

Abstract
Oxidative stress and iron accumulation are tightly associated with neurodegenerative diseases. Mitochondrial ferritin (FtMt) is identified as an iron-storage protein located in the mitochondria, and its role in regulation of iron hemeostasis in neurodegenerative diseases has been reported. However, the role of FtMt in hydrogen peroxide (H2O2)-induced oxidative stress and iron accumulation in neuronal cells has not been studied. Here, we overexpressed FtMt in neuroblastoma SH-SY5Y cells and induced oxidative stress by treating with extracellular H2O2. We found that overexpression of FtMt significantly prevented cell death induced by H2O2, particularly the apoptosis-dependent cell death. The protective effects involved inhibiting the generation of cellular reactive oxygen species, sustaining mitochondrial membrane potential, maintaining the level of anti-apoptotic protein Bcl-2, and inhibiting the activation of pro-apoptotic protein caspase 3. We further explored the mechanism of these protective effects and found that FtMt expression markedly altered iron homeostasis of the H2O2 treated cells as compared to that of controls. The FtMt overexpression significantly reduced cellular labile iron pool (LIP) and protected H2O2-induced elevation on LIP. While in H2O2 treated SH-SY5Y cells, the increased iron uptake and reduced iron release, in correlation with levels of DMT1(-IRE) and ferroportin 1, resulted in heavy iron accumulation, the FtMt overexpressing cells didn’t show any significant changes in levels of iron transport proteins and in the level of LIP. These results implicate a neuroprotective role of FtMt on H2O2-induced oxidative stress, which may provide insights into the treatment of iron accumulation associated neurodegenerative diseases.
Keywords: mitochondrial ferritin, hydrogen peroxide, iron, oxidative stress, labile iron pool
Iron is an essential component for activities of numerous proteins and enzymes that are critical for cell respiration, proliferation and signal transduction [1, 2]. Iron metabolism in the body is strictly controlled, and disruption of iron homeostasis would cause severe diseases. Recently, neurodegenerative disorders that associated with iron accumulation in neurons, such as Parkinson’s and Alzheimer’s diseases, have attracted more and more attention [3-5]. Excess iron is highly toxic because of its ability to form highly reactive hydroxyl radicals in the presence of hydrogen peroxide (H2O2) [6]. The production of these reactive oxygen species (ROS) in neurons can damage cellular macromolecules including proteins, lipids and DNA, and finally the triggered oxidative stress will induce apoptosis of the neurons [7, 8].
Ferritin is a ubiquitous iron-storage protein in the cytosol that forms a spherical shell by 24 subunits and accommodates up to 4500 iron atoms [9, 10]. Most intracellular iron is stored in the cytosol by binding to ferritin. In mammalian cells, two ferritin subtypes have been found, H-ferritin and L-ferritin. The former subtype has ferroxidase activity essential for the storage of free iron in ferritin, while the latter has a nucleation site that is involved in iron-core formation [11, 12]. It has been reported that ferritins exert cellular protective roles against iron-mediated free radical damage induced by a variety of sources [10, 13]. In neuronal cells, elevated ferritin expression has been shown to protect the MPTP-induced experimental PD models well [14]. Therefore, it appears that neuronal cell survival is also dependent on the cellular level of ferritin.
Mitochondrial ferritin (FtMt) is a ferritin type protein targeted to mitochondria, and has been characterized structurally and functionally analogous to the well-characterized cytosolic H-ferritin [15]. FtMt has been shown to modulate cellular iron metabolism dramatically [16-19]. Previous studies suggested that overexpression of FtMt caused redistribution of iron from cytosol to mitochondria [16, 19], thus high levels of FtMt resulted in an iron deficient phenotype in cytosol [20]. The expression of FtMt is restricted to mitochondria of cells of testes, the central nervous system, and some other high oxygen-consumption tissues [21], indicating that the major role of FtMt may be protecting mitochondria from iron-dependent oxidative damage in cells characterized by high metabolic activity and oxygen consumption [22]. Our previous studies have found that FtMt overexpression protected 6-hydroxydopamine-induced dopaminergic cell damage, potentially playing an important neuroprotective role in Parkinson’s Disease [23]. Studies by Campanella et al. revealed a protective role of FtMt in Friedreich ataxia, a disease characterized by mitochondrial iron overload and oxidative damage [24, 25]. FtMt expression also inhibited tumor growth due to cytosolic iron deprivation [26]. The protective role of FtMt against oxidative stress in other disease models has also been suggested [22, 27-30]. These studies demonstrated that FtMt is not only involved in storing cellular iron, but may also play a role in protecting mitochondria from iron-dependent oxidative damage [22-30].
In this study, we aimed to investigate the role that FtMt plays against the oxidative stress to mitochondria induced by H2O2. A recent study by Dev et al. revealed the effect of H2O2 treatment on LIP level and cellular iron-uptake, -storage and -release proteins in the neuroblastoma cell line SH-SY5Y [31]. They found that iron heavily accumulated in SH-SY5Y cells after H2O2 treatment, and iron-release protein FPN1 significantly decreased, whereas iron-uptake protein didn’t change much [31]. Interestingly, they also found the expression of iron-storage protein H-ferritin was decreased, which was not in accordance with the regulation by the iron-regulatory protein (IRP) [32]. However, the functions of the iron-storage protein in mitochondria, FtMt, in H2O2 induced oxidative stress in neuronal cells have not been studied. We hypothesized that FtMt may play a neuroprotective role in H2O2 induced cell stress. Thus, we overexpressed FtMt gene in the neuroblastoma SH-SY5Y cells to see if an increase of FtMt expression can sequester more free iron and counter the H2O2-induced iron accumulation and cell damage. We further investigated its effects on iron metabolism and the mechanisms of neuroprotection in H2O2-induced apoptosis. This study would be useful for understanding the roles of FtMt in neurodegenerative diseases. It may provide insight into discovering new therapeutic methods for treatment of iron overload-related neurodegenerative disorders.
