for example, DERA requires long processing times (hours to days), and is substrate-inhibited.[7] We report a process that accelerates four different enzymes at standard temperature and pressure, but many water-soluble enzymes could be accelerated.
Recently, vortex fluidic devices (VFDs) have been used to accelerate covalent and non-covalent bond formation. VFDs process solutions in thin films by the rapid rotation of a sample tube (Figure 1).[8] Within the thin film, species are subjected to high levels of shear stress, mass transfer, and vibrational energy input at specific rotational speeds. For example, the VFD demonstrated the effective folding of four different proteins within minutes at standard temperature and pressure.[9] The VFD has also been used to improve the synthesis of lidocaine[10] and several other organic transformations.[11] In a continuous flow regime, flow rates of up to 20 mL/min can be achieved to process up to 30 L per day in the current, benchtop configuration. Since VFD processing increased the rates of organic reactions and protein folding, we hypothesized biocatalysis, which requires both reactivity and correct protein fold could benefit.
Figure 1.

The VFD and its Faraday waves. (A) A schematic of the VFD. (B) A VFD sample tube containing 5 mL solution rotating at 3.50 krpm at a tilt angle, θ, of 45°, shows Faraday waves formed within the thin film. Overlaid schematically, such pressure waves can affect the enzyme-substrate complex. (C) This enlargement focuses on VFD-generated Faraday waves. Additional images of Faraday waves can be found in Figure S14.
Control reactions with alkaline phosphatase demonstrated the requirements for high, specific rotational speeds of the VFD to generate a thin film containing the enzyme for accelerated catalysis (Figures S2, S3 and S4). VFD-mediated acceleration of four biocatalysts was compared to identical unprocessed enzyme-substrate solutions for efficient reaction optimization (Figure 2). First, VFD processing times were varied to identify short time periods (10 min to 2 h) suitable for further optimization (Figure 2A). Esterase produced a lower VFD-based enhancement (two-fold) compared to the other three enzymes; esterase also needed longer reactions times due to the enzyme’s requirements for low substrate concentrations.[12] In general, after long time periods, the substrate is expended, and the unprocessed solution can attain similar levels of substrate conversion. Furthermore, VFD-processed solutions display parallel activities to the non-VFD counterparts for the first few minutes before rapid acceleration (e.g., alkaline phosphatase in Figure S9). A similar lag period before rate acceleration has been described previously for ultrasound-accelerated enzyme acceleration.[13]
Figure 2.
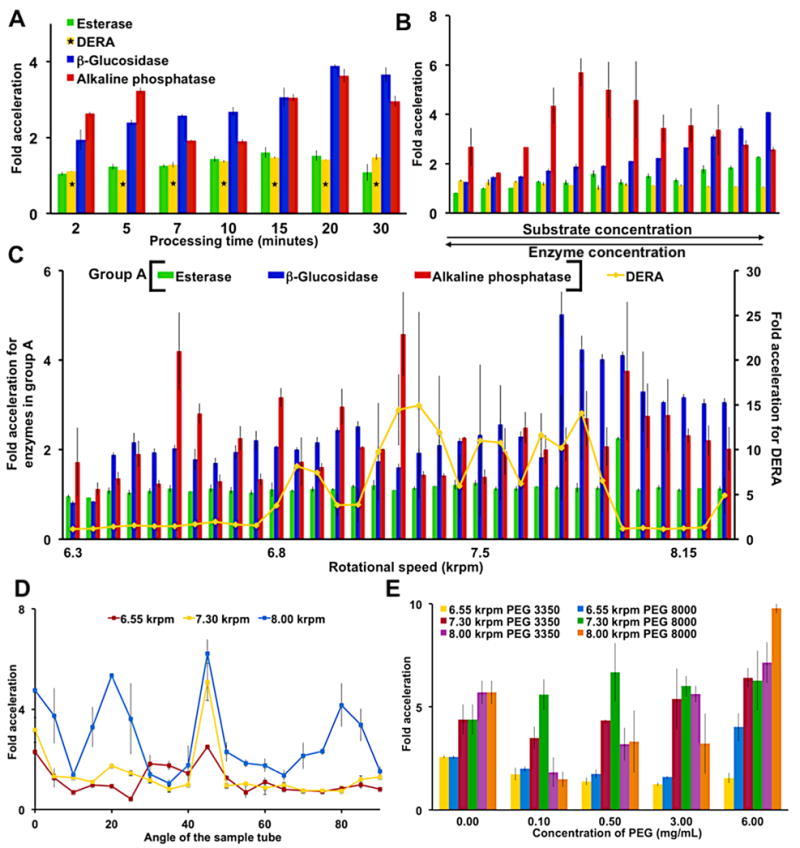
Parameters for accelerated biocatalysis of the four enzymes. Fold acceleration was determined by the ratio of the VFD-mediated substrate conversion to an identical enzyme-substrate solution not treated by the VFD. (A) A time dependent study at a fixed rotational speed (8.00 krpm) reveals processing times for further optimization. As indicated with a DERA required longer reaction times of 60, 80, 100, 120, 140, 160 and 180 min. (B) Simultaneous changes to the substrate and enzyme concentrations at a 8.00 krpm rotational speed mapped the reaction landscape (Table S2A–D). (C) Rotational speed scans in 50 rpm increments identify harmonic oscillations associated with Faraday wave-promoted biocatalysis. Error bars are larger for