

Abstract
Columnaris disease caused by Gram-negative rod Flavobacterium columnare is one of the most common diseases of catfish. F. columnare is also a common problem in other cultured fish species worldwide. F. columnare has three major genomovars; we have sequenced a representative strain from genomovar I (ATCC 49512, which is avirulent in catfish) and genomovar II (94-081, which is highly pathogenic in catfish). Here, we present a comparative analysis of the two genomes. Interestingly, F. columnare ATCC 49512 and 94-081 meet criteria to be considered different species based on the Average Nucleotide Identity (90.71% similar) and DNA–DNA Hybridization (42.6% similar). Genome alignment indicated the two genomes have a large number of rearrangements. However, function-based comparative genomics analysis indicated that the two strains have similar functional capabilities with 2,263 conserved orthologous clusters; strain ATCC 49512 has 290 unique orthologous clusters while strain 94-081 has 391. Both strains carry type I secretion system, type VI secretion system, and type IX secretion system. The two genomes also have similar CRISPR capacities. The F. columnare strain ATCC 49512 genome contains a higher number of insertion sequence families and phage regions, while the F. columnare strain 94-081 genome has more genomic islands and more regulatory gene capacity. Transposon mutagenesis using Tn4351 in pathogenic strain 94-081 yielded six mutants, and experimental infections of fish showed hemolysin and glycine cleavage protein mutants had 15 and 10% mortalities, respectively, while the wild-type strain caused 100% mortalities. Our comparative and mutational analysis yielded important information on classification of genomovars I and II F. columnare as well as potential virulence genes in F. columnare strain 94-081.
Keywords: Flavobacterium columnare, genomovars, comparative genomics, catfish, transposon mutagenesis, virulence, fish health
Introduction
Channel catfish is the most important aquaculture commodity in Mississippi and the largest aquaculture industry in the United States, but it is impacted negatively by columnaris disease caused by Flavobacterium columnare. In the United States, the disease causes losses up to $30 million annually (Shoemaker et al., 2011; Declercq et al., 2013). Columnaris outbreaks may result in high mortality, especially during spring and autumn, and are associated with stressful conditions, including high temperatures, elevated organic loads, crowded ponds, and excessive handling (Wakabayashi, 1991). F. columnare causes acute to chronic infections and affects gills, skin, and fins; in particular, dorsal fin and surrounding skin are often affected, causing “saddleback” lesions. When the disease is acute or subacute, yellowish areas of necrotic tissue can be seen in the gills, resulting in severe gill pathology (Decostere, 2002).
F. columnare was isolated by Davis in 1922 (Davis, 1922) and named Bacillus columnaris. After several reclassifications, a final name was assigned in 1996 (Bernardet et al., 1996). F. columnare is a Gram-negative, long rod in the family Flavobacteriaceae (Davis, 1922; Bernardet et al., 1996). F. columnare demonstrates varying colony morphologies and genetic heterogeneity as well as significant variation in virulence in different fish species. F. columnare strains are assigned into one of three genomovar groups by using 16S rDNA restriction fragment length polymorphism analysis (Triyanto and Wakabayash, 1999).
Flavobacterium columnare ATCC 49512, belonging to genomovar group I (Michel et al., 2002), was isolated from a brown trout skin lesion in France in 1987 (Bernardet, 1989), and it is not virulent to catfish (Soto et al., 2008). F. columnare genomovar II is the most virulent genomovar group in warm water fish species, including catfish (Triyanto and Wakabayash, 1999; Arias et al., 2004; Darwish and Ismaiel, 2005; Olivares-Fuster et al., 2007; Shoemaker et al., 2008; Bullard et al., 2013). However, genomovar I isolates are more virulent than genomovar II isolates in rainbow trout challenges (Evenhuis and LaFrentz, 2016). F. columnare strain 94-081 is in genomovar II and is highly virulent in catfish (Soto et al., 2008; Staroscik et al., 2008; Lawrence et al., 2012).
Flavobacterium johnsoniae UW101 genome was the first completed Flavobacterium genome reported (accession # CP000685) (McBride et al., 2009). Later, the genome of Flavobacterium psychrophilum JIP02/86, the causative agent of cold water disease in salmonid fish, was reported (accession # 511344733) (Duchaud et al., 2007). Flavobacterium branchiophilum FL-15 genome was finished in 2011 (accession # FQ859183) (Touchon et al., 2011), and our group sequenced the complete genome of F. columnare genomovar I strain ATCC 49512 (accession # CP003222.2) (Tekedar et al., 2012). F. columnare genomovar II strain 94-081 was recently sequenced and completed by our group (accession # CP013992.1) (Kumru et al., 2016).
Comparative analysis of complete genomes provides an excellent opportunity to determine unique genomic features. Availability of non-virulent and virulent strains belonging to genomovars I and II should reveal potential virulence factors and mechanisms causing columnaris disease. Here, we report the first comparative analysis of F. columnare strain ATCC 49512 (genomovar I) and strain 94-081 (genomovar II) genomes. Moreover, random transposon mutagenesis in strain 94-081 was conducted to identify potential virulence genes. We expect that the new knowledge gained from this study will clarify the systematic classification of genomovar I and II F. columnare and help elucidate the pathogenesis of columnaris disease.
Materials and Methods
Bacterial Species and Growth Conditions
Bacterial strains and plasmids used in this study are listed in Table 1. F. columnare strain 94-081 was grown at 30°C in FCGM agar plate and FCGM broth with shaking at 200 rpm (Farmer, 2004). E. coli strains were cultured at 37°C in the Luria-Bertani (LB) medium and LB broth with shaking at 200 rpm.
Table 1.
Bacterial strains and plasmids.
| Strains | Reference |
|---|---|
| Flavobacterium columnare 94-081 | Soto et al., 2008 |
| Escherichia coli S17-1 λpir | Simon et al., 1983 |
| Plasmids | |
| pEP4351 (Tn4351 transposon) |
Genomic DNA Extraction and Sequencing
For genomic DNA extraction from F. columnare strain 94-081 and transposon mutants, the CTAB/NaCI protocol (Murray and Thompson, 1980; Wilson, 2001) was used with some modifications. The 1.5 ml of F. columnare strain 94-081 culture was pelleted by centrifugation for 2 m at 12,000 rpm. The supernatant was discarded, the pellet was resuspended in 567 μl of TE buffer, and 20 μl of 10 mg/ml RNAse A was added. After 1 h incubation at 37°C, 40 μl of 10% SDS was added. After gentle mixing, 6 μl of 10 mg/ml proteinase K was added. The sample was incubated for 1 h at 56°C, after which 100 μl of 5 M NaCl was added with thorough mixing. The 80 μl of CTAB/NaCl solution was added, and the sample was incubated at 65°C for 10 m. An equal volume of chloroform/isoamyl alcohol was added followed by centrifugation for 5 m at 14,000 rpm. The aqueous phase was transferred to a fresh tube. The 0.7 ml of phenol/chloroform/isoamyl alcohol was added, followed by gentle mixing and centrifugation for 5 m at 14,000 rpm. The aqueous phase was transferred to a fresh tube. The 0.6 ml of isopropanol was added, and the centrifugation was repeated. Finally, after discarding the supernatant, precipitated DNA was washed by adding 1 ml of 70% ethanol, and centrifugation was conducted for 5 m at 12,000 rpm. The supernatant was removed, and the pellet was dried briefly at room temperature and resuspended in 100 μl of TE buffer. Genomic DNA sequencing was conducted as described (Kumru et al., 2016).
Genome Annotation and Comparative Analysis
F. columnare strain ATCC 49512 and strain 94-081 genome sequence accession numbers are CP003222 and CP013992, respectively. For annotation and coding protein comparison, we applied RAST (Overbeek et al., 2014) and NCBI PGAAP (Angiuoli et al., 2008) annotation tools. For RAST annotation, nucleotide files were uploaded to RAST by default features (RAST annotation scheme: classic RAST, gene caller: RAST, FIGfam version: Release70, automatically fix errors, fix frameshifts, build metabolic model, backfill gaps, turn on debug, and disable replication: yes, verbose level: 0). Mauve multiple genome alignment tool (Darling et al., 2010) was used with default settings for comparison of genomes. To evaluate general genetic similarity, two-way Average Nucleotide Identity (ANI) (Goris et al., 2007) and DNA–DNA hybridization (DDH) assessments (Meier-Kolthoff et al., 2013) were conducted using recommended default settings. A phylogenetic tree using the complete genomes of 22 Flavobacterium genus members was built from their core genomes (as of 04/2017). The core genome gene sets were aligned one by one using MUSCLE (Edgar, 2004) followed by concatenation of the alignments. Alignment results were used to compute Kimura distance matrix, which was used as input for the Neighbor-Joining algorithm as implemented in the PHYLIP package (Felsenstein, 1989).