MATERIALS AND METHODS
Cell lines and H2O2 treatment
The stable FtMt-expressing SH-SY5Y cell line (FtMt-SY5Y) and pcDNA3.1(-) empty vector transfected cell line (vector-SY5Y) were generated as described previously [23]. Briefly, the amplified mouse FtMt cDNA, with a C-terminal hemagglutinin (HA) epitope sequence, was cloned into pcDNA3.1(-) vector to generate construct FtMt-pcDNA3.1(-)[20]. The plasmids of FtMt-pcDNA3.1(-) and empty vector were transfected into SH-SY5Y cells, and stable transfectants were selected [23]. The expression of mouse FtMt protein was confirmed with western blotting by using anti-HA antibody [23]. The endogenous expression of human FtMt in SH-SY5Y cells was very low as compared to the levels of overexpressed mouse FtMt as measured by RT-PCR [23]. The cell viability and apoptotic ratio of FtMt-SY5Y cells had no difference compared to the wild-type (WT) SH-SY5Y cells [23], but the growth of FtMt-SY5Y cells was significantly slower [26].
The WT SH-SY5Y cells, FtMt-SY5Y cells and vector-SY5Y cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fatal bovine serum, 100 U/ml penicillin and 100 U/ml streptomycin. After the cells reaching ~80% confluency, H2O2 were added to a final concentration of 100 μM (or as described in each specific experiment), and cells were then incubated at 37? for 24 h prior to analysis.
Antibodies and Chemicals
Rabbit anti-human β-actin antibody, rabbit anti-rat DMT1(+IRE) antibody, rabbit anti-rat DMT1(-IRE) antibody, anti-mouse FPN1 antibody and anti-rat TfR1 antibody were purchased from ADI (San Antonio, TX, USA); anti-human H-ferritin and L-ferritin antibodies were purchased from Abcam (Cambridge, MA, USA); anti-human caspase 3 antibody, and anti-mouse Bax and Bcl-2 antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); rabbit anti-mouse FtMt polyclonal antibody [21] and mouse anti-human FtMt monoclonal antibody [33] were gifts from Prof. Sonia Levi. The salicylaldehyde isonicotinoyl hydrazone (SIH) was synthesized as described by Ponka et al. [34]. Unless otherwise stated, all chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Western blotting
After H2O2 treatment (100 μM, 24 h), cells were homogenized and lysed with RIPA buffer and protein content was determined by the Bradford assay. Aliquots of cell lysate containing approximately 30 μg of protein were immediately mixed with loading buffer and boiled for 10 minutes. Equal amounts of protein for different cells were loaded, resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a PVDF membrane. The blots were blocked by incubating with 5% nonfat milk in PBS containing 0.1% Tween 20 (PBS-T) for 1 h, and hybridized with primary antibodies. After washing 3 times for 15 minutes each with PBS-T, the blots were incubated for 1 h with peroxidase-coupled secondary antibodies, and detected with the ECL plus Western Blotting Detection Reagents (Pierce Biotechnology, Rockford IL). Six independent experiments were performed for each treatment.
Cell viability assay
Cell viability was assessed by vital dye reduction with 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT) [35]. Briefly, SH-SY5Y cells were seeded in a 96-well plate at a density of 2×104 cells/well. After attachment, H2O2 was added to cell culture in DMEM without serum at final concentrations of 0, 20, 40, 60, 80, 100, 120, 140 μmol/L, and incubated for the subsequent 24 h at 37?. H2O2 treated or untreated cells were incubated in MTT (0.5 mg/mL) for another 3 h, and then cells were lysed with DMSO, and the absorbance at 595 nm was measured with a Bio-Rad model 3550 microplate reader (Richmond, CA). All samples were measured in six independent experiments.
Detection of cell apoptosis
After H2O2 treatment (100 μM, 24 h), cell apoptosis was detected by using annexin V/PI assay by flow cytometry as described previously [23].
Measurement of the labile iron pool (LIP) level
The LIP level was determined by the quenching of calcein fluorescence as described previously [36, 37]. After treatment with or without 100 μM H2O2 for 24 h, the cells were harvested, washed, and resuspended in PBS (pH 7.4) buffer containing calcein-AM (0.25 μM final concentration), and incubated for 30 min at 37?. The excess calcein-AM on the cell surface was washed out 3 times with PBS. The fluorescence intensity of calcein-AM was quantified by a fluorescence spectrophotometer (Hitachi F-4500), at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. When the baseline was stable, SIH (100 μM final concentration) was added, and the increase in fluorescence intensity reflected the levels of calcein-bound iron. Three independent experiments were performed for each treatment.
Detection of intracellular oxidative stress-ROS assay
Intracellular ROS were examined using 2′,7′-dichlorofluorescein diacetate (DCF-DA) as described before [38]. After treatment with or without 100 μM H2O2 for 24 h, cells were harvested, washed, and resuspended in PBS containing DCF-DA (10 μM final concentration), and incubated for 30 min at 37?. Cells were washed twice with PBS, and the fluorescence signals were measured by a fluorescence spectrophotometer with 488 nm excitation and 525 nm emission wavelengths. Three independent experiments were performed for each treatment. The data were expressed as a percentage of the fluorescence relative to the fluorescence of the untreated WT control cells.
Detection of mitochondrial membrane potential (MMP)
Changes in the MMP with or without H2O2 treatment (100 μM, 24 h) of SH-SY5Y cells were determined by measuring the retention of rhodamine 123 using flow cytometry [39]. The uptake of rhodamine123 into mitochondria is an indicator of the MMP. Cells were incubated with rhodamine123 at a final concentration of 5 μM for 30 min at 37?. After washing twice with PBS, fluorescence was recorded at 488 nm excitation and 525 nm emission wavelengths. Three independent experiments were performed for each treatment.
Measurement of iron uptake and release
55Fe (55FeCl3, Perkin-Elmer Life Sciences Company, Wellesley City, MA) solution was prepared by mixing 55FeCl3 with FeSO4 in a molar ratio of 1:10 followed by a 50-fold molar excess of 2-mercaptoethanol and 0.27 M sucrose (pH 6.5) as described previously [40, 41]. The H2O2 treated (100 μM, 24 h) or untreated cells (about 1×106 cells) were washed three times with PBS buffer, and added with 55FeCl3 solution (1 mM) for 30 min at 37? and then washed three times with PBS. The cells were homogenized in buffer containing 1% sodium dodecyl sulphate (SDS), and aliquots of the total cell extract were assayed for released radioactivity with Liquid Scintillation Analyzer (Beckman) and protein concentrations by Lowry method. Three independent experiments were performed for each treatment.
For measurement of iron release, the cells were incubated with 55FeCl3 solution for 30 min and then washed three times with PBS. The cells were then incubated with 1 ml PBS at 37? for 30 min. The supernatant was collected. The cells were homogenized after washing three times with PBS. Both supernatant and cell extract were assayed for radioactivity. Percentage (%) of 55Fe release = (cpm in supernatant) / (cpm in supernatant + cpm in cells) × 100%.