DERA than any other enzyme due to the product’s non-linear, fluorescence calibration curve. (D) Varying the tilt angle of the sample tube identifies 45° as optimal. Thus, a 45° tilt angle was used throughout this report. (E) The addition of PEG dramatically slowed the non-VFD control, but the VFD processed solution demonstrated significant catalytic activity. Error bars indicate the standard deviation around the mean (n=3 with three independent measurements on three different VFDs). With the exception of a single data point requiring 90% confidence limits, all data reported here have no overlapping errors within 95% confidence limits. The concentrations of enzymes and substrates are as follows: alkaline phosphatase (6.77 nM) and its substrate p-nitrophenol phosphate (0.17 mM), β-glucosidase (19.3 nM) and its substrate 4-nitrophenyl β-D-glucopyranoside (7.5 mM), esterase (0.12 nM) and its substrate p-nitrophenol acetate (44 μM) and DERA (7.69 μM) and its fluorogenic substrate (0.52 mM) unless otherwise indicated, and as described in Table S2A-D.
Next, substrate and enzyme concentrations were simultaneously varied for the rapid scanning of reaction space to find effective reaction conditions (Figure 2B). This optimization unexpectedly revealed that VFD-mediated enzyme reactions are less susceptible to substrate inhibition than conventional conditions. For example, β-glucosidase without VFD processing encounters substrate inhibition at around 3.1 mM 4-nitrophenyl β-D-glucopyranoside; VFD processing delays the onset of substrate inhibition until almost a three-fold higher concentration (Figure S10B). With the exception of DERA, the three enzymes tolerated higher concentrations of substrate without losing VFD-mediated acceleration. This decrease in substrate inhibition suggests the VFD increases enzymatic kcat, as further demonstrated below. DERA catalyzed the retro-aldol reaction of a pro-fluorophore at 144 μmol h−1L−1 when processed in the VFD (7.90 krpm rotational speed), compared to 10.7 μmol h−1L−1 under non-VFD conditions. DERA has previously been employed to synthesize high-value, complex, polyoxygenated compounds.[7b] The VFD-mediated DERA reaction achieved an average 15-fold enhancement. Conventional approaches to improving DERA have applied extensive screening[7b] and multiple rounds of error-prone PCR. For example, screening 20,000 colonies yielded a 10-fold increase in DERA activity.[14] Comparing the efforts required to achieve >10-fold acceleration by VFD in several days with conventional protein engineering, highlights the power of the approach reported here.
Enzyme acceleration by the VFD is sensitive to the tilt angle of the sample tube and the viscosity of the solution (Figure 2D–E). A tilt angle of 45° provided the strongest response, as has been previously observed in other VFD experiments.[8b] Second, concentrations of viscous, steric crowding reagents that decrease or terminate enzymatic catalysis in the non VFD-mediated control conditions were overcome in the VFD. Biocatalytic acceleration was achieved, for example, in high concentrations of PEG 8000 (6.00 mg/ml, 0.75 M), a condition that suppresses enzymatic catalysis in non-VFD conditions. Through rapid micro mixing or other associated phenomena, VFD-processed alkaline phosphatase tolerated high concentration of PEG 8000, resulting in ≈9-fold enhancement. The relative indifference to high concentrations of substrate and steric crowding suggests the VFD could be applied to processes requiring complex mixtures and minimal amounts of solvent.