To predict bacterial protein secretion systems, analysis with MacSyFinder was performed using default features (unordered replicon, circular, all available systems, maximal E-value 1.0, maximal independent E-value 0.001, minimal profile coverage 0.5) (Abby et al., 2014). CRISPRFinder was used to detect the clustered regularly interspaced short palindromic repeats (CRISPRs) and specific families of tandem repeats (Grissa et al., 2007). To determine phage elements, PHASTER was used (Arndt et al., 2016). To identify genomic islands (GIs), IslandViewer31, which integrates three different identification approaches (IslandPick, SIGI-HMM, IslandPath-DIMOB), was applied (Hsiao et al., 2003; Waack et al., 2006; Langille et al., 2008; Dhillon et al., 2015). To determine insertion sequences (ISs), ISsaga2 was used (Varani et al., 2011). For identification and analysis of signal transduction regulatory proteins, P2RP3 was used (Barakat et al., 2013). Orthologous genes analysis was performed using OrthoVenn4 with default features (E-value 1e - 5, inflation value 1.5) (Wang et al., 2015). Virulence factors were determined by downloading MvirDB5 (Zhou et al., 2007) and constructing a searchable database in Genomics Workbench 6.5.1 with a cutoff E-value of 10-10.
Random Transposon Mutagenesis
Random transposon mutagenesis in F. columnare 94-081 was performed as described previously with some modifications (McBride and Kempf, 1996; Karlyshev et al., 2000; Karsi et al., 2009). Briefly, F. columnare 94-081 and donor E. coli S17-1 λpir strain with pEP4351 were grown to mid-log phase. Then bacteria were harvested by centrifugation at 12,000 rpm for 2 m. F. columnare and donor E. coli were mixed at 1:2 or 1:4 ratios and spotted onto a filter paper placed on FCGM agar. Following overnight incubation at 30°C, bacteria were removed from the filter paper by washing with FGCM broth, and serial dilutions were prepared. Diluted bacteria were spread on FCGM agar containing erythromycin (10 μg/ml) and colistin (50 μg/ml). During development of the method, lower concentrations of erythromycin were used (1 μg/ml and 5 μg/ml), but some background non-mutant colonies occurred. At 10 μg/ml, erythromycin-resistant F. columnare colonies appeared after 2 days of incubation at 30°C. Tn4351 insertion sites were identified using a single-primer PCR method (Karlyshev et al., 2000; Karsi et al., 2009). Tn4351 ends were amplified using specific left (atcaggcagtatatcccλgg) or right (attgcgctttatctccctgtaa) primers, and amplicons were sequenced using nested left (atcgacctcgtλagacttgg) or right (ggacggacaattλgcλga) primers. Sequences were searched using BLASTX for identification of genes with Tn4351 insertion.
Fish Virulence
This study was conducted by a protocol approved by the Mississippi State University Institutional Animal Care and Use Committee. Virulence of mutants was compared to parent strain 94-081 in channel catfish as described (Karsi et al., 2009). Briefly, 640 specific-pathogen-free (SPF) channel catfish fingerlings (14.65 ± 1.43 cm, 24.75 ± 6.50 g) were stocked into 32 40-L flow-through tanks (flow rate: 1 L/m) at a rate of 20 fish/tank and acclimated for 1 week. Chlorine, dissolved oxygen, and temperature were monitored daily. Four tanks were randomly assigned to each F. columnare mutant (total six mutants), F. columnare strain 94-081, and negative control. Immersion challenge was applied by lowering the water in each tank to 10-L and by adding 100 ml overnight culture (adjusted to equivalent bacterial concentration using optical density at 600 nm). Infection dose (average 1.93 × 107 CFU/ml water) was determined by serial dilution and plate counting. Negative control treatment tanks were exposed to an equal volume of sterile FCGM. After 5 h of incubation under aerated conditions, water flow was restored to each tank, and mortalities were recorded daily for a total of 8 days.
Results
Genome Features
The circular genome size of F. columnare strain ATCC 49512 is 3,162,432 bp (G+C 31.5%), which has 2,632 predicted protein coding genes, 1,121 of which are hypothetical proteins. The genome includes 5 ribosomal RNA operons (5S, 16S, and 23S) (two tandem and three scattered operons), and 74 tRNAs. RAST annotation showed 318 subsystems in the F. columnare strain ATCC 49512 genome (Table 2).
Table 2.
Genome features of F. columnare ATCC 49512 and 94-081.
| Features/Strain | ATCC 49512 | 94-081 |
|---|---|---|
| Status | Completed | Completed |
| Source | Brown Trout | Diseased Catfish |
| Country of isolation | France | United States |
| Date of isolation | 1987 | 1994 |
| Genome size | 3,162,432 | 3,321,600 |
| G+C content (%) | 31.5 | 30.8 |
| Number of total protein | 2,632 | 2,779 |
| Number of total genes | 2,772 | 2,897 |
| Complete rRNAs operons | 5 | 4 |
| Number of total rRNA | 15 | 12 |
| Number of total tRNA | 74 | 74 |
| Number of other RNA | 1 | 3 |
| Number of pseudogene genes | 50 | 29 |
| Number of hypothetical protein | 1,121 | 1,158 |
| Number of subsystems | 318 | 324 |
| Compared function based unique genes | 10 | 34 |
| Sequence-based unique genes | 393 | 412 |
The circular genome size of F. columnare strain 94-081 is 3,321,600 bp. The chromosome is predicted to contain 2,779 protein-coding genes, 1,158 of which encode hypothetical proteins. The genome has 4 ribosomal RNA (5S, 16S, and 23S) operons (two of which are located in tandem), 74 tRNAs, and 3 non-coding RNAs. RAST annotation showed 324 subsystems in the F. columnare strain 94-081 genome (Table 2).
Gene function analysis by RAST showed that strain ATCC 49512 contains 393 unique coding genes, and strain 94-081 has 412 unique coding genes (Figure 1). In particular, genes unique to strain 94-081 encode biosynthetic enzymes for amino acids and derivatives, cofactor synthetic enzymes, cell wall synthesis enzymes, DNA metabolism proteins, membrane transport proteins, cell division proteins, regulation and cell signaling proteins, and carbohydrate metabolism enzymes.
FIGURE 1.

F. columnare ATCC 49512 and F. columnare 94-081 genome subsystems by RAST annotation.
Phylogeny Analysis
Genome alignment by Mauve showed that although the F. columnare ATCC 49512 and F. columnare 94-081 genomes have local synteny of gene clusters, extensive genomic rearrangements are present (Figure 2). Two-way ANI is 90.71%, and DDH approximation is 42.60%. A phylogenetic tree was built based on the core genomes of 22 Flavobacterium strains with complete genomes (Figure 3). The core Flavobacterium genome consisted of 1,025 genes, which encompassed 1,073,527 bp per genome (23,617,594 bp total for all 22 genomes).
FIGURE 2.

Comparison of F. columnare ATCC 49512 (A) and F. columnare 94-081 (B) genomes using MAUVE genome alignment.
FIGURE 3.

Phylogenetic tree based on comparison of the core genomes from all the current complete genomes in the Flavobacterium genus.
Secretion Systems
The F. columnare ATCC 49512 genome has a type I secretion system (T1SS), a partial type II secretion system (T2SS), a partial type III secretion system (T3SS), type IV pilus (T4P; homologous to components of the T2SS), type IV secretion system (T4SS) elements, type VI secretion system subtype 3 (T6SSiii), and type IX secretion system (T9SS). The F. columnare strain 94-081 genome contains a T1SS, T3SS, T4P, T4SS, T6SSiii, and T9SS as a complete operon or partial elements (Table 3).
Table 3.
Predicted protein secretion systems in F. columnare ATCC 49512 and 94-081.
| Secretion systems | Role in the system∗ | Genes | Number of genes available in F. columnare ATCC 49512 | Number of genes available in F. columnare 94-081 |
|---|---|---|---|---|
| T1SS | Mandatory | T1SS-abc | 6 | 6 |
| T1SS-mfp | 3 | 3 | ||
| T1SS-omf | 4 | 3 | ||
| T2SS | Mandatory | T2SS-gspJ | 1 | 0 |
| T3SS | Mandatory | T3SS-sctN | 2 | 2 |
| T4P | Mandatory | T4P-pilB | 1 | 1 |
| T4P-pilC | 1 | 1 | ||
| T4P-pilM | 2 | 1 | ||
| T4P-pilQ | 1 | 1 | ||
| T4SS | Mandatory | T4SS-t4cp2 | 1 | 0 |
| Accessory | T4SS-G-tfc18 | 0 | 1 | |
| T4SS-I-traP | 0 | 1 | ||
| T6SSi | Mandatory | T6SSi-tssH | 2 | 2 |
| T6SSiii | Mandatory | T6SSiii-tssB | 1 | 1 |
| T6SSiii-tssC | 1 | 1 | ||
| T6SSiii-tssD | 4 | 5 | ||
| T6SSiii-tssE | 1 | 1 | ||
| T6SSiii-tssF | 1 | 1 | ||
| T6SSiii-tssG | 1 | 1 | ||
| T6SSiii-tssH | 1 | 1 | ||
| T6SSiii-tssI | 3 | 6 | ||
| T6SSiii-tssK | 1 | 1 | ||
| T6SSiii-tssN | 1 | 1 | ||
| T6SSiii-tssP | 1 | 1 | ||
| T9SS | Mandatory | T9SS-gldK | 1 | 1 |
| T9SS-gldL | 1 | 1 | ||
| T9SS-gldM | 1 | 1 | ||
| T9SS-gldN | 1 | 1 | ||
| T9SS-sprA | 1 | 1 | ||
| T9SS-sprE | 1 | 1 | ||
| T9SS-sprT | 1 | 1 | ||
| Accessory | T9SS-porQ | 2 | 2 | |
| T9SS-porU | 1 | 1 | ||
| T9SS-porV | 1 | 1 | ||
| T9SS-gldJ | 1 | 1 |
∗Mandatory components are essential for system function. Accessory components are not essential for the system.