Statistical analysis
All statistical analyses were completed using SPSS 21.0 software. Results are presented as means ± SD. The statistical analyses of group differences were assessed by a two-way analysis of variance (ANOVA). P-values of <0.05 were considered to be statistically significant; P <0.01 was considered to be remarkably significant.
RESULTS
FtMt overexpression rescued neuronal cell death induced by H2O2 treatment
Previous studies have indicated that FtMt plays an important role in protecting cells from iron-dependent oxidative damage [23, 25, 27, 30], particularly in neurodegenerative diseases [42, 43]. In order to investigate the role of FtMt in hydrogen peroxide (H2O2)-induced neuronal cell damage, a stable FtMt-overexpressing neuroblasma cell line, FtMt-SY5Y, generated previously was used [23]. Wild-type SH-SY5Y cells and pcDNA3.1(-) empty plasmid transfected cells (vector-SY5Y) were used as controls. The expression of FtMt in FtMt-SY5Y cells was confirmed (Fig. 1A). The level of overexpressed mouse FtMt was much higher than that of the endogenous human FtMt. The cell viability of the three cell lines was determined after H2O2 treatment for 24 h. Results showed that H2O2 reduced cell viability in all three cell lines in a concentration-dependent manner (Fig. 1B). However, after H2O2 treatment, at concentrations of 60, 80, 100, 120 and 140 μM, cell viability in FtMt-SY5Y cells was significantly higher than that of the vector-SY5Y cells (P < 0.01, Fig. 1B). With 100 μM H2O2 treatment, the viability of SH-SY5Y and vector-SY5Y cells were decreased to ~40%, whereas the viability of FtMt-SY5Y cells only had a ~11% decrease. 100 μM of H2O2 treatment was then used for the following assays. These results indicated that H2O2 induced cell damage and that overexpression of FtMt in SH-SY5Y cells improved cell viability.
Figure 1. Effects of FtMt on cell viability and apoptosis ratios of cells treated with H2O2.

(A) The expressions of exogenous mouse FtMt (top panel) and endogenous human FtMt (bottom panel) in FtMt overexpressed transfectants (FtMt) were examined by western blot using anti-mouse and anti-human FtMt antibodies, respectively. K562-Mt6 is a human FtMt overexpressing cell line (a gift from Prof. Sonia Levi), which is used as a positive control. (B) The wild-type (WT) SH-SY5Y cells, empty vector transfectants (vector), and FtMt overexpressed transfectants (FtMt) were treated with increasing concentrations (0, 20, 40, 60, 80, 100, 120, 140 μM) of H2O2 for 24 h. Cell viability was measured with the MTT assay. Data were presented as percentage of the cell viability compared with untreated WT control cells ± SD, n=6 (##, p < 0.01 vs. the H2O2-treated vector control cells). (C) The relative apoptotic ratios of control and FtMt-SY5Y cells with or without H2O2 treatment (100 μM, 24 h) were determined by flow-cytometry (Ci and Cii). Data were presented as means ± SD; n=3 (**, p < 0.01 vs. the untreated cells of same genotype; ##, p < 0.01 vs. the H2O2-treated vector controls).
To explore if the rescue effect of FtMt was resulted from increased resistance of FtMt-SY5Y cells against apoptosis induced by H2O2, the apoptotic rates in vector-SY5Y and FtMt-SY5Y cells were quantified by flow cytometry after annexin V and PI staining. After treated with 100 μM H2O2 for 24 h, the ratio of apoptotic vector-SY5Y cells increased from 4.7% to 39.5% (P < 0.01, Fig. 1Cii, indicated by **); the apoptotic ratio of FtMt-SY5Y cells also increased (from 4.9% to 16.5%, P < 0.01). However, when comparing the increases between FtMt overexpressing cells and vector control cells, the FtMt overexpressing cells had significantly lower number of apoptotic cells (P < 0.01, Fig. 1C, indicated by ##). The results inferred that FtMt has a significant anti-apoptotic role in H2O2-induced cell damage.
FtMt maintained Bcl-2/Bax ratio and inhibited the activation of caspase 3
Bcl-2 and Bax are anti-apoptotic and pro-apoptotic proteins, and the Bcl-2/Bax ratio has been widely used to monitor apoptosis [44]. To further confirm if the rescue effect of FtMt on cell death was resulted from decreasing cell apoptosis, the ratios of Bcl-2/Bax for vector-SY5Y and FtMt-SY5Y cells with/without H2O2 treatment were quantified by western blotting. In vector-SY5Y cells, dramatic decreases in Bcl-2 levels and increases in Bax levels were observed after H2O2 treatment as compared to that of the untreated control group, causing obviously reduced Bcl-2/Bax ratios (P < 0.01, Fig. 2A). However, the FtMt over-expressed FtMt-SY5Y cells maintained the levels of both Bcl-2 and Bax, showing a relatively small change in the Bcl-2/Bax ratio (P < 0.05, Fig. 2A). H2O2 treatment also caused caspase 3 activation in SH-SY5Y cells and vector-SY5Y cells, as shown by the increased cleaved-caspase 3 levels (Fig. 2B), whereas the activation wasn’t observed in the FtMt-SY5Y cells. These findings indicate that FtMt significantly maintains the normal level of Bcl-2/Bax and prevents the activation of caspase 3 to protect the cells against H2O2-induced apoptosis, thereby protecting cells from death.
Figure 2. FtMt protected cells from apoptosis by maintaining Bcl-2/Bax ratio and inhibiting activation of caspase 3.

(A) The protein expression levels of Bcl-2 and Bax in H2O2 treated (100 μM, 24 h) or untreated cells were determined by western-blot (right panel), and ratios of Bcl-2/Bax were calculated and presented as mean ± SD (left panel); n=6 (*, P < 0.05 and **, P < 0.01 vs. the untreated cells of same genotype; ##, p < 0.01 vs. the H2O2-treated vector controls). (B) Expression levels of caspase 3 and cleaved-caspase 3 were shown (right panel). Ratios of cleaved-caspase 3/caspase 3 were calculated and presented as mean ± SD (left panel); n=6 (**, P < 0.01 vs. the untreated cells of same genotype; ##, p <0 .01 vs. the H2O2-treated vector controls).
