

Abstract
Metabolic markers associated with the Metabolic Syndrome (MetS) may be affected by interactions between the APOE genotype and plasma fatty acids (FA). In this study, we explored FA-gene interactions between the missense APOE polymorphisms and FA status on metabolic markers in MetS. Plasma FA, blood pressure, insulin sensitivity and lipid concentrations were determined at baseline and following a 12-week randomized, controlled, parallel, dietary FA intervention in 442 adults with MetS (LIPGENE study). FA-APOE gene interactions at baseline and following change in plasma FA were assessed using adjusted general linear models. At baseline E4 carriers had higher plasma concentrations of total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C) and apolipoprotein B (apo B) compared with E2 carriers; and higher TC, LDL-C and apo B compared with E3/E3. Whilst elevated plasma n-3 polyunsaturated FA (PUFA) was associated with a beneficially lower concentration of apo CIII in E2 carriers, a high proportion of plasma C16:0 was associated with insulin resistance in E4 carriers. Following FA intervention, a reduction in plasma long-chain n-3 PUFA was associated with a reduction in apo CII concentration in E2 carriers. Our novel data suggest that individuals with MetS may benefit from personalized dietary interventions based on APOE genotype.
Introduction
The Metabolic Syndrome (MetS) is characterized by a clustering of risk factors related to cardiovascular disease (CVD) and type-II diabetes (T2D), including abdominal obesity, insulin resistance, hypertension, dyslipidemia and inflammation1, 2. Individuals with 4–5 features of MetS have a 3.7-fold increased risk of CVD and 24-fold increased risk of T2D3. The primary goal of clinical management is to reduce risk for metabolic and atherosclerotic disease4. This is achieved by targeting modifiable risk factors such as obesity, physical inactivity and inappropriate diets, in addition to smoking. Unhealthy diets, such as those high in saturated fatty acids (SFA), have been shown to increase low-density lipoprotein cholesterol (LDL-C) and CVD risk5. Whilst a large scale meta-analysis (n = 347,747) conducted in 2010 revealed no association between SFA and risk of stroke or CVD6, recent Cochrane review found a beneficial impact of SFA reduction on CVD risk7. Nevertheless, a reduction in LDL-C and CVD risk has been observed following replacement of SFA with unsaturated FA8–10, with evidence for a greater benefit of polyunsaturated FA (PUFA)7, 11. Given that responsiveness to dietary fat alteration is highly heterogeneous, there is interest in the impact of non-modifiable risk factors, such as genetics, to assist in prevention and treatment of chronic diseases such as MetS.
Of particular interest is the apolipoprotein E (APOE) genotype, a key regulator of lipoprotein metabolism, shown to account for up to 7% of the variance observed in total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-C)12. Polymorphisms in the APOE gene, rs429358 (Cys112Arg) and rs7412 (Arg158Cys), encode three common alleles, ε2 (Cys122 and Cys158), ε3 (Cys112 and Arg158) and ε4 (Arg112 and Arg158), which combine to form 6 genotypes, ε2/ε2, ε2/ε3, ε2/ε4, ε3/ε3, ε3/ε4 and ε4/ε4. Some scientists have reported a higher incidence of the ε4 allele among MetS subjects13, 14. The ε4 allele has been associated with increased TC, LDL-C, coronary artery disease (CAD) mortality and reduced high-density lipoprotein cholesterol (HDL-C) concentrations12, 15–19. However, associations between ε4 and CAD mortality in the 2007 meta-analysis (n = 47,467) were moderate (OR 1.06, 95% CI 0.99–1.13)16. The APOE genotype has also been associated with the development of Alzheimer’s20. To date, intervention studies have suggested that E4 carriers may be more sensitive to dietary cholesterol, total fat, SFA and long chain n-3 PUFA (LC n-3 PUFA, comprising EPA and DHA) modulation21–26. Thus, it has been suggested that the detrimental effects of the APOE genotype might be ameliorated by modulating the type and quantity of dietary fat27. More recently, independent associations between the ε4 allele and CVD risk have been observed among individuals with MetS28.
Although there has been a wealth of interest in the functional impact of polymorphisms at the APOE locus, a limited number of RCT have investigated dietary fat manipulation and APOE genotype, and very few in subjects with the MetS phenotype. Thus, we examined the relationship between the missense APOE polymorphism, habitual FA status and following dietary FA intervention on metabolic markers in a MetS population.
Subjects and Methods
The LIPGENE parallel dietary intervention study was conducted at 8 EU centers: Dublin, Ireland; Reading, UK; Oslo, Norway; Marseille, France; Maastricht, The Netherlands; Cordoba, Spain; Krakow, Poland; and Uppsala, Sweden, as described previously29. The study was conducted according to the Declaration of Helsinki and registered with the US National Library of Medicine ClinicalTrials.gov registry (NCT00429195; 01/30/2007). Ethical approval for the study was granted at each center and informed written consent was obtained from each subject prior to participation.
Participants
A total of 442 of the 486 participants randomized into the LIPGENE study were included in the present analysis owing to missing genotype data for APOE polymorphism in 44 participants. The group consisted of 248 women and 194 men, with a mean (±SEM) age of 54 (±1) years. The inclusion criteria were age 35–70 years, BMI 20–40 kg/m2 and presence of the MetS, as defined by a modified version of the NCEP ATP III criteria, in which participants required at least three of the following: fasting plasma glucose 5·5–7.0 mmol/l, serum triacylglycerol (TAG) ≥ 1·5 mmol/l, serum HDL-cholesterol <1·0 mmol/l in males and <1·3 mmol/l in females, waist circumference >102 cm in males and >88 cm in females, and elevated blood pressure (BP) (systolic BP ≥ 130 mmHg, diastolic BP ≥ 85 mmHg or on prescribed BP-lowering medication).
Study design
Participants recruited to the 12-week study were randomized according to age, sex and fasting plasma glucose using Minimization Programme for Allocating Participants to Clinical Trials (Department of Clinical Epidemiology, The London Hospital Medical College, UK) to one of four isoenergetic diets that differed according to fat quantity and quality: high fat SFA-rich diet, high fat monounsaturated fatty acids (MUFA)-rich diet, low-fat high-complex carbohydrate diet supplemented with 1.24 g/d LC n-3 PUFA (Marinol C-38) or low-fat high-complex carbohydrate diets supplemented with 1 g/d high-oleic acid sunflower oil29. Approximately 120 subjects were assigned to each dietary group. Dietary targets were obtained using a novel dietary exchange model previously described30. Habitual assessment of dietary intake, using a 3-day weighed food record, formed the basis of the isoenergetic (<0.2 kg weight change) dietary fat modification. Dietary change was facilitated via detailed dietetic consultation and re-assessed at weeks 6 and 12 using a 3-day weighed food record to ensure dietary compliance. Prior to the intervention, participants were required to complete a health and lifestyle questionnaire to assess habitual physical activity, smoking status, alcohol intake and socio-demographic status, which remained consistent throughout the study. Anthropometric measures, blood pressure and biological samples were taken with consent, following a 12-hour overnight fast before and after the 12-week intervention. Collection of samples was conducted according to standardized operating procedures to ensure consistency across centers. Detailed recruitment and study procedures have been published previously29.