CRISPR-Cas System
Results of the CRISPRs analysis are summarized in Table 4. The F. columnare ATCC 49512 genome contains two CRISPR loci; one has 43 spacers, and the other has 8 spacers. Direct repeat (DR) lengths are 36 bp. The first CRISPR locus is located near a pseudogene (possibly CRISPR-associated Cas2 genes), and the second CRISPR locus is between genes encoding hypothetical proteins. While the CRISPR-associated Cas1 and possible Cas2 genes are adjacent in the genome, the Cas9 gene is at a different location. A questionable third CRISPR is located near the CRISPR-associated Cas9 gene. The strain ATCC 49512 genome also encodes Cas9 type II and Cas1 type II proteins.
Table 4.
CRISPRs in F. columnare ATCC 49512 and 94-081.
| CRISPR begin | CRISPR end | No. of spacers | DR length | CRISPR length (bp) | |
|---|---|---|---|---|---|
| Flavobacterium columnare 94-081 | |||||
| CRISPR 2 | 320994 | 323320 | 34 | 36 | 2,326 |
| CRISPR 4 | 758381 | 758905 | 7 | 36 | 524 |
| CRISPR 7 | 3165645 | 3165919 | 3 | 26 | 274 |
| Possible CRISPR 1 | 212275 | 212374 | 1 | 32 | 99 |
| Possible CRISPR 3 | 758039 | 758235 | 2 | 36 | 196 |
| Possible CRISPR 5 | 1334949 | 1335043 | 1 | 26 | 94 |
| Possible CRISPR 6 | 2395248 | 2395326 | 1 | 23 | 78 |
| Possible CRISPR 8 | 3207039 | 3207149 | 1 | 29 | 110 |
| Flavobacterium columnare ATCC 49512 | |||||
| CRISPR 1 | 391564 | 394476 | 43 | 36 | 2,912 |
| CRISPR 2 | 1679967 | 1680530 | 8 | 36 | 563 |
| Possible CRISPR 3 | 1994455 | 1994554 | 1 | 23 | 99 |
By comparison, the F. columnare 94-081 genome contains three CRISPR loci with 34, 7, and 3 spacers. DR lengths are 36, 36, and 26 bp, and the sequences are different for each CRISPR locus. The first CRISPR locus is located between CRISPR-associated genes Cas9 and Cas2. The second CRISPR locus is located between genes encoding two hypothetical proteins. The third CRISPR locus interrupts a gene encoding the cell envelope biogenesis protein OmpA (AWN65_RS13895). The F. columnare 94-081 genome has five possible CRISPRs-like structures with only two or three spacers in each (Table 4). Possible CRISPR 5 is located in a gene encoding a hypothetical protein (AWN65_RS05930). Possible CRISPR 3 is located near the second CRISPR locus, and the other possible CRISPRs are located between genes encoding hypothetical proteins. The strain 94-081 genome encodes Cas9 type II, Cas1 type II, and Cas2 type I-II-III proteins.
Phage Sequences
The F. columnare ATCC 49512 genome carries three incomplete phage clusters in different locations on the genome. The lengths of the phage clusters are 6.3, 7, and 11.6 kb. Phage sequences were searched against Flavobacterium genomes using BLASTX, and 31 genes were determined (Table 5). Seven of these genes encode possible virulence factors in MvirDB.
Table 5.
Phage elements in F. columnare ATCC 49512 and 94-081.
| Location | Locus tag | Product | ||
|---|---|---|---|---|
| Flavobacterium columnare strain 94-081 | Region I | 175781-176356 | AWN65_RS00815 | Guanylate kinase |
| 176392-176757 | AWN65_RS00820 | Four helix bundle protein | ||
| 176791-177372 | AWN65_RS00825 | Nicotinic acid mononucleotide adenylyltransferase | ||
| 177495-178397 | AWN65_RS00830 | N-Acetylmuramoyl-L-alanine amidase | ||
| 179325-180776 | AWN65_RS00835 | Nicotinate phosphoribosyltransferase | ||
| 180901-181632 | AWN65_RS00840 | Metallophosphatase | ||
| 181665-183095 | AWN65_RS00845 | ATPase | ||
| 183207-183656 | AWN65_RS00850 | Hypothetical protein | ||
| 183762-184319 | AWN65_RS00855 | RNA 2’-phosphotransferase | ||
| 184558-185193 | AWN65_RS00860 | hypothetical protein | ||
| 185330-185518 | AWN65_RS00865 | Hypothetical protein | ||
| 185635-186198 | AWN65_RS00870 | Hypothetical protein | ||
| 186284-187126 | AWN65_RS00875 | Crystallin J1 | ||
| 187123-187659 | AWN65_RS00880 | Hypothetical protein | ||
| 187656-188486 | AWN65_RS00885 | Phosphoribosylpyrophosphate synthetase | ||
| 188909-189601 | AWN65_RS00890 | NUDIX hydrolase | ||
|
| ||||
| Flavobacterium columnare ATCC 49512 | Region I | 1241010-1241513 | FCOL_RS05470 | Chromosome partitioning protein ParB |
| 1241510-1242790 | FCOL_RS05475 | Phosphoadenosine phosphosulfate sulfurtransferase | ||
| 1242787-1244013 | FCOL_RS05310 | Hypothetical protein | ||
| 1244047-1244736 | FCOL_RS05485 | Hypothetical protein | ||
| 1244733-1245056 | FCOL_RS05490 | Hypothetical protein | ||
| 1245075-1245617 | FCOL_RS05495 | hypothetical protein | ||
| 1245798-1246442 | FCOL_RS05500 | Hypothetical protein | ||
| 1246439-1246918 | FCOL_RS05505 | Peptidoglycan L-alanyl-D-glutamate endopeptidase | ||
| 1246890-1247372 | FCOL_RS05510 | Hypothetical protein | ||
|
|
||||
| Region II | 2941840-2942271 | FCOL_RS14160 | Hypothetical protein | |
| 2942993-2943172 | FCOL_RS12865 | Transposase | ||
| 2943259-2944173 | FCOL_RS12870 | Transposase | ||
| 2944446-2945636 | FCOL_RS12875 | Acyltransferase | ||
| 2945889-2946626 | FCOL_RS12880 | Capsular polysaccharide biosynthesis protein | ||
| 2946753-2947616 | FCOL_RS12885 | ABC transporter permease | ||
| 2947619-2948905 | FCOL_RS12890 | ABC transporter ATP-binding protein | ||
|
|
||||
| Region III | 3022580-3023155 | FCOL_RS13225 | Guanylate kinase | |
| 3023191-3023556 | FCOL_RS13230 | Four helix bundle protein | ||
| 3023645-3024157 | FCOL_RS13235 | Nicotinate-nicotinamide nucleotide adenylyltransferase | ||
| 3024280-3025182 | FCOL_RS13240 | N-Acetylmuramoyl-L-alanine amidase | ||
| 3025996-3027444 | FCOL_RS13245 | Nicotinate phosphoribosyltransferase | ||
| 3027503-3028006 | FCOL_RS13250 | Hypothetical protein | ||
| 3028052-3028456 | FCOL_RS13255 | Hypothetical protein | ||
| 3028541-3029266 | FCOL_RS13260 | Metallophosphatase | ||
| 3029263-3029805 | FCOL_RS13265 | RNA 2’-phosphotransferase | ||
| 3029802-3030464 | FCOL_RS13270 | Hypothetical protein | ||
| 3030527-3031030 | FCOL_RS13275 | RNase III inhibitor | ||
| 3031196-3031753 | FCOL_RS13280 | Hypothetical protein | ||
| 3031753-3032286 | FCOL_RS13285 | Hypothetical protein | ||
| 3032283-3033113 | FCOL_RS13290 | Phosphoribosylpyrophosphate synthetase | ||
| 3033491-3034183 | FCOL_RS13295 | NUDIX hydrolase | ||
The F. columnare 94-081 genome contains one incomplete phage cluster of 13.8 kb with 16 genes, two of which have significant similarity with proteins in MvirDB. Eight of the phage genes were common in both F. columnare genomes. Phage elements in the F. columnare genomes are listed in Table 5.
Genomic Islands and Insertion Elements
The F. columnare ATCC 49512 genome contains eight integrated GIs encoding 71 proteins, 50 of which are hypothetical. On the other hand, the F. columnare 94-081 genome has 15 integrated GIs encoding 162 proteins, 125 of which are hypothetical (Figure 4). Transposase, integrase, and DNA repair protein were encoded in GIs from both strains. Glycerol transferase, IS481 family transposase, and DNA-directed RNA polymerase sigma-70 factor were unique in GIs from strain ATCC 49512, whereas AAA family ATPase, ABC transporter ATP-binding protein, and flagellar motor protein MotB were unique to strain 94-081. The F. columnare ATCC 49512 genome contains 14 IS families with 95 interspersed protein-coding genes, while the F. columnare 94-081 genome has nine IS families with 79 interspersed predicted protein-coding genes (Figure 5).