FtMt attenuated H2O2-induced increases in ROS level and H2O2-induced decreases in mitochondrial membrane potential (MMP)
Evidences have strongly suggested that apoptosis is closely linked to production of damaging ROS during electron transport [7, 45], therefore we determined the effect of FtMt expression on the ROS level, by measuring the ROS-dependent fluorescence of DCF-DA using flow cytometry [38]. The relative ROS levels of different cells with/without H2O2 treatment and their differences are shown in Fig. 3A. The H2O2 treatments led to significant increases in intracellular ROS production in both SH-SY5Y and vector-SY5Y cells as compared to the untreated controls (P < 0.01). In contrast, the ROS level of FtMt-SY5Y cells only increased slightly by H2O2 treatment as compared to the untreated control. This indicates that although the overexpression of FtMt doesn’t alter the intracellular ROS levels in SH-SY5Y cells under normal conditions, it can significantly attenuate the H2O2-induced increase in ROS level, which therefore protects the SH-SY5Y cells from oxidative damage.
Figure 3. FtMt attenuated H2O2-induced increases in ROS level and H2O2-induced decreases in MMP.

(A) Cells treated with or without 100 μM H2O2 for 24 h were detected for ROS production by 2,7-dichlorofluorescein diacetate (DCF-DA) fluorescence. The relative fluorescence level for each group against that of the WT untreated control is expressed as mean ± SD, n=3 (**, P < 0.01 vs. the untreated cells of same genotype; ##, p < 0.01 vs. the H2O2-treated vector controls). (B) The MMP of H2O2 treated (100 μM, 24 h) or untreated cells was detected by the fluorescence of rhodamine 123. Data are presented as means of fluorescence intensity ± SD, n=3 (*, P < 0.05 vs. the untreated cells of same genotype; #, p <0.05 vs. the H2O2-treated vector controls).
The state of MMP is an indicator of the metabolic activity of mitochondria and closely relates to mitochondrial dysfunction and ROS overproduction-induced apoptosis [39]. To examine the underlying protective mechanism of FtMt on mitochondria, the MMP of the three different cell lines was measured. The results showed that H2O2 treatment decreased MMP by approximately 36% and 38% in SH-SY5Y and vector-SY5Y cells (P < 0.05, Fig. 3B), respectively, as compared with the untreated control groups; however, in FtMt-SY5Y cells, the MMP decreased only about 12% (Fig. 3B). These results indicate that FtMt maintains the MMP and protects against the mitochondrial damage induced by H2O2.
FtMt significantly decreased intracellular labile iron pool (LIP) levels
Excess ferrous ion (Fe2+) can result in the generation of ROS, which damages cellular macromolecules including proteins, lipids and DNA, and consequently triggers apoptosis [6-8, 46]. It has been shown that FtMt could mobilize iron into mitochondria and dramatically redistributes intracellular iron [16, 20]. To further clarify FtMt’s protective mechanism on cell death, the LIP levels in different cell lines were quantified by using iron-specific fluorescence probe calcein-AM. Accessible free iron quenched by calcein was quantified and shown in Fig. 4. The FtMt overexpressing cells indeed had lower amount of LIP as compared with the WT SH-SY5Y cells (P < 0.05, indicated by &). H2O2 treatment dramatically raised the LIP levels in WT and vector transfected SH-SY5Y cells (P < 0.01, indicated by **), but it only led to a minor increase in the LIP levels in FtMt overexpressing cells. These results infer that the largely increased LIP in SH-SY5Y cells, following H2O2 treatment, results in the increased ROS and apoptosis as observed above. The protective role of FtMt overexpression on H2O2-induced elevation in LIP is remarkably significant.
Figure 4. FtMt overexpression significantly decreased cellular LIP levels in both H2O2 treated (100 μM, 24 h) and untreated cells.
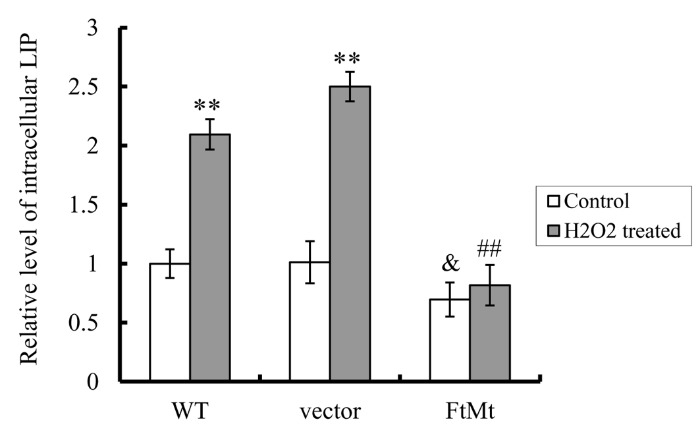
LIP levels were determined by the quenching of calcein fluorescence method using fluorescence spectrophotometer. The relative LIP level as compared to WT untreated control was calculated and presented as mean ± SD; n=3 (**, p < 0.01 vs. the untreated cells of same genotype; &; p < 0.05 vs. the untreated vector controls; ##, p < 0.01 vs. the H2O2-treated vector controls).
FtMt inhibited H2O2-induced elevations in iron uptake and reductions in iron release
Iron-induced oxidative stress can be very destructive because of a positive-feedback loop developed from the release of more free iron from the iron-containing proteins, such as ferritin, heme proteins and Fe-S clusters [7]. As a result, the toxic effect of iron overload in neuronal cells is exacerbated. To clarify the mechanisms of FtMt in regulating intracellular iron homeostasis and the consequent neuroprotective role in neuronal cells, the iron uptake and release were determined by 55Fe isotope tracer experiments in vector-SY5Y cells and FtMt-SY5Y cells after treated with H2O2. The results showed that the cellular 55Fe uptake increased significantly in all cell lines after H2O2 treatment (P < 0.01, Fig. 5A), but the increased amount in FtMt-SY5Y cells is much less than that of the vector-SY5Y controls (P < 0.05, indicated by ##). The 55Fe release in the control vector-SY5Y cells decreased significantly (P<0.01, Fig. 5B), whereas iron release in FtMt-SY5Y cells only slightly decreased. These results suggest that H2O2 causes cells to uptake more iron, but release less; therefore, iron accumulates in cytosol. However, when FtMt overexpresses, it partially counteracts these effects and maintained a relatively low cellular iron level, protecting cells from damage.
Figure 5. FtMt overexpression attenuated the H2O2-induced increase of 55Fe uptake and decrease of 55Fe release.