Biochemical analysis
Plasma TC, HDL-cholesterol, LDL-cholesterol, TAG, non-esterified fatty acids (NEFA) and glucose concentrations were analyzed using enzymatic colorimetric methods (Instrumentation Laboratory, Warrington, UK; WAKO NEFA C kit, Alpha Laboratories, Hampshire, UK). Assays were used for quantification of plasma concentrations of apolipoproteins AI, B and E (Behring Werke AG, Marburg, Germany), triacylglycerol-rich lipoprotein (TRL) apo B48, and apo CIII and apo CII (Diasys, Bouffe´mont, France). Plasma FA were extracted and transmethylated with borontrifluoride in methanol and fatty acid methyl esters (FAME) of plasma fatty acids analysed using a Shimadzu GC-14A gas liquid chromatograph fitted with a Shimadzu C-r6A integrator and a 25 M BP 21 polar aluminium silica column (Shimadzu, Japan). Fatty acids were identified using FAME standards (Sigma-Aldrich Company Ltd, Dorset, England); see ref. 31 for detailed description of method. Enzyme-linked immunosorbent assay (ELISA) was used to determine C-reactive protein (CRP) (BioCheck Inc., Foster City, CA, USA). Serum insulin was determined by solid-phase, two-site fluoro-immunometric assay (Wallac Oy, Turku, Finland). Homeostasis model assessment (HOMA-IR) was calculated as: [(fasting plasma glucose × fasting plasma insulin)/22.5]32. All samples were analyzed centrally.
DNA extraction and genotyping
DNA was extracted from the buffy coat of whole fasted blood using the AutoPure LS automated system (Gentra Systems Inc, Minneapolis, MN). Samples with low-yield (<10 ng) were subjected to whole-gene amplification using the REPLI-g kit (Qiagen Ltd, West Sussex, UK). The SNP rs7412 and rs429358 were genotyped according to LIPGENE protocol by Illumina Inc. (San Diego, CA), with use of the Golden Gate Assay on a BeadStation 500 G system (Illumina Inc, San Diego, CA). Adherence to Hardy–Weinberg equilibrium at each SNP locus was determined using the chi2 test with 1 degree of freedom; SNPs were in accordance with the Hardy–Weinberg equilibrium.
Statistical analysis
Data are expressed as means ± SEM. All data were checked for skewness and kurtosis and where necessary normalized by log (WC, NEFA, HDL-C, TC-HDL-C ratio, TAG, apo CIII, glucose, insulin, HOMAIR and CRP) and square root (apo E and apo CII) transformation. Associations between APOE genotype and biomarkers were determined using a general linear model (GLM) with adjustment for age, sex, center and BMI. Where a significant overall genotype effect was observed (P ≤ 0.05) a post hoc test (Bonferroni) was applied to determine between genotype differences.
Nutrient-gene interactions were determined using the adjusted GLM (as described) but with the addition of a genotype × fatty acid (% of total plasma lipids) interaction term (see Supplementary Table 1 for list of FA evaluated). Where a significant nutrient-gene interaction was found with the FA evaluated as a continuous variable, the result was verified by dichotomizing the dataset by median plasma FA concentrations (as a categorical variable) to compare the effect of genotype within subjects who had similar habitual diets, e.g. within the higher intake (above median) or lower intake (below median) groups, using a post hoc test (Bonferroni).
The interactions between genotype and plasma FA (SFA, total PUFA and LC n-3 PUFA) on each biochemical variable, following dietary intervention, was assessed by using 0% change in plasma FA (% of total plasma lipids) to dichotomize subjects, and then using the resulting groups as fixed factors in a GLM (i.e. reduction in plasma FA and increase in plasma FA). This method provides an objective assessment of change in nutrient intake, irrespective of errors in dietary reporting and study compliance. Furthermore, splitting the data according to diet allocation (i.e. divided into four) would have led to insufficient carrier numbers in each group. Using change in plasma FA more clearly distinguishes the impact of actual dietary change on outcome variables. The plasma FA examined were those manipulated in the LIPGENE intervention: SFA, MUFA and LC n-3 PUFA. Non-parametric data were transformed using log (Y + a), where a is the minimum constant (triglyceride-rich lipoprotein cholesterol fraction (TRL-C), apo A1, apo CII, apo CIII, apo E, glucose and CRP)33. As previously, the interaction term genotype × FA was added to a GLM, with the biochemical variable as the response variable and the respective pre-intervention variable as a covariate. Additional covariates added to the model were age, sex, center and weight change [post intervention weight (kg) – pre intervention weight (kg)]. Where a significant overall genotype effect was observed (P ≤ 0.05) a post hoc test (Bonferroni) was applied to determine between and within group differences. Statistical analysis was performed using Minitab for Windows (version 16, Coventry, UK). Post-hoc power analyses were conducted using G*Power34.
Results
Genotypes were reported as E2 carriers, comprising (E2/E2 n = 3, and E2/E3, n = 43), E4 carriers (E4/E4 n = 12 and E4/E3 n = 103), homozygous E3/E3 (n = 264) and E2/E4 carriers (n = 17).
Genotype frequency
Genotype and allele frequency according to LIPGENE center are shown in Table 1. A geographic cline can be seen with respect to allele frequency, with the most southerly country expressing the lowest frequency of the ε4 allele and the most northerly country greater than 2-fold higher (E3/E4 and E4/E4 combined frequency, 8.6% for Cordoba, Spain versus 22.8% for Oslo, Norway).
Table 1.
Frequency of APOE genotype by LIPGENE Dietary Fatty Acid Intervention Study center (n = 442).
| All | Norway | Sweden | Ireland | Netherlands | UK | Poland | France | Spain | |
|---|---|---|---|---|---|---|---|---|---|
| Genotype (n, %) | |||||||||
| E2/E2 | 3 (0.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.2) | 1 (1.7) | 0 (0.0) | 0 (0.0) | 1 (1.4) |
| E2/E3 | 43 (9.7) | 5 (8.7) | 4 (8.3) | 7 (12.0) | 7 (15.9) | 5 (8.6) | 7 (10.3) | 2 (5.1) | 6 (8.5) |
| E2/E4 | 17 (3.8) | 3 (5.2) | 3 (6.2) | 1 (1.7) | 1 (2.2) | 2 (3.4) | 2 (2.9) | 2 (5.1) | 3 (4.2) |
| E3/E3 | 264 (59.7) | 27 (47.3) | 26 (54.1) | 37 (63.8) | 26 (59.0) | 26 (44.8) | 42 (61.8) | 28 (71.8) | 52 (74.2) |
| E3/E4 | 103 (23.3) | 21 (36.8) | 15 (31.2) | 10 (17.2) | 7 (15.9) | 21 (36.2) | 16 (23.5) | 6 (15.3) | 7 (10.0) |
| E4/E4 | 12 (2.7) | 1 (1.7) | 0 (0.0) | 3 (5.1) | 2 (4.5) | 3 (5.1) | 1 (1.4) | 1 (2.5) | 1 (1.4) |
| E2 carriersa | 46 (10.4) | 5 (8.7) | 4 (8.3) | 7 (12.0) | 8 (18.1) | 6 (10.3) | 7 (10.2) | 2 (5.1) | 7 (10.0) |
| E4 carriersb | 115 (26.0) | 22 (38.6) | 15 (31.2) | 13 (22.4) | 9 (20.4) | 24 (41.3) | 17 (24.9) | 7 (17.9) | 8 (11.4) |
| ALL | 442 | 57 | 48 | 58 | 44 | 58 | 68 | 39 | 70 |
| Allele frequency (%) | |||||||||
| ε2 | 7.5 | 7.0 | 7.3 | 6.9 | 11.4 | 7.8 | 6.6 | 6.3 | 7.9 |
| ε3 | 76.2 | 70.2 | 74.0 | 78.4 | 75.0 | 67.2 | 78.8 | 81.3 | 83.6 |
| ε4 | 16.3 | 22.8 | 18.8 | 14.7 | 13.6 | 25.0 | 14.7 | 12.6 | 8.6 |
Values are n (%). aGenotype groups combined; E2 carriers represent E2/E2 and E2/E3. bGenotype groups combined; E4 carriers represent E4/3 and E4/E4.