FIGURE 4.

Genomic islands in the genomes of the F. columnare strain ATCC 49512 (A) and strain 94-081 (B). Red: integrated prediction methods; orange: SIGI-HMM prediction methods; blue: IslandPath-DIMOB prediction method.
FIGURE 5.

Insertion sequences (ISs) in the genomes of F. columnare strains ATCC 49512 and strain 94–081.
Regulatory Systems
The F. columnare strain ATCC 49512 genome encodes 32 predicted two-component system proteins, 58 transcription factor proteins, and 7 other DNA-binding proteins. The F. columnare strain 94-081 genome encodes 32 two-component system proteins, 74 transcription factor proteins, and 6 other DNA-binding proteins (Table 6). Although they have the same number of two-component systems, the distribution of histidine kinases (HK), response regulators (RR), and phosphotransferase proteins (PP) is different between the two strains. Strain 94-081 has more regulatory capacity than strain ATCC 49512 primarily due to the number of transcriptional regulator (TR) proteins.
Table 6.
Regulatory systems in F. columnare ATCC 49512 and 94-081.
| F. columnare | Predicted regulatory proteins |
|||||||
|---|---|---|---|---|---|---|---|---|
| Two component systems |
Transcription factors |
Other DNA-binding proteins |
||||||
| HK | RR | PP | TR | OCS | RR | SF | ODP | |
| 94-081 | 13 | 15 | 4 | 49 | 9 | 9 | 7 | 6 |
| ATCC 49512 | 15 | 14 | 3 | 35 | 7 | 9 | 7 | 7 |
HK, histidine kinases; RR, response regulators; PR, phosphotransferase proteins; TR, transcriptional regulators; OCS, one-component systems; SF, sigma factors; ODP, other DNA-binding proteins.
Each of the two strains encodes unique regulatory proteins; strain ATCC 49512 encodes a unique transposase, cupin, PAS/PAC sensor signal transduction HK, and integration host factor subunit beta. Strain 94-081 encodes unique sensor HK, AAA family ATPase, ATP-binding protein, and HxIR family TR.
Orthology Analysis
Orthology analysis by OrthoVenn showed that the F. columnare strain ATCC 49512 and strain 94-081 genomes contained 2,263 intersecting orthologous clusters. Strain ATCC 49512 has 290 unique orthologous clusters, which is composed of 274 single copy genes and 16 clusters with ≥2 paralogs. Strain 94-081 has 391 unique orthologous clusters with 370 single copy genes and 21 clusters with ≥2 paralogs. In its unique clusters, 28 of the F. columnare ATCC 49512 genes encode hypothetical proteins, and 18 encode transposases. Proteins unique tp strain ATCC 49512 included two Rhs element Vgr proteins, two peptidoglycan L-alanyl-D-glutamate endopeptidases, and one RHS repeat-associated core domain protein. In the F. columnare 94-081 genome, 90 of the total unique proteins were hypothetical, two were TRs, two were XRE family TRs, and two were endonucleases.
Virulence Factors
Protein sequences of strains ATCC 49512 and 94-081 were searched against MvirDB. Results indicated that strain ATCC 49512 encoded a high number of proteins matching transposases in MvirDB, while strain 94-081 encoded a large number of proteins matching hypothetical proteins in the virulence database. MvirDB searches with a cutoff E-value of 10-10 indicated that F. columnare strain ATCC 49512 encodes 567 potential virulence proteins and F. columnare 94-081 encodes 592 potential virulence proteins (Supplementary Tables 1, 2). Proteins matches in the MvirDB database by both strains include 16S rRNA dimethyltransferase, 23S rRNA methyltransferase RlmB, an ABS transporter, a cold shock protein, gliding motility lipoproteins GldJ and GldK, molecular chaperones DnaJ, DnaK, GroEL, and HtpG, and secretion protein HlyD. In strain ATCC 49512, unique MvirDB matches include cell envelope biogenesis protein OmpA, multidrug ABC transporter permease/ATPase, multi-sensor hybrid HK, and sigma-54-dependent Fis family TR. Unique MvirDB matches from strain 94-081 include a hemolysin, λ family ATPase, Clp protease ClpC, an efflux transporter periplasmic adaptor subunit, flagellar motor protein MotB, and transcription factors.
Mutational Analysis
Using erythromycin at 10 μg/ml, our conjugation yielded six F. columnare strain 94-081 Tn4351 mutants (Table 7). All six colonies contained transposon insertions; no background non-mutant colonies occurred. Wild-type strain 94-081 caused 100% mortality in catfish fingerlings, while FcMut01 [F. columnare Tn4351 chalcone isomerase (CHI) insertion mutant] and FcMut02 (hemolysin mutant) had averages of 20 and 15% mortalities, respectively. Additionally, FcMut03 (anhydro-N-acetylmuramic acid kinase mutant) had an average of 40% mortalities, and FcMut04 (glycine dehydrogenase mutant) had an average of 10% mortalities. FcMut05 and FcMut06 had similar mortalities to the wild-type strain (Figure 6).
Table 7.
Tn4351 insertion mutants of F. columnare 94-081.
| Mutants | Transposon end | Locus tag | Gene ID |
|---|---|---|---|
| FcMut01 | F | AWN65_RS03755 | Chalcone isomerase |
| FcMut02 | R | AWN65_RS11020 | Hemolysin |
| FcMut03 | F | AWN65_RS03750 | Anhydro-N-acetylmuramic acid kinase (AnmK) |
| FcMut04 | R | AWN65_RS11110 | Glycine cleavage system protein P (GcvP) |
| FcMut05 | R | AWN65_RS03940 | Transcriptional regulator TetR |
| FcMut06 | R | AWN65_RS04445 | Peptidoglycan-binding protein LysM |
FIGURE 6.
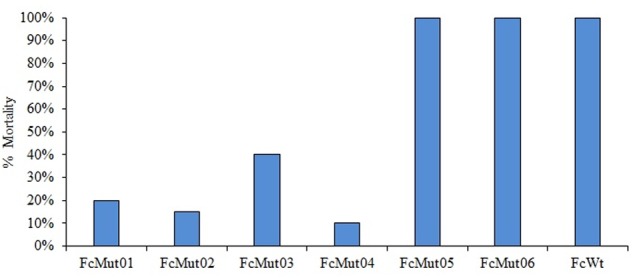
Mean percent mortalities resulting from experimental infection of channel catfish by F. columnare strain 94-081 and strain 94-081 Tn4351 insertion mutants (FcMut01: chalcone isomerase; FcMut02: hemolysin; FcMut03: anhydro-N-acetylmuramic acid kinase (AnmK); FcMut04: glycine cleavage system protein P (GcvP); FcMut05: transcriptional regulator TetR, FcMut06: peptidoglycan-binding protein LysM.
Discussion
F. columnare strains are divided into three genomovar groups based on their colony morphology and genetic heterogeneity (Triyanto and Wakabayash, 1999). The purpose of this study was to compare the genomes of F. columnare strain ATCC 49512 (genomovar I) and F. columnare strain 94-081 (genomovar II) (Tekedar et al., 2012; Kumru et al., 2016). Evaluation of both strains in catfish by immersion challenge showed that F. columnare strain ATCC 49512 was not virulent in catfish, while F. columnare strain 94-081 was highly virulent (Soto et al., 2008). Among the three genomovar groups, genomovar II contains the most F. columnare strains that are virulent to catfish (Triyanto and Wakabayash, 1999; Arias et al., 2004; Darwish and Ismaiel, 2005; Olivares-Fuster et al., 2007; Shoemaker et al., 2008; Bullard et al., 2013).
F. columnare strain ATCC 49512 and strain 94-081 are the first complete genomes in their corresponding genomovars; the genomovar III group does not have a representative complete genome yet. F. columnare genomes are bigger than F. psychrophilum (2,860,382 bp) but smaller than F. branchiophilum (3,559,884 bp) and F. johnsoniae (6,096,872 bp). In F. columnare, the strain 94-081 genome (3,331,600 bp) is 169 kb bigger than the strain ATCC 49512 genome (3,162,432 bp). Furthermore, strain 94-081 has 147 more predicted protein-coding genes and has one less ribosomal RNA operon compared to strain ATCC 49512 (Table 2). RAST annotation showed that both genomovars share similar subsystems (Figure 1).
Genome alignment by MAUVE showed local synteny but very different overall alignments. ANI between the two strains (90.71%) and DDH determination (42.60%) indicate that the two genomovars could be considered different species. The accepted ANI cutoff to define two strains as the same species is greater than 95% (Goris et al., 2007). Similarly, the recommended DDH cutoff to define strains as a species is >70% (Meier-Kolthoff et al., 2013, 2014). Phylogenetic analysis of all the available complete genomes in the Flavobacterium genus shows that F. columnare genomovar I and genomovar II strains cluster separately, but they are more closely related to each other than other species (Figure 3). F. indicum GPTSA100-9 and F. psychrophilum are the closest species to F. columnare. Based on these criteria, our results suggest these two strains could be considered different species.