Detailed determinations were described in “Materials and methods”. (A) 55Fe uptake levels were shown as iron concentrations. Data represent means ± SD; n=3 (*, P <0.05, and **, P < 0.01 vs. the untreated cells of same genotype; #, p < 0.05 vs. the H2O2-treated vector controls). (B) 55Fe release levels were the percentages of iron in supernatant of cell culture against total iron (sum of supernatant and cell lysate). Data represent means±SD; n=3 (**, P < 0.01 vs. the untreated cells of same genotype; ##, p < 0.01 vs. the H2O2-treated vector controls).
FtMt maintained iron transport proteins at relative steady levels
To explore the protective function of FtMt on oxidative damage at molecular level, we examined the alterations of iron transport proteins induced by H2O2 treatment in different cell lines. In SH-SY5Y and vector-SY5Y cells, following H2O2 treatment, the iron efflux protein ferroportin 1 (FPN1) significantly decreased (P < 0.05, Fig. 6A). The iron uptake proteins, transferrin receptor 1 (TfR1) and divalent metal transporter 1 with iron responsive element (DMT1(+IRE)), didn’t show significant changes (Fig. 6B and 6C), but DMT1 without iron responsive element (DMT1(-IRE)) increased dramatically (P < 0.01, Fig. 6D). The changes in FPN1 and DMT1(-IRE) may account for the observed intracellular iron accumulation in SH-SY5Y cells caused by H2O2 treatment [31].
Figure 6. FtMt repressed the alterations of iron release and uptake proteins induced by H2O2 treatment (100 μM, 24 h).

FPN1 (A), TfR1 (B), DMT1(+IRE) (C), DMT1(-IRE) (D), L-ferritin (E) and H-ferritin (F) levels were determined by western blots. A representative blot image for each protein and its respective β-actin was shown. The expression levels in different groups were calculated by normalizing the specific bands to their respective β-actin bands. The relative expression levels as compared to WT untreated control group were calculated and presented as means ± SD, n=6 (*, p <0.05 vs. the untreated cells of same genotype; &, p < 0.05 vs. the untreated vector controls; #, p < 0.05 and ##, p < 0.01 vs. the H2O2-treated vector controls).
In contrast to SH-SY5Y cells and vector-SY5Y cells, H2O2 treatment didn’t significantly alter FPN1, TfR1, DMT1(+IRE) and DMT1(-IRE) levels in FtMt-SY5Y cells (Fig. 6A-D). The expression levels of TfR1, DMT1(+IRE) and DMT1(-IRE) in H2O2 treated FtMt-SY5Y cells were significantly different from that in treated vector-SY5Y cells (indicated by # or ##, P < 0.05 or 0.01, respectively, Fig. 6B-D). These results demonstrated that FtMt overexpressing cells maintained the iron transport proteins at relatively steady levels, protecting them from damages induced by H2O2.
In addition, we found that, prior to H2O2 treatment, the FtMt-SY5Y cells alone had lower FPN1 expression (P < 0.05, indicated by &, Fig. 6A) and higher TfR1 expression (P < 0.05) than the untreated vector-SY5Y controls. These findings were consistent with our previous observations [23] and may attribute to cellular regulations in response to low LIP level caused by FtMt overexpression.
FtMt inhibited H2O2-induced reductions in cytosolic ferritin levels
The effects of H2O2 treatment on cytosolic ferritin levels in different cells were also determined. As shown in Fig. 6E, L-ferritin levels in SH-SY5Y and vector-SY5Y cells significantly decreased after H2O2 treatment (P < 0.01). However, the FtMt overexpressing cells had low levels of cytosolic L-ferritin, despite with or without H2O2 treatment. The H-ferritin levels in all three cells decreased slightly, but not statistically significant (Fig. 6F). These results indicated that H2O2 induced downregulations of cytosolic ferritins, particularly L-ferritin, and that FtMt overexpression counteracted these effects. The FtMt-SY5Y cells had low levels of cytosolic ferritins, which were consistent with their low intracellular LIP levels, attesting the iron redistributing function of FtMt.
DISCUSSION
At normal condition, cells were in a balance between ROS and antioxidant. Once the balance was disrupted, cells would be exposed to oxidative stress damage. ROS destroys the enzymes in cytosol, induces lipid peroxidation, decreases MMP, and further leads to cell apoptosis. Iron is one of the important activators that lead to oxidative stress damage [6, 46]. Thus, the iron level in the body is under strict control, and dysregulation of iron homeostasis could cause severe diseases. FtMt, as an iron-storage protein in mitochondria, has been reported to be involved in iron redistribution from cytosol to mitochondria [15, 16, 20]. Recently, FtMt was mainly recognized for its protective role in diseases associated with iron-dependent oxidative damage [23-25, 27-29], although that FtMt sensitized cells to oxidative stress was also reported [47]. Our previous studies have indicated the neuroprotective role of FtMt in Parkinson’s disease and Alzheimer’s disease [23, 27]. In this study, we explored the effects of FtMt on H2O2 induced neuronal cell damage and further investigated FtMt’s functions at the molecular level.
An FtMt overexpressing neuroblastoma SH-SY5Y cell line is used to better investigate the functions of FtMt. Our results indicated that overexpression of FtMt significantly protected neuroblastoma cells from H2O2 induced apoptosis, which was due to the restriction effects of FtMt on the increase of intracellular ROS and the decrease of MMP, thus protecting cells from oxidative stress-mediated apoptosis. On the molecular level, we found that H2O2 decreased the expression of Bcl-2 and increased the level of Bax dramatically in SH-SY5Y and vector-SY5Y cells, while the changes of Bcl-2/Bax ratio in FtMt-SY5Y cells were much smaller. Our results also showed that caspase 3, another important member of the apoptotic family, was activated by H2O2 treatment in SH-SY5Y and vector-SY5Y cells, leading to caspase 3-dependent cell apoptosis. However, in the FtMt overexpressing cells, no obvious increase in cleaved-caspase 3 level was observed.
It has been reported that H2O2 can modulate cellular iron metabolism [48, 49]. It affects cellular iron acquisition by both IRP1-dependent and -independent mechanisms, and modulates intracellular iron distribution at a time-dependent manner by unknown mechanisms [48]. Consistent with this, our results showed that after H2O2 treatment, cellular LIP levels greatly increased in SH-SY5Y and vector-SY5Y cells, reaching more than 2-times of the untreated cells. This change may attribute to two aspects. One is the observed higher iron uptake and lower release, correlating with lower expression of iron efflux protein FPN1 and higher iron absorption protein DMT1(-IRE); the other reason might be the internal iron release from various intracellular iron-containing sources, including ferritin, induced by oxidative stress after H2O2 treatment [47, 48, 50].