The dependence on rotational speeds was also specific to each enzyme (Figure 2C and S11). Such requirements likely reflect differences in enzyme size, structure and dynamics. Esterase for example was highly dependent on a single rotational speed for enhanced activity. When processing esterase under VFD-mediated conditions, the only rotational speed to generate an enhancement was 8.00-krpm; at all other rotational speeds, the enzyme behaved similarly to non-VFD-mediated conditions. To map out the fine details of such resonances, a high-resolution scan of rotational speeds examined the acceleration of alkaline phosphatase and β-glucosidase (Figure 3). The rotational landscapes are intricate with little overlap of optimal rotational speeds. Device-specific variations in rotational landscapes were also observed, likely due to differences between device bearings and components (e.g., the Teflon collar, which wears due to friction from the sample tube); thus, Figure 3 depicts two enzymes processed by a single VFD. In addressing this issue of wear, and avoiding variable vibrations, we turned to 3D printing. Fabricating the collar out of high-density ABS plastic allowed an interchangeable sleeve to be incorporated. Changing the insert upon wear insures reproducibility of the reported experiments (Figure S19).
Figure 3.
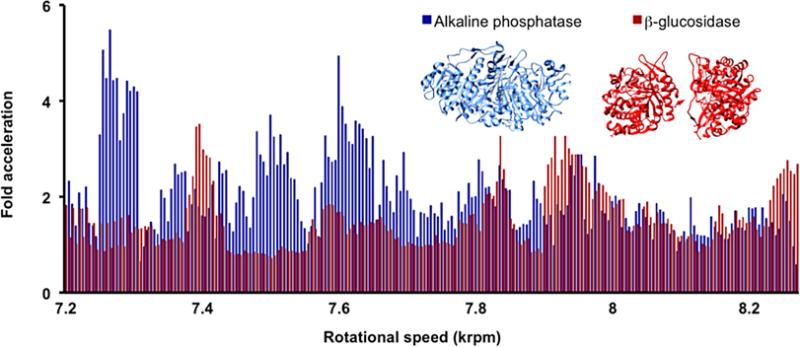
The rotational landscape of β-glucosidase and alkaline phosphatase. Though the two enzymes have similar levels of response at some rotational speeds, distinctly different rotational landscapes are revealed. The results demonstrate the enzyme specificity of VFD-mediated acceleration. Each data point represents the mean (n=2) for a 10 min reaction at the indicated rotational speeds. The alkaline phosphatase enzyme-substrate solution used Fast alkaline phosphatase (6.77 nM) and p-nitrophenol phosphate solution (0.17 mM) whilst the β−glucosidase enzyme-substrate system used β-glucosidase (19.3 nM) and 4-nitrophenyl β-D-glucopyranoside (7.5 mM).
Michaelis-Menten-based experiments were performed with β−glucosidase, and the kinetic constants for both VFD-processed and non-VFD conditions were derived (Table 1). The kcat in the VFD-mediated reaction was around 2.5-fold faster than the non-VFD reaction (Figure 3). A lower Michaelis-Menten constant (Km) was also obtained for the VFD-processed enzyme-substrate solution; 2.50 mM compared to 3.76 mM for a non-VFD mediated reaction. The decrease in Km demonstrates the higher affinity for the β-glucosidase-substrate interaction under VFD-mediated conditions. The increase of kcat and the decrease in Km leads to an increase in enzyme efficiency (kcat/Km) of around 3.5-fold for the VFD-mediated reaction.
Table 1.