Subject characteristics
In Table 2, subject anthropometry and fasted lipid profiles are reported according to APOE genotype. E2 carriers had significantly lower baseline plasma concentrations of TC, LDL-C and apo B when compared with the E4 carriers; and lower TC, LDL-C and apo B compared to the E3/E3 group. TC:HDL-C ratio was significantly higher in E4 carriers compared with E2 carriers and apo E in E2 carriers (compared with E3/E3 and E4 carriers). Apo E was also significantly higher in E2/E4 carriers when compared with E4 carriers. In addition, E4 carriers had significantly higher TRL-C and lower CRP than E3/E3. There was no evidence of a genotype-dependant difference in baseline anthropometry, blood pressure or markers of insulin resistance. Plasma FA values (% total plasma FA) and dietary fat intakes (g) according to APOE genotype are reported in Supplementary Tables 2 and 3; no significant differences according to genotype were observed. E2/E4 carriers were removed from subsequent analysis due to their low population frequency.
Table 2.
Effect of the APOE genotype on anthropometric variables, blood pressure, fasted plasma- and serum- profiles in the LIPGENE Dietary Fatty Acid Intervention Study (n = 442; males n = 194, females n = 248).
| All (n = 442) | E2 carriers (n = 46) | E3/E3 (n = 264) | E4 carriers (n = 115) | E2/E4 (n = 17) | P | |
|---|---|---|---|---|---|---|
| Age (y) | 55 ± 1 | 56 ± 1 | 54 ± 1 | 55 ± 1 | 53 ± 2 | 0.026 |
| BMI (kg/m2) | 31.6 ± 1.0 | 32.6 ± 0.7 | 32.6 ± 0.3 | 32.4 ± 0.5 | 31.6 ± 1.1 | 0.316 |
| Waist (cm) | 104 ± 3 | 107 ± 2 | 106 ± 1 | 105 ± 1 | 104 ± 2 | 0.894 |
| Diastolic BP (mm Hg) | 86 ± 2 | 87 ± 1 | 86 ± 1 | 86 ± 1 | 87 ± 2 | 0.729 |
| Systolic BP (mm Hg) | 137 ± 4 | 137 ± 2 | 139 ± 1 | 140 ± 1 | 137 ± 4 | 0.744 |
| TC (mmol/L) | 5.15 ± 0.69 | 4.98 ± 0.16a | 5.38 ± 0.06b | 5.53 ± 0.09b | 5.03 ± 0.21 | 0.015 |
| LDL-C (mmol/L) | 3.08 ± 0.11 | 2.67 ± 0.16a | 3.27 ± 0.06b | 3.57 ± 0.09b | 3.14 ± 0.18 | 0.001 |
| NEFA (µmol/L) | 610 ± 27 | 675 ± 37 | 597 ± 14 | 641 ± 23 | 593 ± 58 | 0.129 |
| HDL-C (mmol/L) | 1.08 ± 0.04 | 1.13 ± 0.05 | 1.11 ± 0.02 | 1.08 ± 0.02 | 1.14 ± 0.09 | 0.234 |
| TC:HDL-C ratio | 5.12 ± 0.20 | 4.65 ± 0.21a | 5.05 ± 0.07 | 5.38 ± 0.14b | 4.92 ± 0.49 | 0.035 |
| TRL-C (mmol/L) | 1.82 ± 0.13 | 0.46 ± 0.05 | 0.34 ± 0.01a | 0.45 ± 0.04b | 0.47 ± 0.10 | 0.493 |
| TAG (mmol/L) | 0.47 ± 0.05 | 1.91 ± 0.16 | 1.71 ± 0.05 | 1.94 ± 0.10 | 1.86 ± 0.18 | 0.371 |
| TRL-TAG (mmol/L) | 0.93 ± 0.09 | 0.89 ± 0.09 | 0.82 ± 0.03 | 0.98 ± 0.07 | 0.89 ± 0.15 | 0.286 |
| Apo AI (mg/L) | 1.37 ± 0.03 | 1.43 ± 0.04 | 1.41 ± 0.02 | 1.38 ± 0.02 | 1.42 ± 0.07 | 0.590 |
| Apo B (mg/L) | 0.95 ± 0.02 | 0.88 ± 0.04a | 1.03 ± 0.01b | 1.09 ± 0.02b | 0.90 ± 0.04 | < 0.001 |
| Apo B48 (mg/L) | 0.85 ± 0.13 | 0.67 ± 0.10 | 0.79 ± 0.06 | 0.78 ± 0.09 | 1.25 ± 0.33 | 0.197 |
| TRL apo B (mg/L) | 56.7 ± 6.1 | 51.1 ± 7.3 | 44.9 ± 2.4 | 59.5 ± 5.4 | 60.6 ± 11.7 | 0.283 |
| Apo CII (mg/L) | 46.75 ± 2.2 | 49.0 ± 3.0 | 44.7 ± 1.0 | 45.7 ± 1.8 | 52.1 ± 4.6 | 0.353 |
| Apo CIII (mg/L) | 152 ± 7 | 169 ± 10 | 157 ± 3 | 159 ± 5 | 159 ± 12 | 0.777 |
| Apo E (mg/L) | 43.8 ± 3.11 | 55.1 ± 4.64a,c | 39.9 ± 0.73b,c | 38.0 ± 1.37b | 50.4 ± 1.98c | < 0.001 |
| Glucose (mmol/L) | 5.92 ± 0.21 | 6.04 ± 0.11 | 5.91 ± 0.06 | 6.10 ± 0.12 | 5.92 ± 0.25 | 0.113 |
| Insulin (µIU/mL) | 9.0 ± 1.5 | 9.7 ± 0.9 | 10.1 ± 0.4 | 10.4 ± 0.6 | 9.0 ± 1.5 | 0.996 |
| HOMA-IR | 2.51 ± 0.56 | 2.64 ± 0.25 | 2.65 ± 0.10 | 2.87 ± 0.19 | 2.51 ± 0.57 | 0.892 |
| CRP (mg/L) | 5.18 ± 0.20 | 5.32 ± 0.77 | 5.51 ± 0.24a | 4.48 ± 0.41b | 4.45 ± 0.41 | 0.028 |
Abbreviations: BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pressure; TC, total cholesterol; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; NEFA, non-esterified fatty acid; TAG, triacylglycerol; TRL-C, triglyceride rich lipoprotein cholesterol fraction; TRL-TG, triglyceride rich lipoprotein triglyceride fraction; CRP, C-reactive protein. Values are means ± s.e.m. Models were adjusted for center, gender, age and BMI. Where P for genotype <0.05, a post-hoc Bonferroni test were used to determine a between group effect. Superscript letters a and b denote significant differences in means (P < 0.05).
Habitual plasma FA and genotype interactions at baseline
Significant gene-nutrient interactions between APOE and plasma SFA (C14:0, C16:0, C18:0), MUFA (C16:1, C18:1, C20:1), n-3 PUFA (C18:4, C20:5, C22:5, C22:6) and n-6 PUFA (C18:2, C18:3, C20:3, C20:4 and C22:4), evaluated as a continuous variable (as a % of total FA), on fasting metabolic biomarkers are reported in Table 3. Where a significant interaction was observed, data were dichotomized by median to give categorical data: total SFA (31.1%), C14:0 (1.76%), C16:0 (25.7%), C18:0 (4.1%), total MUFA (28.5%), C16:1 (1.20%), C18:1 (26.5%), C20:1 (0.21%), total n-6 PUFA (35.1%), total n-3 PUFA (3.47%), C20:5 (eicosapentanoic acid, EPA) (0.72%), C22:6 (docosapentanoic acid, DPA) (0.39%) and C22:5 (docosahexanoic acid, DHA) (2.04%) to determine the effect of specific genotypes in subjects with similar plasma FA concentrations based on habitual diet.