The reduction of FPN1 is consistent with the newly published study by Dev, et al. [31], in which the role of extracellular H2O2 in regulation of iron homeostasis-related genes was exhaustively investigated. They concluded that the substantially reduced FPN1 may be responsible for iron accumulation in the H2O2 treated cells, whereas the alterations of other iron-transport proteins were not obvious. However, they only determined total DMT1 levels, which didn’t alter much. Here, we found that the DMT1(+IRE) showed a statistically insignificant decrease following H2O2 treatment, which was likely due to the increased iron-binding activity of IRP1 as reported previously [47, 50]. But interestingly, we found that the IRP-independent DMT1(-IRE) expressed to a significantly higher level. DMT1(-IRE) is an important protein involved in iron transport, however, its molecular mechanism was still unknown [51, 52]. We hypothesized that the increased expression of DMT1(-IRE) on cell membrane may account for the increase of iron absorption observed in our 55Fe isotope tracer experiments; in addition, the DMT1 on membrane of cell organelles, such as membranes of lysosome and mitochondria [53, 54], may also elevate, likely playing important roles in iron redistribution.
In FtMt-SY5Y cells, we found the LIP levels significantly reduced. However, it seems that the iron acquisition increased and iron release decreased as indicated by the higher TfR1 and lower FPN1 expression. These suggested that the increased iron influx was preferentially transferred into mitochondria and incorporated into FtMt rather than into cytosol [16, 20], which explains the decreased cytosolic LIP even with higher iron acquisition and lower iron release. Despite the differences of FtMt-SY5Y cells from WT SH-SY5Y on iron metabolism, the over-expressed FtMt protected H2O2-induced elevation in LIP in FtMt cells. Similar to the WT or vector controls, H2O2 treatment indeed induced higher iron uptake and lower iron release in FtMt-SY5Y cells, however, their LIP level didn’t increase significantly because of the overexpressed FtMt. Consistent with these, both the iron-transport proteins and the cytosolic iron-storage proteins were not altered notably by the extracellular H2O2 treatment. These all indicated that overexpression of FtMt can cause dramatic redistribution of cellular iron, even at a severe condition, such as extracellular H2O2 treatment.
In summary, our results showed that FtMt played an important role in rescuing cellular damage induced by H2O2, which was achieved by regulating iron metabolism (Fig. 7). In SH-SY5Y cells, H2O2 treatment causes an increase in DMT1(-IRE) expression and decrease in FPN1 expression, resulting in the increased intracellular LIP level. The increased LIP level induces the generation of ROS, which subsequently lead cells to apoptosis, which possibly involves the decrease of Bcl-2/Bax ratio and the activation of apoptosis signal pathway protein caspase 3. In the FtMt overexpressing cells, a great amount of free iron was captured and stored in FtMt, resulting in a substantial decrease in LIP level. Therefore, under H2O2 treatment, the FtMt overexpressing cells decreased the production of ROS, conserved MMP, maintained the anti-apoptotic protein Bcl-2 level, and inhibited the activation of caspase 3. Consequently, FtMt protected cells from oxidative damage.
Figure 7. A schematic representation of the proposed neuroprotective mechanism of FtMt on H2O2-induced neuronal cell damage.
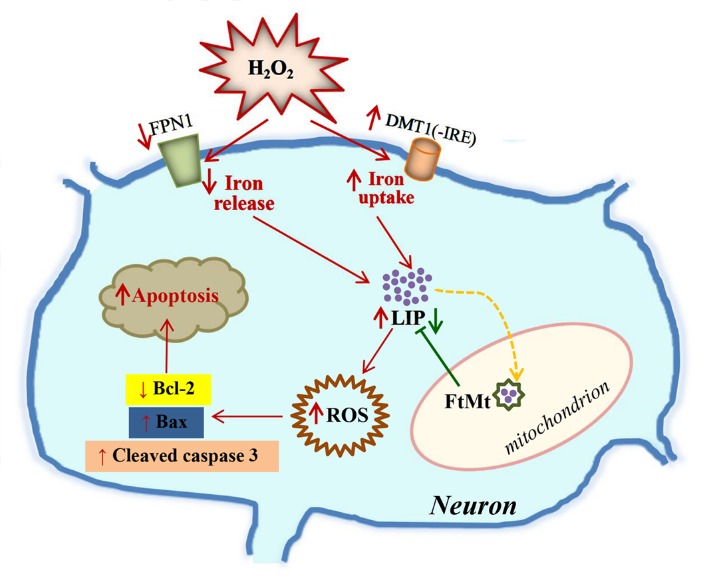
Extracellular H2O2 changes the expression of iron-related proteins, particularly iron-release protein FPN1 and iron-uptake protein DMT1(-IRE), resulting in an increase in the intracellular LIP level. The free iron may donate electrons for the generation of ROS, and then cell is triggered to begin the process of programmed cell death, which involves the signalings of Bcl-2, Bax and caspase 3. Alternatively, the overexpressed FtMt may withdraw iron from the cytoplasmic pool, decreasing LIP level. This in turn attenuates H2O2-induced neurotoxicity and reduces oxidative damage. The damaging effects caused by H2O2 treatment were indicated with red arrows, while the protective effect of FtMt on LIP level was indicated by a green arrow.
Thus, our current study revealed a protective role of FtMt in neuronal cells against H2O2 induced oxidative damage, which was achieved by modulating the homeostasis of iron metabolism. This observation is consistent with the study by Al-Qenaei et al., in which a defensive effect of FtMt against H2O2 treatment in Jurkat T cells was implicated [30]. Since dysregulation of iron homeostasis, together with oxidative stress, has been largely demonstrated in several neurodegenerative disorders [3-6, 43], modulation of FtMt expression may prevent or cure these diseases. This process can be greatly facilitated by investigating the mechanisms of regulation of FtMt expression [55, 56]. This study may provide insight into the development of novel effective strategies for treatment and prevention of neurodegenerative diseases caused by iron-dependent oxidative damage.
Acknowledgements
The work was supported in part by the National Natural Science Foundation of China [grant numbers 31300898, 31271473 and 31520103908]; and the Natural Science Foundation of Hebei Normal University [grant number L2013B12].