Bacterial secretion systems are necessary for bacterial growth, virulence, and competition (Green and Mecsas, 2016), and there are several types of bacterial secretion systems (Gerlach and Hensel, 2007; Sato et al., 2010; Dalbey and Kuhn, 2012; Xu and Luo, 2013). The F. columnare strain ATCC 49512 and strain 94-081 genomes both have complete T1SS, T6SSiii, and T9SS as well as other partial secretion system elements (Table 3). Proteases, lipases, hemolysins, and leukotoxins are secreted by T1SS in pathogens (Akatsuka et al., 1995; Dirix et al., 2004; Bleves et al., 2010; Kanonenberg et al., 2013). Strain ATCC 49512 encodes more T1SS structural proteins than strain 94-081.
Type VI secretion system has three subtypes, including a new subtype unique to the Bacteroidetes first described as T6SSiii in F. johnsoniae (Russell et al., 2014). T6SS contributes to virulence of Gram-negative bacteria by transferring toxins to host cells or preventing the growth of other bacteria (Zoued et al., 2014; Aubert et al., 2015; Costa et al., 2015). Although both strains have a T6SSiii, the number of TssD (Hcp) and TssI (VgrG) secretion proteins encoded in the two strains is different (Table 3). TssD (Hcp) and TssI (VgrG) are bacteriophage-related proteins interacting with non-overlapping sets of effectors. While Hcp is ring-shaped and interacts with effectors within its pore, VgrG is a phage tail spike-like protein that interacts with effectors via conserved adaptor domains. Strain 94-081 has more VgrG and Hcp proteins than strain ATCC 49512, which could contribute to genomovar II pathogenicity in catfish.
Type IX secretion system or PorSS was identified first in Porphyromonas gingivalis and F. johnsoniae; this secretion system is also common in the Bacteroidetes phylum (McBride, 2001; Sato et al., 2010; McBride and Zhu, 2013; Kharade and McBride, 2015). In the Bacteroidetes phylum, T9SS is a protein carriage system for bacteria, and it is important for surface motility protein secretion and adhesion in F. johnsoniae (Shrivastava et al., 2013). Both genomovar strains have similar T9SS and motility proteins.
In prokaryotes, CRISPR systems are the “memory” component of a genetic adaptive immune system (Touchon et al., 2011). Cas (CRISPR-associated family genes) system provides the mechanism of prokaryotic resistance to foreign DNA (Mojica et al., 2005; Pourcel et al., 2005; Makarova et al., 2006). CRISPR-Cas proteins may control gene expression and regulate bacterial virulence (Hatoum-Aslan and Marraffini, 2014; Westra et al., 2014). Generally, up to 16 CRISPR clusters have been identified in prokaryotic genomes with the same or different numbers of DRs, which can vary in size between 24 to 47 bp (Mojica et al., 1995; Jansen et al., 2002; Bolotin et al., 2005; Barrangou et al., 2007). Spacer sequences are usually between 0.6 and 2.5X DR size, and spacers are the CRISPR immunity targets (Stern et al., 2010; Brodt et al., 2011). Some CRISPR systems are classified as “possible” because their DRs are not 100% identical (Grissa et al., 2007). Strain 94-081 has eight CRISPR loci (including five “possible”), and strain ATCC 49512 has three (one “possible”). Although strain 94-081 has more CRISPR loci than strain ATCC 49512, the total number of DR in CRISPR is very similar between to the two strains. Strain 94-081 has more CRISPR-Cas proteins, but the significance of this in bacterial pathogenesis is unknown.
Phages and prophages (lysogenic bacteriophage inserted into bacterial chromosomes and plasmids) contribute to bacterial environmental adaptation, antibiotic resistance, or pathogenicity. Bacteriophages can carry bacterial virulence genes, and chromosomal integration can inactivate bacterial genes (Casjens, 2003; Chibani-Chennoufi et al., 2004; Coates and Hu, 2007; Zhou et al., 2011; Arndt et al., 2016). Bacteriophages can be inactive, and some are activated in host cells to express virulence functions (Casjens, 2003; Coates and Hu, 2007; Zhou et al., 2011; Arndt et al., 2016). Strain ATCC 49512 has more prophage regions and proteins than strain 94-081, which may allow some unique environmental adaptations.
Genomic islands are clustered genes in prokaryotic genomes, and they play a major role in microbial genome evolution. They are commonly considered to originate from horizontal gene transfer and can encompass large genomic regions. Often they encode adhesion proteins, toxins, T3SSs, iron uptake proteins, antibiotic resistance proteins, or virulence factors (Hacker and Carniel, 2001; Whittle et al., 2002; Dobrindt et al., 2004; Gal-Mor and Finlay, 2006; Langille et al., 2010). IslandViewer3 integrates three of the most accurate GI prediction methods, IslandPick, IslandPath-DIMOB, and SIGI-HMM, which utilize different prediction methods (Hsiao et al., 2003; Waack et al., 2006; Langille et al., 2008; Langille and Brinkman, 2009; Dhillon et al., 2015). Strain 94-081 has more GIs, most of which encode hypothetical proteins. Twenty-four proteins encoded in GIs from strain 94-081 have significant matches with virulence proteins in MvirDB, while 15 of the GI proteins in strain ATCC 49512 have matches with proteins in MvirDB.
Insertion sequences are self-directed mobile genetic elements that contribute to horizontal gene transfer and genome organization (Varani et al., 2011). IS elements range from 0.7 to 3.5 kb in size, but they are usually less than 2.5 kb (Mahillon and Chandler, 1998; Siguier et al., 2006). IS elements appear to contribute to evolution of pathogenic bacterial genomes (Moran and Plague, 2004; Song et al., 2010; Schmitz-Esser et al., 2011; Varani et al., 2011), and movement of IS in the genome can contribute to antibiotic resistance (Tan, 1999; Wagner, 2006). Strain ATCC 49512 has more IS families and genes than strain 94-081, possibly indicating increased horizontal gene exchange has occurred.
Transcriptional regulation by transcription factors and two-component system proteins is important for microbial adaptation (Barakat et al., 2013). Two-component signal transduction systems (TCS) consist of HKs and a RR. These systems modulate gene expression in response to changing environmental conditions, including pathogenic events such as invasion of host cells, biofilm formation, and resistance to antibiotics (Ernst et al., 1999; Laub, 2011; Dsouza et al., 2014; Kadowaki et al., 2016). Other types of transcriptional regulatory factors include sigma factors (SF) and one component systems (OCS) (Osterberg et al., 2011; Barakat et al., 2013). The F. columnare 94-081 genome contains 18 more transcription factor proteins and two component system elements than the F. columnare ATCC 49512 genome, suggesting a more complex regulatory network.
Orthologous genes (clusters of genes derived from a common ancestor) (Fitch, 1970) are useful for comparative analysis of genome functional pathways. Orthologous cluster comparison can also be helpful for phylogenetic analysis (Henikoff et al., 1997; Mushegian et al., 1998; Wang et al., 2015). Interestingly, even though ANI and DDH analysis indicate strains ATCC 49512 and 94-081 could be considered different species, and genome alignment indicates extensive genomic rearrangements, strain ATCC 49512 and strain 94-081 have a large core of orthologous clusters (2,263 total). This indicates that despite their genomic differences, ATCC 49512 and 94-081 are functionally similar. Therefore, phenotypic differentiation of genomovar I and genomovar II strains may not be feasible. Interestingly, the relatively low number of unique proteins to genomovar II strains mediate a large difference in virulence in catfish; further study of these proteins is warranted. Eleven of the unique proteins from strain 94-081 are encoded in GIs, and three of the unique proteins from strain ATCC 49512 are encoded by GIs, suggesting horizontal gene transfer as the source.
Identification of virulence factors is important for understanding bacterial pathogenesis and host/pathogen interactions (Wu et al., 2008). Comparison of the strains’ predicted proteomes against MvirDB indicated a similar number of potential virulence genes (567 for strain ATCC 49512 and 592 for strain 94-081). GIs frequently carry virulence genes, and many of the virulence genes unique to genomovar II are in GIs (Zhou et al., 2007). Some of the unique predicted virulence proteins from genomovar II are involved in secretion systems and regulatory proteins.
A random transposon mutagenesis method was adapted to F. columnare genomovar II strain 94-081. Previously, transfer of plasmid pCP29 by conjugation into strain 94-081 was reported (Staroscik et al., 2008), but to our knowledge the current study is the first report of transposon mutagenesis in this strain. Similar to previously reported transposon mutagenesis using Tn4351 in F. columnare (Staroscik et al., 2008), the efficiency of mutagenesis was low (a total of six mutants were obtained from a single conjugation), so refinement of this method is warranted. We used a higher concentration of erythromycin (10 μg/ml) than previously reported (Staroscik et al., 2008), which eliminated background colonies. Virulence of the mutants was compared to parent strain 94-081 using an established model with channel catfish. Three of the mutants caused <20% mortalities (compared to 100% mortalities for wild-type strain). One of these had an insertion in a gene encoding CHI, which is an enzyme involved in flavone/flavanone degradation in bacteria. The human intestinal anaerobic bacterium Eubacterium ramulus expresses this enzyme (Braune et al., 2016), but it has not been implicated in virulence. Another mutation that caused attenuation was in a gene encoding a hemolysin, which can be an important virulence factor (Rowe and Welch, 1994; Zhang and Austin, 2000). In F. psychrophilum, hemolytic activity is contact dependent and mediated by a thermolabile enzyme (Hogfors-Ronnholm and Wiklund, 2010). The third mutant was in glycine cleavage system protein P (gcvP). The glycine cleavage system was linked to virulence in the fish pathogen Edwardsiella ictaluri, where the enzyme was shown to be involved in neutrophil and serum resistance (Karsi et al., 2009; Dahal et al., 2013).