Table 3.
Significant associations and interactions for baseline plasma FA (%FA) and APOE genotype on metabolic variables in the LIPGENE Dietary Fatty Acid Intervention Study (n = 416).
| E2 carriers | E3/E3 | E4 carriers | P | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Low FA | High FA | Low FA | High FA | Low FA | High FA | FA | Genotype | FA × Genotype | |
| C16:0 | (n = 21) | (n = 25) | (n = 137) | (n = 122) | (n = 53) | (n = 58) | |||
| HOMAIR | 2.21 ± 0.26 | 2.98 ± 0.40 | 2.51 ± 0.13 | 2.78 ± 0.15 | 2.35 ± 0.26a | 3.41 ± 0.26b | < 0.001 | 0.747 | 0.018 |
| Insulin (mmol/L) | 8.18 ± 0.82 | 10.9 ± 1.41 | 9.61 ± 0.48 | 10.4 ± 0.53 | 8.70 ± 0.89a | 12.2 ± 0.87b | 0.001 | 0.641 | 0.033 |
| N-6 PUFA | (n = 25) | (n = 21) | (n = 123) | (n = 126) | (n = 59) | (n = 52) | |||
| Apo E (mg/L) | 65.8 ± 7.52a,c | 42.3 ± 3.04b | 41.9 ± 1.17a,d | 38.0 ± 0.88b | 41.1 ± 1.97a,d | 34.7 ± 1.91b | < 0.001 | < 0.001 | 0.012 |
| TRL-C (mmol/L) | 0.63 ± 0.08a,c | 0.25 ± 0.03b | 0.43 ± 0.02a,d | 0.25 ± 0.01b | 0.58 ± 0.06a,d | 0.32 ± 0.03b | < 0.001 | 0.003 | 0.029 |
| C20:1 | (n = 20) | (n = 26) | (n = 128) | (n = 131) | (n = 58) | (n = 53) | |||
| Apo E (mg/L) | 69.6 ± 9.46a,c | 44.1 ± 2.28b | 42.1 ± 1.16d | 37.6 ± 0.86 | 38.5 ± 1.93d | 37.6 ± 2.1 | 0.001 | < 0.001 | 0.004 |
| N-3 PUFA | (n = 20) | (n = 26) | (n = 136) | (n = 123) | (n = 60) | (n = 51) | |||
| Apo CIII (mg/L) | 190 ± 17.7a | 154 ± 11.4b | 156 ± 4.11 | 158 ± 4.40 | 158 ± 7.91 | 159 ± 5.73 | 0.005 | 0.458 | 0.022 |
| Apo E (mg/L) | 63.6 ± 9.44a,c | 48.6 ± 3.62b,c | 39.9 ± 1.01d | 39.8 ± 1.07 | 39.4 ± 2.12d | 36.3 ± 1.67d | 0.001 | < 0.001 | 0.037 |
| NEFA (µmol/L) | 585 ± 53.2 | 745 ± 46.2 | 596 ± 21.4 | 595 ± 18.2 | 674 ± 34.9 | 600 ± 28.4 | 0.200 | 0.148 | 0.042 |
| EPA/C20:5 (n-3) | (n = 20) | (n = 26) | (n = 136) | (n = 123) | (n = 50) | (n = 61) | |||
| Apo CIII (mg/L) | 182 ± 17.9a | 160 ± 11.8b | 153 ± 3.74 | 162 ± 4.80 | 157 ± 8.71 | 160 ± 5.95 | 0.001 | 0.365 | 0.041 |
| Apo E (mg/L) | 66.9 ± 9.2a,c | 46.1 ± 3.5b | 40.2 ± 1.1d | 39.5 ± 1.0 | 40.4 ± 2.5d | 36.1 ± 1.5 | < 0.001 | < 0.001 | 0.002 |
| DHA/C22:6 (n-3) | (n = 20) | (n = 26) | (n = 122) | (n = 137) | (n = 65) | (n = 46) | |||
| Apo E (mg/L) | 55.1 ± 6.4c | 55.1 ± 6.4 | 48.0 ± 0.9d | 41.5 ± 1.1 | 39.5 ± 2.0d | 35.9 ± 1.8 | 0.029 | < 0.001 | 0.020 |
Abbreviations: DHA, docosahexanoic acid; EPA, eicosapentanoic acid; FA, fatty acid; HOMA-IR, homeostasis model assessment of insulin resistance; NEFA, non-esterified fatty acid; PUFA, polyunsaturated fatty acid TRL-C, triglyceride rich lipoprotein cholesterol fraction. Values are means ± s.e.m. Data were analyzed using general linear models with adjustment for age, baseline BMI, baseline alcohol intake (g/d), sex, exercise level index, center and smoking status. Where P for plasma FA × genotype <0.05, a post hoc Bonferroni test used to determine between group effects (low FA, less than median plasma FA; high FA, greater than median plasma FA). Superscript letters a and b denote significant differences between low and high FA within each genotype; letters c and d significant differences between genotypes within each FA group, P < 0.05.
At baseline, significant interactions between APOE and plasma n-6 PUFA were observed for TRL-C. Whilst all three genotype groups had significantly higher TRL-C concentrations in the low plasma n-6 PUFA group, compared with the high plasma n-6 PUFA group, E2 carriers with low plasma n-6 had significantly higher TRL-C than E3/E3 and E4 carriers.
The APOE genotype also influenced apo E and apo CIII concentrations according to plasma n-6 PUFA and n-3 PUFA, and plasma n-3 PUFA respectively. Lower plasma n-3 PUFA, n-6 PUFA, EPA and C20:1 was associated with significantly higher apo E concentrations in E2 carriers. The same interaction was observed with the E3/E3 genotype for n-6 PUFA. In the low plasma n-3 PUFA, n-6 PUFA, EPA, DHA and C20:1 groups, E2 carriers had significantly higher concentrations of apo E than E3/E3 and E4 carriers. For apo CIII, dichotomization according to median FA revealed significantly lower concentrations of apo CIII with a higher proportion plasma total n-3 PUFA and EPA in E2 carriers only (Fig. 1B).
Figure 1.
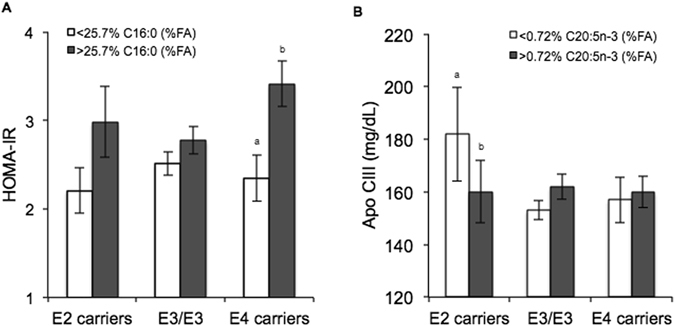
Effect of the APOE genotype and </> median baseline plasma fatty acid concentration on (A) HOMA-IR (APOE × C16:0 interaction, P = 0.018) and (B) apo CIII (APOE × C20:5n-3 interaction, P = 0.041) in metabolic syndrome subjects. Values are means ± s.e.m. Letters (a and b) are used to denote significant differences between plasma fatty acid groups within the same genotype, P < 0.05 using post hoc Bonferroni.
Another key finding in the present study was the variation in insulin resistance according to APOE genotype and plasma total SFA; nutrient-gene interactions were observed for both HOMA-IR and insulin. FA analysis revealed that E4 carriers with higher proportions of plasma C16:0 had significantly higher HOMA-IR and insulin than those with lower plasma C16:0 (Fig. 1A). Interaction between C18:0 and APOE on HOMA-IR and insulin approached significance.