References
- [1].Andrews SC (1998). Iron storage in bacteria. Adv Microb Physiol, 40: 281-351 [DOI] [PubMed] [Google Scholar]
- [2].Ponka P (1999). Cellular iron metabolism. Kidney Int Suppl, 69: S2-11 [DOI] [PubMed] [Google Scholar]
- [3].Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol, 13: 1045-1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ayton S, Lei P (2014). Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. Biomed Res Int, 2014: 581256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Langkammer C, Ropele S, Pirpamer L, Fazekas F, Schmidt R (2014). MRI for iron mapping in Alzheimer’s disease. Neurodegener Dis, 13: 189-191 [DOI] [PubMed] [Google Scholar]
- [6].Halliwell B, Gutteridge JM (1990). Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol, 186: 1-85 [DOI] [PubMed] [Google Scholar]
- [7].Eaton JW, Qian M (2002). Molecular bases of cellular iron toxicity. Free Radic Biol Med, 32: 833-840 [DOI] [PubMed] [Google Scholar]
- [8].Ray PD, Huang BW, Tsuji Y (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal, 24: 981-990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Harrison PM, Arosio P (1996). The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta, 1275: 161-203 [DOI] [PubMed] [Google Scholar]
- [10].Arosio P, Levi S (2002). Ferritin, iron homeostasis, and oxidative damage. Free Radic Biol Med, 33: 457-463 [DOI] [PubMed] [Google Scholar]
- [11].Chasteen ND, Harrison PM (1999). Mineralization in ferritin: an efficient means of iron storage. J Struct Biol, 126: 182-194 [DOI] [PubMed] [Google Scholar]
- [12].Lawson DM, Artymiuk PJ, Yewdall SJ, Smith JM, Livingstone JC, Treffry A, et al. (1991). Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature, 349: 541-544 [DOI] [PubMed] [Google Scholar]
- [13].Arosio P, Ingrassia R, Cavadini P (2009). Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta, 1790: 589-599 [DOI] [PubMed] [Google Scholar]
- [14].Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, et al. (2003). Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson’s disease. Neuron, 37: 899-909 [DOI] [PubMed] [Google Scholar]
- [15].Levi S, Corsi B, Bosisio M, Invernizzi R, Volz A, Sanford D, et al. (2001). A human mitochondrial ferritin encoded by an intronless gene. J Biol Chem, 276: 24437-24440 [DOI] [PubMed] [Google Scholar]
- [16].Corsi B, Cozzi A, Arosio P, Drysdale J, Santambrogio P, Campanella A, et al. (2002). Human mitochondrial ferritin expressed in HeLa cells incorporates iron and affects cellular iron metabolism. J Biol Chem, 277: 22430-22437 [DOI] [PubMed] [Google Scholar]
- [17].Cazzola M, Invernizzi R, Bergamaschi G, Levi S, Corsi B, Travaglino E, et al. (2003). Mitochondrial ferritin expression in erythroid cells from patients with sideroblastic anemia. Blood, 101: 1996-2000 [DOI] [PubMed] [Google Scholar]
- [18].Drysdale J, Arosio P, Invernizzi R, Cazzola M, Volz A, Corsi B, et al. (2002). Mitochondrial ferritin: a new player in iron metabolism. Blood Cells Mol Dis, 29: 376-383 [DOI] [PubMed] [Google Scholar]
- [19].Santambrogio P, Erba BG, Campanella A, Cozzi A, Causarano V, Cremonesi L, et al. (2011). Over-expression of mitochondrial ferritin affects the JAK2/STAT5 pathway in K562 cells and causes mitochondrial iron accumulation. Haematologica, 96: 1424-1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nie G, Sheftel AD, Kim SF, Ponka P (2005). Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood, 105: 2161-2167 [DOI] [PubMed] [Google Scholar]
- [21].Santambrogio P, Biasiotto G, Sanvito F, Olivieri S, Arosio P, Levi S (2007). Mitochondrial ferritin expression in adult mouse tissues. J Histochem Cytochem, 55: 1129-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cocco E, Porrini V, Derosas M, Nardi V, Biasiotto G, Maccarinelli F, et al. (2013). Protective effect of mitochondrial ferritin on cytosolic iron dysregulation induced by doxorubicin in HeLa cells. Mol Biol Rep, 40: 6757-6764 [DOI] [PubMed] [Google Scholar]
- [23].Shi ZH, Nie G, Duan XL, Rouault T, Wu WS, Ning B, et al. (2010). Neuroprotective mechanism of mitochondrial ferritin on 6-hydroxydopamine-induced dopaminergic cell damage: implication for neuroprotection in Parkinson’s disease. Antioxid Redox Signal, 13: 783-796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Campanella A, Isaya G, O’Neill HA, Santambrogio P, Cozzi A, Arosio P, et al. (2004). The expression of human mitochondrial ferritin rescues respiratory function in frataxin-deficient yeast. Hum Mol Genet, 13: 2279-2288 [DOI] [PubMed] [Google Scholar]
- [25].Campanella A, Rovelli E, Santambrogio P, Cozzi A, Taroni F, Levi S (2009). Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: hypothesis for a protective role in Friedreich ataxia. Hum Mol Genet, 18: 1-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shi ZH, Shi FF, Wang YQ, Sheftel AD, Nie G, Zhao YS, et al. (2015). Mitochondrial ferritin, a new target for inhibiting neuronal tumor cell proliferation. Cell Mol Life Sci, 72: 983-997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wu WS, Zhao YS, Shi ZH, Chang SY, Nie GJ, Duan XL, et al. (2013). Mitochondrial ferritin attenuates beta-amyloid-induced neurotoxicity: reduction in oxidative damage through the Erk/P38 mitogen-activated protein kinase pathways. Antioxid Redox Signal, 18: 158-169 [DOI] [PubMed] [Google Scholar]
- [28].Wang L, Yang H, Zhao S, Sato H, Konishi Y, Beach TG, et al. (2011). Expression and localization of mitochondrial ferritin mRNA in Alzheimer’s disease cerebral cortex. PLoS One, 6: e22325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].You LH, Li Z, Duan XL, Zhao BL, Chang YZ, Shi ZH (2016). Mitochondrial ferritin suppresses MPTP-induced cell damage by regulating iron metabolism and attenuating oxidative stress. Brain Res, 1642: 33-42 [DOI] [PubMed] [Google Scholar]