Mutation of the gene encoding anhydro-N-acetylmuramic acid kinase (anmK) in 94-081 also caused attenuation, but mean percent mortalities were higher (>40%). In Francisella, the anmK gene is necessary for full virulence, but it is not required for intracellular growth (Ludu et al., 2008). Two F. columnare 94-081 mutants were not attenuated. One of the mutations was in a TetR family TR, which typically function as transcriptional repressors and can regulate antibiotic resistance, catabolic pathways, quorum sensing, and virulence of other pathogenic bacteria (Krushkal et al., 2011; Cuthbertson and Nodwell, 2013). The 94-081 lysM mutant was also not attenuated; LysM is a peptidoglycan-binding protein that is widely distributed in prokaryotes. In some bacteria, LysM mediates attachment to extracellular matrix (Downer et al., 2002).
In summary, comparison of the genomes from F. columnare genomovar I strain ATCC 49512 and genomovar II strain 94-081 provided some evidence that the two genomovars could be considered separate species (based on ANI and DDH analysis). However, orthology analysis revealed a largely conserved core genome, indicating that phenotypically the two genomovars are very similar. In particular, core metabolic functions are similar between the two genomovars. Both genomovars have CRISPR-Cas systems and evidence of horizontal gene acquisition, and despite local syntenic regions, a large number of genomic rearrangements are present between the two. For the first time, we report transposon mutagenesis of a genomovar II strain; this tool along with the genome sequence and an established infection model in catfish will enable future elucidation of mechanisms of pathogenesis in this important fish pathogen.
Author Contributions
Designed the experiments: SK, HT, NG, GW, ML, and AK. Conducted comparative genomics analysis: SK and HT. Transposon mutagenesis and fish challenge were performed by NG. The manuscript was written by SK, HT, ML, and AK. All authors read and accepted the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank EDGAR team for helping us to create the phylogenetic tree.
Funding. SK is supported by a fellowship from the Ministry of National Education of Turkey. This work was supported by the United States Department of Agriculture, National Institute of Food and Agriculture (2006-35600-16571), USDA-ARS CRIS project 31000-012-00D, and the College of Veterinary Medicine, Mississippi State University.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01375/full#supplementary-material
References
- Abby S. S., Neron B., Menager H., Touchon M., Rocha E. P. (2014). MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS ONE 9:e110726 10.1371/journal.pone.0110726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akatsuka H., Kawai E., Omori K., Shibatani T. (1995). The three genes lipB, lipC, and lipD involved in the extracellular secretion of the Serratia marcescens lipase which lacks an N-terminal signal peptide. J. Bacteriol. 177 6381–6389. 10.1128/jb.177.22.6381-6389.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angiuoli S. V., Gussman A., Klimke W., Cochrane G., Field D., Garrity G., et al. (2008). Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS 12 137–141. 10.1089/omi.2008.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias C. R., Welker T. L., Shoemaker C. A., Abernathy J. W., Klesius P. H. (2004). Genetic fingerprinting of Flavobacterium columnare isolates from cultured fish. J. Appl. Microbiol. 97 421–428. 10.1111/j.1365-2672.2004.02314.x [DOI] [PubMed] [Google Scholar]
- Arndt D., Grant J. R., Marcu A., Sajed T., Pon A., Liang Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44 W16–W21. 10.1093/nar/gkw387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert D. F., Hu S., Valvano M. A. (2015). Quantification of type VI secretion system activity in macrophages infected with Burkholderia cenocepacia. Microbiology 161 2161–2173. 10.1099/mic.0.000174 [DOI] [PubMed] [Google Scholar]
- Barakat M., Ortet P., Whitworth D. E. (2013). P2RP: a Web-based framework for the identification and analysis of regulatory proteins in prokaryotic genomes. BMC Genomics 14:269 10.1186/1471-2164-14-269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., et al. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315 1709–1712. 10.1126/science.1138140 [DOI] [PubMed] [Google Scholar]
- Bernardet J. F. (1989). ’Flexibacter columnaris’: first description in France and comparison with bacterial strains from other origins Dis. Aquat. Organ. 6 37–44. 10.3354/dao006037 [DOI] [Google Scholar]
- Bernardet J. F., Segers P., Vancanneyt M., Berthe F., Kersters K., Vandamme P. (1996). Cutting a Gordian knot: emended classification and description of the genus Flavobacterium, emended description of the family Flavobacteriaceae, and proposal of Flavobacterium hydatis norn. nov. (Basonym, Cytophaga aquatilis Strohl and Tait 1978). Int. J. Syst. Bacteriol. 46 128–148. 10.1099/00207713-46-1-128 [DOI] [Google Scholar]
- Bleves S., Viarre V., Salacha R., Michel G. P., Filloux A., Voulhoux R. (2010). Protein secretion systems in Pseudomonas aeruginosa: a wealth of pathogenic weapons. Int. J. Med. Microbiol. 300 534–543. 10.1016/j.ijmm.2010.08.005 [DOI] [PubMed] [Google Scholar]
- Bolotin A., Quinquis B., Sorokin A., Ehrlich S. D. (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151(Pt 8), 2551–2561. 10.1099/mic.0.28048-0 [DOI] [PubMed] [Google Scholar]
- Braune A., Engst W., Elsinghorst P. W., Furtmann N., Bajorath J., Gutschow M., et al. (2016). Chalcone isomerase from Eubacterium ramulus catalyzes the ring contraction of flavanonols. J. Bacteriol. 198 2965–2974. 10.1128/JB.00490-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodt A., Lurie-Weinberger M. N., Gophna U. (2011). CRISPR loci reveal networks of gene exchange in archaea. Biol. Direct 6:65 10.1186/1745-6150-6-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard S. A., Mohammed H., Arias C. R. (2013). First record of the fish pathogen Flavobacterium columnare genomovar II from bluegill, Lepomis macrochirus (Rafinesque), with observations on associated lesions. J. Fish. Dis. 36 447–451. 10.1111/jfd.12005 [DOI] [PubMed] [Google Scholar]
- Casjens S. (2003). Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 49 277–300. 10.1046/j.1365-2958.2003.03580.x [DOI] [PubMed] [Google Scholar]
- Chibani-Chennoufi S., Bruttin A., Dillmann M. L., Brussow H. (2004). Phage-host interaction: an ecological perspective. J. Bacteriol. 186 3677–3686. 10.1128/JB.186.12.3677-3686.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates A. R., Hu Y. (2007). Novel approaches to developing new antibiotics for bacterial infections. Br. J. Pharmacol. 152 1147–1154. 10.1038/sj.bjp.0707432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa T. R., Felisberto-Rodrigues C., Meir A., Prevost M. S., Redzej A., Trokter M., et al. (2015). Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 13 343–359. 10.1038/nrmicro3456 [DOI] [PubMed] [Google Scholar]
- Cuthbertson L., Nodwell J. R. (2013). The TetR family of regulators. Microbiol. Mol. Biol. Rev. 77 440–475. 10.1128/MMBR.00018-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahal N., Abdelhamed H., Lu J., Karsi A., Lawrence M. L. (2013). Tricarboxylic acid cycle and one-carbon metabolism pathways are important in Edwardsiella ictaluri virulence. PLoS ONE 8:e65973 10.1371/journal.pone.0065973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalbey R. E., Kuhn A. (2012). Protein traffic in Gram-negative bacteria–how exported and secreted proteins find their way. FEMS Microbiol. Rev. 36 1023–1045. 10.1111/j.1574-6976.2012.00327.x [DOI] [PubMed] [Google Scholar]
- Darling A. E., Mau B., Perna N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwish A. M., Ismaiel A. A. (2005). Genetic diversity of Flavobacterium columnare examined by restriction fragment length polymorphism and sequencing of the 16S ribosomal RNA gene and the 16S-23S rDNA spacer. Mol. Cell. Probes 19 267–274. 10.1016/j.mcp.2005.04.003 [DOI] [PubMed] [Google Scholar]
- Davis H. S. (1922). A new bacterial disease of fresh-water fishes. Bull. U. S. Bur. Fish. 38 261–280. 10.5962/bhl.title.49773 [DOI] [Google Scholar]
- Declercq A. M., Haesebrouck F., Broeck W. V. D., Bossier P., Decostere A. (2013). Columnaris disease in fish: a review with emphasis on bacterium-host interactions. Vet. Res. 44:27 10.1186/1297-9716-44-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decostere A. (2002). Flavobacterium columnare infections in fish: the agent and its adhesion to the gill tissue. Verh. K. Acad. Geneeskd. Belg 64 421–430. [PubMed] [Google Scholar]
- Dhillon B. K., Laird M. R., Shay J. A., Winsor G. L., Lo R., Nizam F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43 W104–W108. 10.1093/nar/gkv401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirix G., Monsieurs P., Dombrecht B., Daniels R., Marchal K., Vanderleyden J., et al. (2004). Peptide signal molecules and bacteriocins in Gram-negative bacteria: a genome-wide in silico screening for peptides containing a double-glycine leader sequence and their cognate transporters. Peptides 25 1425–1440. 10.1016/j.peptides.2003.10.028 [DOI] [PubMed] [Google Scholar]