Nutrient-gene interactions were observed between total n-3 PUFA and APOE for plasma NEFA and plasma SFA and APOE on CRP (data not shown); however, no specific differences between genotypes were detected after post-hoc analysis.
Plasma FA and genotype effects following FA dietary intervention
Significant gene-nutrient interactions between APOE and change in plasma SFA, PUFA and LC n-3 PUFA, evaluated as a continuous variable (as a % of total FA), on metabolic biomarkers were only observed for LC n-3 PUFA (Table 4). An increase in LC n-3 PUFA was represented by a >0% change in LC n-3 PUFA whereas a decrease in LC n-3 PUFA was represented by a <0% change in LC n-3 PUFA (categorical data). Plasma FA’s changed in all subjects.
Table 4.
Effects of significant change in plasma LC n-3 PUFA (%FA) and APOE genotype interactions on metabolic variables following the LIPGENE Dietary Fatty Acid Intervention Study (n = 351).
| E2 carriers | E3/E3 | E4 carriers | P | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Decrease LC n-3 (n = 15) | Increase LC n-3 (n = 26) | Decrease LC n-3 (n = 75) | Increase LC n-3 (n = 142) | Decrease LC n-3 (n = 29) | Increase LC n-3 (n = 64) | FA | Genotype | FA × Genotype | |
| TRL-C (mmol/L) | −0.05 ± 0.05 | 0.09 ± 0.09c | 0.03 ± 0.02 | −0.02 ± 0.02d | 0.07 ± 0.04 | −0.02 ± 0.04 | 0.561 | 0.077 | 0.021 |
| Apo CII (mg/L) | −6.37 ± 3.08a,c | 1.19 ± 2.41b | 1.69 ± 1.32 | −1.45 ± 0.72 | 3.13 ± 2.07d | −0.15 ± 1.21 | 0.259 | 0.112 | 0.002 |
| Apo E (mg/L) | −3.95 ± 1.52 | 1.58 ± 2.34c | 0.52 ± 0.93 | −1.97 ± 0.77d | 0.03 ± 1.68 | −1.41 ± 1.47d | 0.228 | 0.161 | 0.025 |
Abbreviations: FA, fatty acid; LC n-3 PUFA, long-chain omega-3 polyunsaturated fatty acid (comprising EPA and DHA); decrease LC n-3, less than 0% change in plasma LC n-3 PUFA; increase LC n-3, greater than 0% change in plasma LC n-3 PUFA; TRL-C, triglyceride rich lipoprotein cholesterol fraction. Values are means ± s.e.m. Data were analyzed using general linear models with adjustment for age, sex, center, change in weight (week 12 – baseline) and the respective pre-intervention variable. Where P for plasma FA × genotype <0.05, a post hoc Bonferroni test used to determine between group effects. Superscript letters a and b denote significant differences between low and high FA within each genotype; letters c and d significant differences between genotypes within each FA group (P < 0.05).
Following dietary FA intervention, significant interactions were observed between APOE genotype and change in LC n-3 PUFA for TRL-C. Whereas an increase in the proportion of plasma LC n-3 PUFA was associated with an increase in TRL-C concentrations in E2 carriers, a reduction in TRL-C was observed in the E3/E3 genotype.
The APOE genotype also influenced the apo CII and apo E response to change in LC n-3 PUFA. Apo CII concentrations reduced in E2 carriers following a reduction in plasma LC n-3 PUFA, however, they increased when plasma LC n-3 PUFA was raised. In E4 carriers, a significantly higher apo CII concentration was observed in response to a reduction in the proportion of plasma LC n-3 PUFA than in E2 carriers (Fig. 2). For apo E, E2 carriers displayed an increase in apo E concentration following an increase in plasma LC n-3 PUFA, whereas reductions were observed in E3/E3 and E4 carriers.
Figure 2.
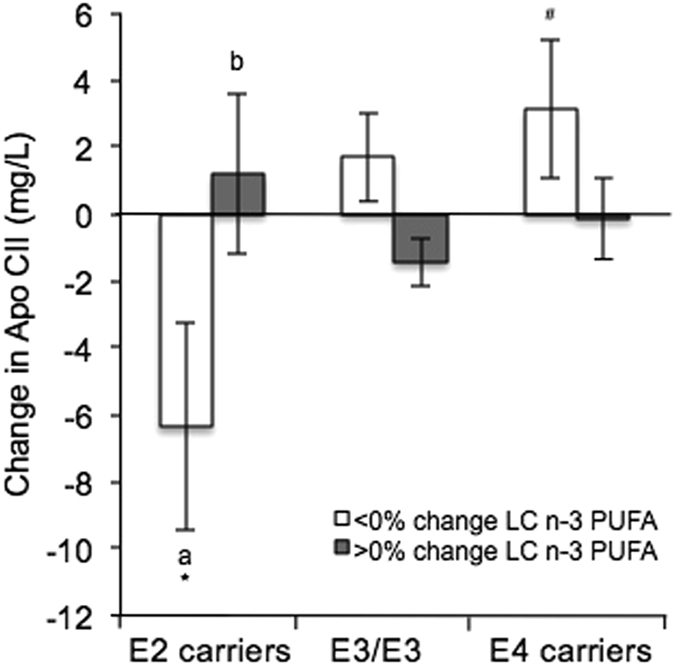
Effect of the APOE genotype and change in plasma LC n-3 PUFA concentration on change in apo CII (APOE × LC n-3 PUFA interaction, P = 0.041) following the LIPGENE Dietary Fatty Acid Intervention in metabolic syndrome subjects. Values are means ± s.e.m. Letters (a and b) are used to denote significant differences between plasma fatty acid groups within the same genotype; symbols (* and #) are used to denote significant differences between genotypes within the same fatty acid group, P < 0.05 using post hoc Bonferroni.
Discussion
A beneficial impact of increasing plasma LC n-3 PUFA on apo CII concentrations in E2 carriers, and a detrimental association between high plasma SFA and insulin resistance in E4 carriers at baseline was observed. To our knowledge, this is the first study to examine the impact of change in plasma FA, from dietary fat intervention, on lipids and insulin resistance in a MetS population.
The allele distribution in the LIPGENE cohort was similar to that reported in previous Caucasian populations35, as was the geographical cline observed for greater frequency of the ε4 allele and lower plasma apo E concentrations in Northern Europeans compared with Southern Europeans36–38. In the European Atherosclerosis Research Study, a “clear-cut” gradient for ε4 allele frequency which followed the coronary heart disease (CHD) mortality gradient was reported38. The 10-year incidence of CHD is 2.68 times greater in Northern than in Southern Europeans39 and it has been proposed that the differential effects of the ε4 and ε2 alleles may contribute to this difference in risk across Europe40. In a recent systematic review, Khan et al. reported a positive association between APOE genotype and TC (P trend: 2 × 10−152), with highest TC in E4 carriers, which support our observations from the LIPGENE cohort (E2 < E3/E3 < E4)41. Circulating LDL-C and apo B concentrations were also significantly greater in E4 carriers at baseline compared with E2 carriers, as observed previously in MetS populations14. However, there were no differences in baseline plasma FA or dietary fat intake.