Michaelis-Menten parameters for the VFD vs. non-VFD mediated processing of β-glucosidase.*
| Parameter | Non VFD-mediated reaction | VFD-mediated rate acceleration |
|---|---|---|
| Vmax (nM s−1) | 128 ± 5.71 | 309 ± 52.4 |
| Km (mM) | 3.76 ± 0.15 | 2.50 ± 0.44 |
| kcat (s−1) | 13.4 ± 0.59 | 32.1 ± 5.45 |
| kcat/Km (mM−1s−1) | 3.55 ± 0.19 | 13.32 ± 4.03 |
For this VFD-process, β-glucosidase (9.26 nM), a rotational speed of 7.60 krpm was used, as this provided the most consistent enhancement over sustained time periods. Error indicates the standard deviation around the mean (n=3). There was no overlapping error at 95% confidence limits. The raw data was fitted to a Michaelis-Menten plot and analyzed using a least squares fitting (LSF) approach (Figures S17 and S17).
We hypothesize that enzymes are accelerated in the VFD from the instantaneous pressure changes generated by Faraday waves. Three possible mechanisms could harness such pressures. Firstly, transient pressurization of the active site around the substrate could occur. In this situation, a decrease in the active site volume through pressurization[15] could increase the turnover number of the enzyme; such enhancement follows from the Van’t Hoff equation.[16] Secondly, pressure-induced protein conformational changes could occur at accelerated rates.[17] As enzymatic catalysis correlates with protein motion, faster protein motions could accelerate catalysis by contributing to the rate-determining process.[17] Thirdly, enzymatic catalysis requires a fine balance between protein stability and conformational flexibility.[18] Pressure-driven conformational changes may increase enzyme activity through β and α-relaxations.[19] These small changes can lead to the acquisition of protein conformations more suited for catalysis.[18]
The rotational landscape is specific for each enzyme studied, and appears to result from enzyme-specific preferences. Single-molecule experiments have elucidated the range of speeds and conformations required for enzymatic catalysis, which are specific for each enzyme.[20] The range of acceleration observed here falls within the expected range of enzyme speeds uncovered through such experiments. Thus, the VFD-driven rate acceleration could simply shift the distribution of enzyme conformational states to favor catalytic events. In future experiments, shaped Faraday waves with specific timing could provide further control and enhancement of biocatalysis.
In conclusion, we have demonstrated that the VFD effectively accelerates four enzymes, and establish a general, simple method to make biocatalysis more practical. VFD-mediated rate acceleration could find broad applicability in chemical transformations at industrial and laboratory scales.
Supplementary Material
Acknowledgments
JB thanks the Taihi Hong Memorial foundation and Kritika Mohan for photography. LM acknowledges the Miguel Velez Scholarship at UCI. GW gratefully acknowledges the National Institute of General Medical Sciences of the NIH (1RO1-GM100700-01). CR acknowledges the Australian Research Council and the Government of South Australia for their financial support during this project.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Aehle W. Enzymes in Industry. WILEY-VCH; Weinheim: 2004. [Google Scholar]; b) Nestl BM, Nebel BA, Hauer B. Curr Opin Chem Biol. 2011;15:187–193. doi: 10.1016/j.cbpa.2010.11.019. [DOI] [PubMed] [Google Scholar]; c) DiCosimo R, McAuliffe J, Poulose AJ, Bohlmann G. Chem Soc Rev. 2013;42:6437–6474. doi: 10.1039/c3cs35506c. [DOI] [PubMed] [Google Scholar]; d) Pollard DJ, Woodley JM. Trends Biotechnol. 2007;25:66–73. doi: 10.1016/j.tibtech.2006.12.005. [DOI] [PubMed] [Google Scholar]; e) Choi JM, Han SS, Kim HS. Biotechnol Adv. 2015;33:1443–1454. doi: 10.1016/j.biotechadv.2015.02.014. [DOI] [PubMed] [Google Scholar]