- [30].Al-Qenaei A, Yiakouvaki A, Reelfs O, Santambrogio P, Levi S, Hall ND, et al. (2014). Role of intracellular labile iron, ferritin, and antioxidant defence in resistance of chronically adapted Jurkat T cells to hydrogen peroxide. Free Radic Biol Med, 68: 87-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dev S, Kumari S, Singh N, Kumar Bal S, Seth P, Mukhopadhyay CK (2015). Role of extracellular Hydrogen peroxide in regulation of iron homeostasis genes in neuronal cells: Implication in iron accumulation. Free Radic Biol Med, 86: 78-89 [DOI] [PubMed] [Google Scholar]
- [32].Butt J, Kim HY, Basilion JP, Cohen S, Iwai K, Philpott CC, et al. (1996). Differences in the RNA binding sites of iron regulatory proteins and potential target diversity. Proc Natl Acad Sci U S A, 93: 4345-4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Koeppen AH, Ramirez RL, Becker AB, Bjork ST, Levi S, Santambrogio P, et al. (2015). The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS One, 10: e0116396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ponka P, Borova J, Neuwirt J, Fuchs O, Necas E (1979). A study of intracellular iron metabolism using pyridoxal isonicotinoyl hydrazone and other synthetic chelating agents. Biochim Biophys Acta, 586: 278-297 [DOI] [PubMed] [Google Scholar]
- [35].Harada J, Sugimoto M (1997). Polyamines prevent apoptotic cell death in cultured cerebellar granule neurons. Brain Res, 753: 251-259 [DOI] [PubMed] [Google Scholar]
- [36].Prus E, Fibach E (2008). Flow cytometry measurement of the labile iron pool in human hematopoietic cells. Cytometry A, 73: 22-27 [DOI] [PubMed] [Google Scholar]
- [37].Epsztejn S, Kakhlon O, Glickstein H, Breuer W, Cabantchik I (1997). Fluorescence analysis of the labile iron pool of mammalian cells. Anal Biochem, 248: 31-40 [DOI] [PubMed] [Google Scholar]
- [38].Guo S, Bezard E, Zhao B (2005). Protective effect of green tea polyphenols on the SH-SY5Y cells against 6-OHDA induced apoptosis through ROS-NO pathway. Free Radic Biol Med, 39: 682-695 [DOI] [PubMed] [Google Scholar]
- [39].Zhang Y, Zhao B (2003). Green tea polyphenols enhance sodium nitroprusside-induced neurotoxicity in human neuroblastoma SH-SY5Y cells. J Neurochem, 86: 1189-1200 [DOI] [PubMed] [Google Scholar]
- [40].Kong WN, Zhao SE, Duan XL, Yang Z, Qian ZM, Chang YZ (2008). Decreased DMT1 and increased ferroportin 1 expression is the mechanisms of reduced iron retention in macrophages by erythropoietin in rats. J Cell Biochem, 104: 629-641 [DOI] [PubMed] [Google Scholar]
- [41].Chen Y, Qian ZM, Du J, Duan X, Chang Y, Wang Q, et al. (2005). Iron loading inhibits ferroportin1 expression in PC12 cells. Neurochem Int, 47: 507-513 [DOI] [PubMed] [Google Scholar]
- [42].Yang H, Yang M, Guan H, Liu Z, Zhao S, Takeuchi S, et al. (2013). Mitochondrial ferritin in neurodegenerative diseases. Neurosci Res, 77: 1-7 [DOI] [PubMed] [Google Scholar]
- [43].Gao G, Chang YZ (2014). Mitochondrial ferritin in the regulation of brain iron homeostasis and neurodegenerative diseases. Front Pharmacol, 5: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tortosa A, Lopez E, Ferrer I (1997). Bcl-2 and Bax proteins in Lewy bodies from patients with Parkinson’s disease and Diffuse Lewy body disease. Neurosci Lett, 238: 78-80 [DOI] [PubMed] [Google Scholar]
- [45].Jing Y, Dai J, Chalmers-Redman RM, Tatton WG, Waxman S (1999). Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood, 94: 2102-2111 [PubMed] [Google Scholar]
- [46].Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR (2004). Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci, 5: 863-873 [DOI] [PubMed] [Google Scholar]
- [47].Lu Z, Nie G, Li Y, Soe-Lin S, Tao Y, Cao Y, et al. (2009). Overexpression of mitochondrial ferritin sensitizes cells to oxidative stress via an iron-mediated mechanism. Antioxid Redox Signal, 11: 1791-1803 [DOI] [PubMed] [Google Scholar]
- [48].Caltagirone A, Weiss G, Pantopoulos K (2001). Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J Biol Chem, 276: 19738-19745 [DOI] [PubMed] [Google Scholar]
- [49].Duarte TL, Jones GD (2007). Vitamin C modulation of H2O2-induced damage and iron homeostasis in human cells. Free Radic Biol Med, 43: 1165-1175 [DOI] [PubMed] [Google Scholar]
- [50].Deb S, Johnson EE, Robalinho-Teixeira RL, Wessling-Resnick M (2009). Modulation of intracellular iron levels by oxidative stress implicates a novel role for iron in signal transduction. Biometals, 22: 855-862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Courville P, Chaloupka R, Cellier MF (2006). Recent progress in structure-function analyses of Nramp proton-dependent metal-ion transporters. Biochem Cell Biol, 84: 960-978 [DOI] [PubMed] [Google Scholar]
- [52].Mims MP, Prchal JT (2005). Divalent metal transporter 1. Hematology, 10: 339-345 [DOI] [PubMed] [Google Scholar]
- [53].Wolff NA, Garrick LM, Zhao L, Garrick MD, Thevenod F (2014). Mitochondria represent another locale for the divalent metal transporter 1 (DMT1). Channels (Austin), 8: 458-466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tabuchi M, Yoshimori T, Yamaguchi K, Yoshida T, Kishi F (2000). Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J Biol Chem, 275: 22220-22228 [DOI] [PubMed] [Google Scholar]
- [55].Guaraldo M, Santambrogio P, Rovelli E, Di Savino A, Saglio G, Cittaro D, et al. (2016). Characterization of human mitochondrial ferritin promoter: identification of transcription factors and evidences of epigenetic control. Sci Rep, 6: 33432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yang H, Guan H, Yang M, Liu Z, Takeuchi S, Yanagisawa D, et al. (2015). Upregulation of mitochondrial ferritin by proinflammatory cytokines: implications for a role in Alzheimer’s disease. J Alzheimers Dis, 45: 797-811 [DOI] [PubMed] [Google Scholar]