In terms of receptor affinity it is now recognized that there are no major differences between the E3 and E4 protein isoform binding to the LDL-receptor (LDL-R)42, yet E4 subjects are often found to have higher TC and LDL-C. A possible explanation for this anomaly is a preferential association of the E4 protein with TRL particles (as opposed to HDL in E3/E3)43. This is proposed to result in more apo E per TRL particle in E4 carriers which increases competition at the LDL-R44, resulting in less uptake of LDL and increased circulating plasma cholesterol. Additionally, VLDL remnant lipolytic conversion to LDL is reported to be at a faster rate in E4 subjects43. Impaired recycling of TRL-derived apoE4 has also been associated with cholesterol accumulation45. In contrast the LDL-R binding affinity of the E2 isoform is less than 2% of the strength of E3 and E4 resulting in slower clearance of VLDL and dietary derived chylomicron (CM) remnants from the blood and typically lower TC and LDL-C concentrations46, 47. The decreased rate of LDL formation and subsequent up-regulation of LDL-R in E2 carriers results in increased circulating TAG35.
At baseline, there were no significant differences in TRL-C between genotypes, however a significant nutrient-gene interaction was observed for plasma n-6 PUFA, with E2 carriers having significantly higher TRL-C at lower intakes of n-6 PUFA than E3/E3 and E4 carriers. In addition to serum TAG, E2 carriers have been reported to have higher concentrations of circulating TRL-C compared with the E3/E3 wild-type19, as was observed in the LIPGENE cohort. A possible explanation of this is the slower CM remnant and VLDL clearance in E2 carriers. In the present study, higher plasma n-6 PUFA was associated with lower TRL-C concentrations, in all genotype groups. This is suggestive of a beneficial impact of plasma n-6 PUFA on plasma cholesterol concentrations. Whilst an interaction was observed between change in LC n-3 PUFA and APOE genotype for TRL-C (following FA dietary intervention), significant differences between groups were not identified. Previous analysis of the LIPGENE data found a beneficial impact of dietary LC n-3 PUFA on LDL density48.
Up to 30% of the variability in serum apo E concentrations has been attributed to the APOE polymorphism, with highest concentrations in E2 carriers and the lowest in E4 carriers, however concentrations are also influenced by sex, age and geography37, 49. Apo E is located on HDL-C, CM and intermediate-density lipoproteins (IDL) and binds to the LDL-R. Due to the impaired binding affinity of APOE2 protein, there is reduced uptake of CM and IDL (compared with E3 and E4), resulting in higher circulating apo E concentrations. In the present study, E2 carriers had significantly higher plasma apo E concentrations when compared with both APOE3 and E4 carriers. Our data confirms that the APOE locus is a major determinant of the plasma apo E concentration even in the presence of MetS. Kypreos et al. reported that plasma apo E has anti-inflammatory properties50 which, in addition to lower TC concentration, may contribute towards the 20% decreased risk of CHD observed in E2 carriers. At baseline, high plasma total n-3 and n-6 PUFA levels were associated with lower concentration of apo E in E2 carriers. However, an increase in LC n-3 PUFA following the LIPGENE dietary FA intervention raised apo E concentrations in E2 carriers, compared with a reduction observed in E4 carriers and E3/E3.
Apo CIII concentrations were significantly lower in E2 individuals with high concentrations of plasma total n-3 PUFA, n-6 PUFA and EPA. Apo CIII is an inhibitor of lipoprotein lipase (LPL) and TRL catabolism51; thus, increased concentrations will promote increased circulating TAG. The TAG lowering effect of LC n-3 PUFA is well documented26, and may be associated with increased LPL activity and gene expression and/or a reduction in apo CIII52. Given that elevated TAG is an independent risk factor for CVD53, these findings suggest a benefit of increased plasma n-3 PUFA in E2 carriers, particularly as this group often present with elevated plasma TAG. When exposed to a lower proportion of plasma n-3 PUFA/ EPA, E2 carriers had significantly more apo CIII than E4 carriers. Whereas interactions between PPARα and APOC-III genotypes and LC n-3 PUFA have been shown to modulate apo CIII concentrations54, 55, the interaction with APOE is novel. Following the LIPGENE dietary FA intervention a significant APOE × LC n-3 PUFA was observed for apo CII. In contrast to apo CIII, apo CII activates LPL and increases TAG catabolism. In E2 carriers, an increase in plasma LC n-3 PUFA was associated with an increase in apo CII concentrations, compared with a decrease when LC n-3 PUFA intake reduced. This provides further evidence for a beneficial impact of LC n-3 PUFA in E2 carriers in relation to TAG metabolism.
An interesting finding in the present analysis was the detrimental association between high plasma C16:0 on markers of insulin resistance, defined by HOMA-IR > 2.656, in E4 carriers. Although E4 carriers with low plasma C16:0 at baseline were not ‘insulin resistant’ (HOMA-IR, =2.35), those with high plasma C16:0 had a 31% greater HOMA-IR. This represents a novel finding and may suggest that E4 carriers with MetS are particularly sensitive to the detrimental metabolic effects of high palmitic acid (C16:0) levels. It is of note that increased plasma C16:0 has been associated with risk of type 2 diabetes, which could be in part due to increased insulin resistance57; our findings indicate that this relationship is amplified in E4 carriers. Previous studies have also shown a negative impact of diets rich in C16:0 and SFA on insulin sensitivity index (SI) in overweight individuals58, 59. However, several studies including the LIPGENE study, found no impact of reducing SFA on SI 29. The lack of effect found in the primary LIPGENE analysis highlights the importance of the APOE genotype (E4) on insulin-glucose homeostasis in metabolically challenged individuals.
At baseline in the current study, there were no genotype-dependent differences on insulin resistance, supporting previous reports by Ragogna et al.60. The frequency of APOE genotypes were also compared across HOMA-IR quintiles in the Framingham cohort, with no genotypic association reported61. However, others reported a significant genotype-obesity interaction for glucose and insulin, whereby obese E4 men (BMI ≥ 30 kg/m2) had higher fasted glucose and insulin compared to both non-obese men and non apoE4 obese subjects62. It has been suggested that the APOE-obesity interaction may intensify insulin resistance in men. A potential mechanism for this is accelerated lipid peroxidation due to increased TC and LDL-C, and reduced LDL diameter in obese E4 carriers62.
Evaluation of the interactions between APOE genotype and FA on metabolic markers was not the primary aim of LIPGENE; thus, the use of a retrospective genotyping approach was unavoidable and could be seen as a potential limitation. Inevitably this resulted in uneven group sizes, although reasonable total numbers were available in each genotype group and adequate study power was achieved. Based on the observed group size and differences in LDL-C between E2 and E4, and E3/E3 and E4 at baseline, the powers (1-β) achieved were 0.99 (effect size = 0.87, critical t = 1.97, P = 0.05) and 0.76 (effect size = 0.30, critical t = 1.97, P = 0.05) respectively. The power achieved for differences in HOMAIR between low and high plasma C16:0 in E4 carriers was 0.82 (effect size = 0.55, critical t = 1.98, P = 0.05) and for differences in apo CII between E2 and E4 carriers following LC n-3 PUFA reduction the power achieved was 0.72 (effect size = 0.83, critical t = 2.02, P = 0.05). A strength of the present analysis was the use of plasma FA to explore nutrient-gene interactions in the APOE gene; plasma FA concentrations indicate the specific exposure of cells to FA and provide insight into potential mechanisms, whereas dietary intake is prone to misreporting.
Plasma SFA, MUFA and PUFA can be informative with respect to dietary FA intakes and positive correlations have been observed between reported dietary total PUFA and LC n-3 PUFA intake (%TE) and corresponding FA in blood lipid fractions63. However, correlations between dietary and plasma SFA and MUFA are weak63. Given that plasma FA can also be synthesized de novo from endogenous fatty acids and non-fatty acid sources, associations found between plasma SFA and measured outcomes may not only reflect dietary intake of FA. A potential limitation of this analysis is that subject groupings may alter for each FA investigated such that the effects of a given FA may be confounded by the presence of higher or lower quantities of other FA. However, this method does enable us to evaluate the impact of changes in plasma FA on concomitant changes in outcome measures. Furthermore, analysis irrespective of diet group allocation increases the sample size and statistical power, and reduces the confounding influence of non-adherence to dietary intervention.