- 2.a) Renata H, Wang ZJ, Arnold FH. Angew Chem Int Edit. 2015;54:3351–3367. doi: 10.1002/anie.201409470. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang ZJ, Renata H, Peck NE, Farwell CC, Coelho PS, Arnold FH. Angew Chem Int Edit. 2014;126:6928–6931. doi: 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Siegel JB, Smith AL, Poust S, Wargacki AJ, Bar-Even A, Louw C, Shen BW, Eiben CB, Tran HM, Noor E, Gallaher JL, Bale J, Yoshikuni Y, Gelb MH, Keasling JD, Stoddard BL, Lidstrom ME, Baker D. P Natl Acad Sci USA. 2015;112:3704–3709. doi: 10.1073/pnas.1500545112. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wallace S, Balskus EP. Angew Chem Int Edit. 2015;54:7106–7109. doi: 10.1002/anie.201502185. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC. Nat Commun. 2015;6 doi: 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Bommarius AS, Riebel BR. Biocatalysis. Wiley-VCH; Weinheim: 2004. [Google Scholar]; b) Koeller KM, Wong CH. Nature. 2001;409:232–240. doi: 10.1038/35051706. [DOI] [PubMed] [Google Scholar]; c) Schramm VL. Annu Rev Biochem. 1998;67:693–720. doi: 10.1146/annurev.biochem.67.1.693. [DOI] [PubMed] [Google Scholar]
- 4.Stroppolo ME, Falconi M, Caccuri AM, Desideri A. CMLS, Cell Mol Life Sci. 2001;58:1451–1460. doi: 10.1007/PL00000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 5th. W H Freeman; New York: 2008. [Google Scholar]
- 6.Wolfenden R, Snider MJ. Accounts Chem Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 7.a) Subrizi F, Crucianelli M, Grossi V, Passacantando M, Botta G, Antiochia R, Saladino R. ACS Catalysis. 2014;4:3059–3068. [Google Scholar]; b) Greenberg WA, Varvak A, Hanson SR, Wong K, Huang H, Chen P, Burk MJ. P Natl Acad Sci USA. 2004;101:5788–5793. doi: 10.1073/pnas.0307563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Britton J, Dalziel SB, Raston CL. Green Chem. 2016;18:2193–2200. [Google Scholar]; b) Britton J, Dalziel SB, Raston CL. RSC Adv. 2015;5:1655–1660. [Google Scholar]
- 9.Yuan TZ, Ormonde CFG, Kudlacek ST, Kunche S, Smith JN, Brown WA, Pugliese KM, Olsen TJ, Iftikhar M, Raston CL, Weiss GA. ChemBioChem. 2015;16:393–396. doi: 10.1002/cbic.201402427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Britton J, Chalker JM, Raston CL. Chem Eur J. 2015;21:10660–10665. doi: 10.1002/chem.201501785. [DOI] [PubMed] [Google Scholar]
- 11.a) Yasmin L, Chen X, Stubbs KA, Raston CL. Sci Rep. 2013;3 doi: 10.1038/srep02282. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Britton J, Raston CL. RSC Adv. 2014;4:49850–49854. [Google Scholar]; c) Britton J, Raston CL. RSC Adv. 2015;5:2276–2280. [Google Scholar]
- 12.Menger FM, Ladika M. J Am Chem Soc. 1987;109:3145–3146. [Google Scholar]
- 13.Zhu K, Liu H, Han P, Wei P. Frontiers of Chemical Engineering in China. 2010;4:367–371. [Google Scholar]
- 14.Jennewein S, Schürmann M, Wolberg M, Hilker I, Luiten R, Wubbolts M, Mink D. Biotechnology Journal. 2006;1:537–548. doi: 10.1002/biot.200600020. [DOI] [PubMed] [Google Scholar]
- 15.Hillson N, Onuchic JN, García AE. P Natl Acad Sci USA. 1999;96:14848–14853. doi: 10.1073/pnas.96.26.14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuster CJT, Scheeren HW. In High Pressure Chemistry. Wiley-VCH; Weinheim: 2007. [Google Scholar]
- 17.Hay S, Scrutton NS. Nat Chem. 2012;4:161–168. doi: 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- 18.Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, Kern D. Nature. 2007;450:913–916. doi: 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- 19.Shrestha UR, Bhowmik D, Copley JRD, Tyagi M, Leão JB, Chu X-Q. P Natl Acad Sci USA. 2015;112:13886–13891. doi: 10.1073/pnas.1514478112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Sims PC, Moody IS, Choi Y, Dong C, Iftikhar M, Corso BL, Gul OT, Collins PG, Weiss GA. J Am Chem Soc. 2013;135:7861–7868. doi: 10.1021/ja311604j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xie S. Single Mol. 2001;2:229–236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.