- Dobrindt U., Hochhut B., Hentschel U., Hacker J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2 414–424. 10.1038/nrmicro884 [DOI] [PubMed] [Google Scholar]
- Downer R., Roche F., Park P. W., Mecham R. P., Foster T. J. (2002). The elastin-binding protein of Staphylococcus aureus (EbpS) is expressed at the cell surface as an integral membrane protein and not as a cell wall-associated protein. J. Biol. Chem. 277 243–250. 10.1074/jbc.M107621200 [DOI] [PubMed] [Google Scholar]
- Dsouza M., Taylor M. W., Turner S. J., Aislabie J. (2014). Genome-based comparative analyses of Antarctic and temperate species of Paenibacillus. PLoS ONE 9:e108009 10.1371/journal.pone.0108009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchaud E., Boussaha M., Loux V., Bernardet J. F., Michel C., Kerouault B., et al. (2007). Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat. Biotechnol. 25 763–769. 10.1038/nbt1313 [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R. K., Guina T., Miller S. I. (1999). How intracellular bacteria survive: surface modifications that promote resistance to host innate immune responses. J. Infect. Dis. 179(Suppl. 2), S326–S330. 10.1086/513850 [DOI] [PubMed] [Google Scholar]
- Evenhuis J. P., LaFrentz B. R. (2016). Virulence of Flavobacterium columnare genomovars in rainbow trout Oncorhynchus mykiss. Dis. Aquat. Organ. 120 217–224. 10.3354/dao03027 [DOI] [PubMed] [Google Scholar]
- Farmer B. (2004). Improved Methods for the Isolation and Characterization of Flavobacterium columnare. Baton Rouge, LA: Louisiana State University. [Google Scholar]
- Felsenstein J. (1989). PHYLIP - phylogeny inference package (version 3.2). Cladistics 5 164–166. [Google Scholar]
- Fitch W. M. (1970). Distinguishing homologous from analogous proteins. Syst. Zool. 19 99–113. 10.2307/2412448 [DOI] [PubMed] [Google Scholar]
- Gal-Mor O., Finlay B. B. (2006). Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol. 8 1707–1719. 10.1111/j.1462-5822.2006.00794.x [DOI] [PubMed] [Google Scholar]
- Gerlach R. G., Hensel M. (2007). Protein secretion systems and adhesins: the molecular armory of Gram-negative pathogens. Int. J. Med. Microbiol. 297 401–415. 10.1016/j.ijmm.2007.03.017 [DOI] [PubMed] [Google Scholar]
- Goris J., Konstantinidis K. T., Klappenbach J. A., Coenye T., Vandamme P., Tiedje J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57(Pt 1), 81–91. 10.1099/ijs.0.64483-0 [DOI] [PubMed] [Google Scholar]
- Green E. R., Mecsas J. (2016). Bacterial secretion systems: an overview. Microbiol. Spectr. 4. 10.1128/microbiolspec.VMBF-0012-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I., Vergnaud G., Pourcel C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35 W52–W57. 10.1093/nar/gkm360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker J., Carniel E. (2001). Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep. 2 376–381. 10.1093/embo-reports/kve097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatoum-Aslan A., Marraffini L. A. (2014). Impact of CRISPR immunity on the emergence and virulence of bacterial pathogens. Curr. Opin. Microbiol. 17 82–90. 10.1016/j.mib.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S., Greene E. A., Pietrokovski S., Bork P., Attwood T. K., Hood L. (1997). Gene families: the taxonomy of protein paralogs and chimeras. Science 278 609–614. 10.1126/science.278.5338.609 [DOI] [PubMed] [Google Scholar]
- Hogfors-Ronnholm E., Wiklund T. (2010). Hemolytic activity in Flavobacterium psychrophilum is a contact-dependent, two-step mechanism and differently expressed in smooth and rough phenotypes. Microb. Pathog. 49 369–375. 10.1016/j.micpath.2010.08.002 [DOI] [PubMed] [Google Scholar]
- Hsiao W., Wan I., Jones S. J., Brinkman F. S. (2003). IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19 418–420. 10.1093/bioinformatics/btg004 [DOI] [PubMed] [Google Scholar]
- Jansen R., Embden J. D., Gaastra W., Schouls L. M. (2002). Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 43 1565–1575. 10.1046/j.1365-2958.2002.02839.x [DOI] [PubMed] [Google Scholar]
- Kadowaki T., Yukitake H., Naito M., Sato K., Kikuchi Y., Kondo Y., et al. (2016). A two-component system regulates gene expression of the type IX secretion component proteins via an ECF sigma factor. Sci. Rep. 6:23288 10.1038/srep23288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanonenberg K., Schwarz C. K., Schmitt L. (2013). Type I secretion systems - a story of appendices. Res. Microbiol. 164 596–604. 10.1016/j.resmic.2013.03.011 [DOI] [PubMed] [Google Scholar]
- Karlyshev A. V., Pallen M. J., Wren B. W. (2000). Single-primer PCR procedure for rapid identification of transposon insertion sites. Biotechniques 28 1078 1080 1082. [DOI] [PubMed] [Google Scholar]
- Karsi A., Gulsoy N., Corb E., Dumpala P. R., Lawrence M. L. (2009). High-throughput bioluminescence-based mutant screening strategy for identification of bacterial virulence genes. Appl. Environ. Microbiol. 75 2166–2175. 10.1128/AEM.02449-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharade S. S., McBride M. J. (2015). Flavobacterium johnsoniae PorV is required for secretion of a subset of proteins targeted to the type IX secretion system. J. Bacteriol. 197 147–158. 10.1128/JB.02085-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krushkal J., Sontineni S., Leang C., Qu Y., Adkins R. M., Lovley D. R. (2011). Genome diversity of the TetR family of transcriptional regulators in a metal-reducing bacterial family Geobacteraceae and other microbial species. OMICS 15 495–506. 10.1089/omi.2010.0117 [DOI] [PubMed] [Google Scholar]
- Kumru S., Tekedar H. C., Waldbieser G. C., Karsi A., Lawrence M. L. (2016). Genome Sequence of the fish pathogen Flavobacterium columnare genomovar II strain 94-081. Genome Announc. 4:e00430-16. 10.1128/genomeA.00430-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille M. G., Brinkman F. S. (2009). IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25 664–665. 10.1093/bioinformatics/btp030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille M. G., Hsiao W. W., Brinkman F. S. (2008). Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 9:329 10.1186/1471-2105-9-329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille M. G., Hsiao W. W., Brinkman F. S. (2010). Detecting genomic islands using bioinformatics approaches. Nat. Rev. Microbiol. 8 373–382. 10.1038/nrmicro2350 [DOI] [PubMed] [Google Scholar]
- Laub M. T. (2011). “The role of two-component signal transduction systems in bacterial stress responses,” in Bacterial Stress Responses, 2 Edn, eds Storz G., Hengge R. (Washington, DC: ASM Pres; ), 45–58. [Google Scholar]
- Lawrence M. L., Karsi A., Tekedar H. C., Banes M. M., Gibbs D. (2012). Comparative Genomics and Transcriptomics of Flavobacterium columnare Isolates from Genomovars I and II. Available at: http://web.abo.fi/konferens/flavobacterium2012/pdf/Lawrence.pdf [Google Scholar]
- Ludu J. S., de Bruin O. M., Duplantis B. N., Schmerk C. L., Chou A. Y., Elkins K. L., et al. (2008). The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J. Bacteriol. 190 4584–4595. 10.1128/JB.00198-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahillon J., Chandler M. (1998). Insertion sequences. Microbiol. Mol. Biol. Rev. 62 725–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova K. S., Grishin N. V., Shabalina S. A., Wolf Y. I., Koonin E. V. (2006). A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 1:7 10.1186/1745-6150-1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride M. J. (2001). Bacterial gliding motility: multiple mechanisms for cell movement over surfaces. Annu. Rev. Microbiol. 55 49–75. 10.1146/annurev.micro.55.1.49 [DOI] [PubMed] [Google Scholar]
- McBride M. J., Kempf M. J. (1996). Development of techniques for the genetic manipulation of the gliding bacterium Cytophaga johnsonae. J. Bacteriol. 178 583–590. 10.1128/jb.178.3.583-590.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride M. J., Xie G., Martens E. C., Lapidus A., Henrissat B., Rhodes R. G., et al. (2009). Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 75 6864–6875. 10.1128/AEM.01495-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride M. J., Zhu Y. (2013). Gliding motility and Por secretion system genes are widespread among members of the phylum Bacteroidetes. J. Bacteriol. 195 270–278. 10.1128/JB.01962-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Kolthoff J. P., Auch A. F., Klenk H. P., Goker M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60 10.1186/1471-2105-14-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Kolthoff J. P., Klenk H. P., Goker M. (2014). Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 64(Pt 2), 352–356. 10.1099/ijs.0.056994-0 [DOI] [PubMed] [Google Scholar]