In conclusion, we observed that a high proportion of plasma palmitic acid was associated with greater insulin resistance in E4 carriers with MetS, whereas increasing plasma LC n-3 PUFA beneficially increased apo CII and reduced apo CIII concentrations in E2 carriers, which may confer TAG lowering benefits. These interactions represent novel findings, which should be explored further. In the context of personalized nutrition, our data suggest that individuals with MetS may benefit from personalized dietary interventions based on APOE genotype.
Electronic supplementary material
Supplementary Information - APOE genotype in the Metabolic Syndrome
Acknowledgements
The authors would like to thank Dr. Karen Ayres from the Department of Mathematics and Statistics at the University of Reading for her advice on the statistical analysis. The data used in this analysis was derived from the LIPGENE study, funded by the European Commission Framework Programme 6 (contract no. FOOD-CT-2003-505944) between 2004–2009.
Author Contributions
The authors’ responsibilities were as follows: H.M.R., J.A.L., W.H.S., C.A.D., J.L., U.R., E.E.B., C.A.D. and A.D. study design and supervision of data collection; A.C.T., C.M., B.K. data collection; R.F. statistical analysis; R.F., A.L.C. and J.A.L. writing of the manuscript. All authors reviewed and edited the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05802-2
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu. Rev. Med. 2005;56:45–62. doi: 10.1146/annurev.med.56.082103.104751. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome a summary of the evidence. Diabetes Care. 2005;28:1769–1778. doi: 10.2337/diacare.28.7.1769. [DOI] [PubMed] [Google Scholar]
- 3.Sattar N, et al. Metabolic syndrome with and without C-reactive protein as a predictor of coronary heart disease and diabetes in the West of Scotland Coronary Prevention Study. Circulation. 2003;108:414–419. doi: 10.1161/01.CIR.0000080897.52664.94. [DOI] [PubMed] [Google Scholar]
- 4.Grundy SM, et al. Diagnosis and management of the metabolic syndrome an American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Circulation. 2005;112:2735–2752. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- 5.de Oliveira Otto MC, et al. Dietary intake of saturated fat by food source and incident cardiovascular disease: the Multi-Ethnic Study of Atherosclerosis. Am. J. Clin. Nutr. 2012;96:397–404. doi: 10.3945/ajcn.112.037770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siri-Tarino, P. W., Sun, Q., Hu, F. B. & Krauss, R. M. Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. The American journal of clinical nutrition, ajcn. 27725 (2010). [DOI] [PMC free article] [PubMed]
- 7.Hooper, L., Martin, N., Abdelhamid, A. & Davey Smith, G. Reduction in saturated fat intake for cardiovascular disease. Cochrane Database Syst Rev6 (2015). [DOI] [PubMed]
- 8.Mozaffarian D, Micha R, Wallace S. Effects on coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7:e1000252. doi: 10.1371/journal.pmed.1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Astrup A, et al. The role of reducing intakes of saturated fat in the prevention of cardiovascular disease: where does the evidence stand in 2010? Am. J. Clin. Nutr. 2011;93:684–688. doi: 10.3945/ajcn.110.004622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillingham LG, Harris-Janz S, Jones PJ. Dietary monounsaturated fatty acids are protective against metabolic syndrome and cardiovascular disease risk factors. Lipids. 2011;46:209–228. doi: 10.1007/s11745-010-3524-y. [DOI] [PubMed] [Google Scholar]
- 11.Jakobsen MU, et al. Major types of dietary fat and risk of coronary heart disease: a pooled analysis of 11 cohort studies. Am. J. Clin. Nutr. 2009;89:1425–1432. doi: 10.3945/ajcn.2008.27124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson PW, Schaefer EJ, Larson MG, Ordovas JM. Apolipoprotein E alleles and risk of coronary disease. A meta-analysis. Arterioscler. Thromb. Vasc. Biol. 1996;16:1250–1255. doi: 10.1161/01.ATV.16.10.1250. [DOI] [PubMed] [Google Scholar]
- 13.Ferreira D, et al. Association of apoliprotein E polymorphisms and metabolic syndrome in subjects with extreme obesity. Clin. Chim. Acta. 2011;412:1559–1562. doi: 10.1016/j.cca.2011.04.035. [DOI] [PubMed] [Google Scholar]
- 14.Tao MH, et al. Different associations of apolipoprotein E polymorphism with metabolic syndrome by sex in an elderly Chinese population. Metabolism. 2011;60:1488–1496. doi: 10.1016/j.metabol.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Song Y, Stampfer MJ, Liu S. Meta-analysis: apolipoprotein E genotypes and risk for coronary heart disease. Ann. Intern. Med. 2004;141:137–147. doi: 10.7326/0003-4819-141-2-200407200-00013. [DOI] [PubMed] [Google Scholar]
- 16.Bennet AM, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. 2007;298:1300–1311. doi: 10.1001/jama.298.11.1300. [DOI] [PubMed] [Google Scholar]
- 17.Waterworth DM, et al. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2010;30:2264–2276. doi: 10.1161/ATVBAHA.109.201020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Povel CM, Boer JM, Imholz S, Dollé ME, Feskens EJ. Genetic variants in lipid metabolism are independently associated with multiple features of the metabolic syndrome. Lipids Health Dis. 2011;10:118. doi: 10.1186/1476-511X-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dallongeville J, Lussier-Cacan S, Davignon J. Modulation of plasma triglyceride levels by apoE phenotype: a meta-analysis. J. Lipid Res. 1992;33:447–454. [PubMed] [Google Scholar]
- 20.Bu G, Apolipoprotein E. and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masson LF, McNeill G, Avenell A. Genetic variation and the lipid response to dietary intervention: a systematic review. Am. J. Clin. Nutr. 2003;77:1098–1111. doi: 10.1093/ajcn/77.5.1098. [DOI] [PubMed] [Google Scholar]
- 22.Carvalho-Wells AL, Jackson KG, Lockyer S, Lovegrove JA, Minihane AM. APOE genotype influences triglyceride and C-reactive protein responses to altered dietary fat intake in UK adults. Am. J. Clin. Nutr. 2012;96:1447–1453. doi: 10.3945/ajcn.112.043240. [DOI] [PubMed] [Google Scholar]
- 23.Olano-Martin E, et al. Contribution of apolipoprotein E genotype and docosahexaenoic acid to the LDL-cholesterol response to fish oil. Atherosclerosis. 2010;209:104–110. doi: 10.1016/j.atherosclerosis.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 24.Tikkanen M, Huttunen JK, Pajukanta PE, Pietinen P. Apolipoprotein E polymorphism and dietary plasma cholesterol response. Can. J. Cardiol. 1995;11:93G–96G. [PubMed] [Google Scholar]
- 25.Lehtimäki T, Moilanen T, Solakivi T, Laippala P, Ehnholm C. Cholesterol-rich diet induced changes in plasma lipids in relation to apolipoprotein E phenotype in healthy students. Ann. Med. 1992;24:61–66. doi: 10.3109/07853899209164146. [DOI] [PubMed] [Google Scholar]
- 26.Minihane AM, et al. ApoE polymorphism and fish oil supplementation in subjects with an atherogenic lipoprotein phenotype. Arterioscler. Thromb. Vasc. Biol. 2000;20:1990–1997. doi: 10.1161/01.ATV.20.8.1990. [DOI] [PubMed] [Google Scholar]
- 27.Ordovas JM, et al. Gene-diet interaction in determining plasma lipid response to dietary intervention. Atherosclerosis. 1995;118:S11–S27. doi: 10.1016/0021-9150(95)90069-1. [DOI] [PubMed] [Google Scholar]
- 28.Teixeira AA, et al. Diversity of Apolipoprotein E genetic polymorphism significance on cardiovascular risk is determined by the presence of Metabolic Syndrome among hypertensive patients. Lipids. Health. Dis. 2014;13:174. doi: 10.1186/1476-511X-13-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tierney A, et al. Effects of dietary fat modification on insulin sensitivity and on other risk factors of the metabolic syndrome–LIPGENE: a European randomized dietary intervention study. Int. J. Obes. 2011;35:800–809. doi: 10.1038/ijo.2010.209. [DOI] [PubMed] [Google Scholar]
- 30.Shaw DI, et al. LIPGENE food-exchange model for alteration of dietary fat quantity and quality in free-living participants from eight European countries. Br. J. Nutr. 2009;101:750–759. doi: 10.1017/S0007114508039962. [DOI] [PubMed] [Google Scholar]
- 31.Phillips CM, et al. Complement component 3 polymorphisms interact with polyunsaturated fatty acids to modulate risk of metabolic syndrome. The American journal of clinical nutrition. 2009;90:1665–1673. doi: 10.3945/ajcn.2009.28101. [DOI] [PubMed] [Google Scholar]
- 32.Matthews D, et al. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 33.Abu-Bader, S. H. Advanced and multivariate statistical methods for social science research with a complete SPSS guide. (Lyceum Books, 2010).