- Michel C., Messiaen S., Bernardet J. F. (2002). Muscle infections in imported neon tetra, Paracheirodon innesi Myers: limited occurrence of microsporidia and predominance of severe forms of columnaris disease caused by an Asian genomovar of Flavobacterium columnare. J. Fish Dis. 25 253–256. 10.1046/j.1365-2761.2002.00364.x [DOI] [Google Scholar]
- Mojica F. J., Diez-Villasenor C., Garcia-Martinez J., Soria E. (2005). Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 60 174–182. 10.1007/s00239-004-0046-3 [DOI] [PubMed] [Google Scholar]
- Mojica F. J., Ferrer C., Juez G., Rodriguez-Valera F. (1995). Long stretches of short tandem repeats are present in the largest replicons of the Archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol. Microbiol. 17 85–93. 10.1111/j.1365-2958.1995.mmi_17010085.x [DOI] [PubMed] [Google Scholar]
- Moran N. A., Plague G. R. (2004). Genomic changes following host restriction in bacteria. Curr. Opin. Genet. Dev. 14 627–633. 10.1016/j.gde.2004.09.003 [DOI] [PubMed] [Google Scholar]
- Murray M. G., Thompson W. F. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8 4321–4325. 10.1093/nar/8.19.4321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushegian A. R., Garey J. R., Martin J., Liu L. X. (1998). Large-scale taxonomic profiling of eukaryotic model organisms: a comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res. 8 590–598. 10.1101/gr.8.6.590 [DOI] [PubMed] [Google Scholar]
- Olivares-Fuster O., Baker J. L., Terhune J. S., Shoemaker C. A., Klesius P. H., Arias C. R. (2007). Host-specific association between Flavobacterium columnare genomovars and fish species. Syst. Appl. Microbiol. 30 624–633. 10.1016/j.syapm.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Osterberg S., del Peso-Santos T., Shingler V. (2011). Regulation of alternative sigma factor use. Annu. Rev. Microbiol. 65 37–55. 10.1146/annurev.micro.112408.134219 [DOI] [PubMed] [Google Scholar]
- Overbeek R., Olson R., Pusch G. D., Olsen G. J., Davis J. J., Disz T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42 206–214. 10.1093/nar/gkt1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel C., Salvignol G., Vergnaud G. (2005). CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151(Pt 3), 653–663. 10.1099/mic.0.27437-0 [DOI] [PubMed] [Google Scholar]
- Rowe G. E., Welch R. A. (1994). Assays of hemolytic toxins. Methods Enzymol. 235 657–667. 10.1016/0076-6879(94)35179-1 [DOI] [PubMed] [Google Scholar]
- Russell A. B., Wexler A. G., Harding B. N., Whitney J. C., Bohn A. J., Goo Y. A., et al. (2014). A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell Host Microbe 16 227–236. 10.1016/j.chom.2014.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K., Naito M., Yukitake H., Hirakawa H., Shoji M., McBride M. J., et al. (2010). A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 107 276–281. 10.1073/pnas.0912010107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz-Esser S., Penz T., Spang A., Horn M. (2011). A bacterial genome in transition–an exceptional enrichment of IS elements but lack of evidence for recent transposition in the symbiont Amoebophilus asiaticus. BMC Evol. Biol. 11:270 10.1186/1471-2148-11-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker C. A., Klesius P. H., Drennan J. D., Evans J. J. (2011). Efficacy of a modified live Flavobacterium columnare vaccine in fish. Fish Shellfish Immunol. 30 304–308. 10.1016/j.fsi.2010.11.001 [DOI] [PubMed] [Google Scholar]
- Shoemaker C. A., Olivares-Fuster O., Arias C. R., Klesius P. H. (2008). Flavobacterium columnare genomovar influences mortality in channel catfish (Ictalurus punctatus). Vet. Microbiol. 127 353–359. 10.1016/j.vetmic.2007.09.003 [DOI] [PubMed] [Google Scholar]
- Shrivastava A., Johnston J. J., van Baaren J. M., McBride M. J. (2013). Flavobacterium johnsoniae GldK, GldL, GldM, and SprA are required for secretion of the cell surface gliding motility adhesins SprB and RemA. J. Bacteriol. 195 3201–3212. 10.1128/JB.00333-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P., Filee J., Chandler M. (2006). Insertion sequences in prokaryotic genomes. Curr. Opin. Microbiol. 9 526–531. 10.1016/j.mib.2006.08.005 [DOI] [PubMed] [Google Scholar]
- Simon R., Priefer U., Puhler A. (1983). A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1 784–791. 10.1038/nbt1183-784 [DOI] [Google Scholar]
- Song H., Hwang J., Yi H., Ulrich R. L., Yu Y., Nierman W. C., et al. (2010). The early stage of bacterial genome-reductive evolution in the host. PLoS Pathog. 6:e1000922 10.1371/journal.ppat.1000922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto E., Mauel M. J., Karsi A., Lawrence M. L. (2008). Genetic and virulence characterization of Flavobacterium columnare from channel catfish (Ictalurus punctatus). J. Appl. Microbiol. 104 1302–1310. 10.1111/j.1365-2672.2007.03632.x [DOI] [PubMed] [Google Scholar]
- Staroscik A. M., Hunnicutt D. W., Archibald K. E., Nelson D. R. (2008). Development of methods for the genetic manipulation of Flavobacterium columnare. BMC Microbiol. 8:115 10.1186/1471-2180-8-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A., Keren L., Wurtzel O., Amitai G., Sorek R. (2010). Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 26 335–340. 10.1016/j.tig.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H. M. (1999). Bacterial catabolic transposons. Appl. Microbiol. Biotechnol. 51 1–12. 10.1007/s002530051356 [DOI] [PubMed] [Google Scholar]
- Tekedar H. C., Karsi A., Gillaspy A. F., Dyer D. W., Benton N. R., Zaitshik J., et al. (2012). Genome sequence of the fish pathogen Flavobacterium columnare ATCC 49512. J. Bacteriol. 194 2763–2764. 10.1128/JB.00281-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchon M., Barbier P., Bernardet J. F., Loux V., Vacherie B., Barbe V., et al. (2011). Complete genome sequence of the fish pathogen Flavobacterium branchiophilum. Appl. Environ. Microbiol. 77 7656–7662. 10.1128/AEM.05625-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triyanto, Wakabayash H. (1999). Genotypic diversity of strains of Flavobacterium columnare from diseased fishes. Fish Pathol. 34 65–71. 10.3147/jsfp.34.65 [DOI] [Google Scholar]
- Varani A. M., Siguier P., Gourbeyre E., Charneau V., Chandler M. (2011). ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 12:R30 10.1186/gb-2011-12-3-r30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waack S., Keller O., Asper R., Brodag T., Damm C., Fricke W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142 10.1186/1471-2105-7-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. (2006). Cooperation is fleeting in the world of transposable elements. PLoS Comput. Biol. 2:e162 10.1371/journal.pcbi.0020162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi H. (1991). Effect of environmental conditions on the infectivity of Flexibacter columnaris to fish. J. Fish Dis. 14 279–290. 10.1111/j.1365-2761.1991.tb00825.x [DOI] [Google Scholar]
- Wang Y., Coleman-Derr D., Chen G., Gu Y. Q. (2015). OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 43 W78–W84. 10.1093/nar/gkv487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra E. R., Buckling A., Fineran P. C. (2014). CRISPR-Cas systems: beyond adaptive immunity. Nat. Rev. Microbiol. 12 317–326. 10.1038/nrmicro3241 [DOI] [PubMed] [Google Scholar]
- Whittle G., Shoemaker N. B., Salyers A. A. (2002). The role of Bacteroides conjugative transposons in the dissemination of antibiotic resistance genes. Cell Mol. Life. Sci. 59 2044–2054. 10.1007/s000180200004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K. (2001). Preparation of genomic DNA from bacteria. Curr. Protoc. Mol. Biol. Chapter 2: Unit 2.4. 10.1002/0471142727.mb0204s56 [DOI] [PubMed] [Google Scholar]
- Wu H. J., Wang A. H., Jennings M. P. (2008). Discovery of virulence factors of pathogenic bacteria. Curr. Opin. Chem. Biol. 12 93–101. 10.1016/j.cbpa.2008.01.023 [DOI] [PubMed] [Google Scholar]
- Xu L., Luo Z. Q. (2013). Cell biology of infection by Legionella pneumophila. Microbes Infect. 15 157–167. 10.1016/j.micinf.2012.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. H., Austin B. (2000). Pathogenicity of Vibrio harveyi to salmonids. J. Fish Dis. 23 93–102. 10.1046/j.1365-2761.2000.00214.x [DOI] [Google Scholar]
- Zhou C. E., Smith J., Lam M., Zemla A., Dyer M. D., Slezak T. (2007). MvirDB–a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res. 35 D391–D394. 10.1093/nar/gkl791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Liang Y., Lynch K. H., Dennis J. J., Wishart D. S. (2011). PHAST: a fast phage search tool. Nucleic Acids Res. 39 W347–W352. 10.1093/nar/gkr485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoued A., Brunet Y. R., Durand E., Aschtgen M. S., Logger L., Douzi B., et al. (2014). Architecture and assembly of the Type VI secretion system. Biochim. Biophys. Acta 1843 1664–1673. 10.1016/j.bbamcr.2014.03.018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.