- 34.Faul F, Erdfelder E, Lang A-G, Buchner A. G* Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior research methods. 2007;39:175–191. doi: 10.3758/BF03193146. [DOI] [PubMed] [Google Scholar]
- 35.Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1988;8:1–21. doi: 10.1161/01.ATV.8.1.1. [DOI] [PubMed] [Google Scholar]
- 36.Eichner JE, et al. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am. J. Epidemiol. 2002;155:487–495. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- 37.Schiele F, et al. Apolipoprotein E serum concentration and polymorphism in six European countries: the ApoEurope Project. Atherosclerosis. 2000;152:475–488. doi: 10.1016/S0021-9150(99)00501-8. [DOI] [PubMed] [Google Scholar]
- 38.Tiret L, et al. ApoE polymorphism and predisposition to coronary heart disease in youths of different European populations. The EARS Study. European Atherosclerosis Research Study. Arterioscler. Thromb. Vasc. Biol. 1994;14:1617–1624. doi: 10.1161/01.ATV.14.10.1617. [DOI] [PubMed] [Google Scholar]
- 39.Menotti A, Lanti M, Puddu P, Kromhout D. Coronary heart disease incidence in northern and southern European populations: a reanalysis of the seven countries study for a European coronary risk chart. Heart. 2000;84:238–244. doi: 10.1136/heart.84.3.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conroy R, et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur. Heart J. 2003;24:987–1003. doi: 10.1016/S0195-668X(03)00114-3. [DOI] [PubMed] [Google Scholar]
- 41.Khan TA, et al. Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: Systematic review and meta-analysis of 14 015 stroke cases and pooled analysis of primary biomarker data from up to 60 883 individuals. Int. J. Epidemiol. 2013;42:475–492. doi: 10.1093/ije/dyt034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minihane A, Jofre-Monseny L, Olano-Martin E, Rimbach G. ApoE genotype, cardiovascular risk and responsiveness to dietary fat manipulation. Proc. Nutr. Soc. 2007;66:183–197. doi: 10.1017/S0029665107005435. [DOI] [PubMed] [Google Scholar]
- 43.Gregg R, et al. Abnormal in vivo metabolism of apolipoprotein E4 in humans. J. Clin. Invest. 1986;78:815. doi: 10.1172/JCI112645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson KG, Maitin V, Leake DS, Yaqoob P, Williams CM. Saturated fat-induced changes in Sf 60–400 particle composition reduces uptake of LDL by HepG2 cells. J. Lipid Res. 2006;47:393–403. doi: 10.1194/jlr.M500382-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Heeren J, Beisiegel U, Grewal T. Apolipoprotein E recycling implications for dyslipidemia and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006;26:442–448. doi: 10.1161/01.ATV.0000201282.64751.47. [DOI] [PubMed] [Google Scholar]
- 46.Siest G, et al. Apolipoprotein E: an important gene and protein to follow in laboratory medicine. Clin. Chem. 1995;41:1068–1086. [PubMed] [Google Scholar]
- 47.Weintraub MS, Eisenberg S, Breslow JL. Dietary fat clearance in normal subjects is regulated by genetic variation in apolipoprotein E. J. Clin. Invest. 1987;80:1571. doi: 10.1172/JCI113243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hartwich J, et al. Lipoprotein profile, plasma ischemia modified albumin and LDL density change in the course of postprandial lipemia. Insights from the LIPGENE study. Scand. J. Clin. Lab. Invest. 2010;70:201–208. doi: 10.3109/00365511003663630. [DOI] [PubMed] [Google Scholar]
- 49.Haddy N, et al. The importance of plasma apolipoprotein E concentration in addition to its common polymorphism on inter-individual variation in lipid levels: results from Apo Europe. Eur. J. Hum. Genet. 2002;10:841–850. doi: 10.1038/sj.ejhg.5200864. [DOI] [PubMed] [Google Scholar]
- 50.Kypreos KE, et al. Mechanisms of obesity and related pathologies: role of apolipoprotein E in the development of obesity. FEBS journal. 2009;276:5720–5728. doi: 10.1111/j.1742-4658.2009.07301.x. [DOI] [PubMed] [Google Scholar]
- 51.McConathy W, et al. Inhibition of lipoprotein lipase activity by synthetic peptides of apolipoprotein C-III. J. Lipid Res. 1992;33:995–1003. [PubMed] [Google Scholar]
- 52.Khan S, et al. Dietary long-chain n-3 PUFAs increase LPL gene expression in adipose tissue of subjects with an atherogenic lipoprotein phenotype. J. Lipid Res. 2002;43:979–985. [PubMed] [Google Scholar]
- 53.McBride PE. Triglycerides and risk for coronary heart disease. JAMA. 2007;298:336–338. doi: 10.1001/jama.298.3.336. [DOI] [PubMed] [Google Scholar]
- 54.Tai ES, et al. Polyunsaturated fatty acids interact with the PPARA-L162V polymorphism to affect plasma triglyceride and apolipoprotein C-III concentrations in the Framingham Heart Study. The Journal of nutrition. 2005;135:397–403. doi: 10.1093/jn/135.3.397. [DOI] [PubMed] [Google Scholar]
- 55.Oliviero O, et al. Apolipoprotein C-III, n-3 Polyunsaturated Fatty Acids, and “Insulin-Resistant” T−455C APOC3 Gene Polymorphism in Heart Disease Patients: Example of Gene-Diet Interaction. Clin. Chem. 2005;51:360–367. doi: 10.1373/clinchem.2004.040477. [DOI] [PubMed] [Google Scholar]
- 56.Bach-Ngohou K, et al. Apolipoprotein E kinetics: influence of insulin resistance and type 2 diabetes. Int. J. Obes. Relat. Metab. Disord. 2002;26:1451–1458. doi: 10.1038/sj.ijo.0802149. [DOI] [PubMed] [Google Scholar]
- 57.Harris WS, et al. Red Blood Cell Fatty Acids and Incident Diabetes Mellitus in the Women’s Health Initiative Memory Study. PloS one. 2016;11:e0147894. doi: 10.1371/journal.pone.0147894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lovejoy JC, Most MM, Lefevre M, Greenway FL, Rood JC. Effect of diets enriched in almonds on insulin action and serum lipids in adults with normal glucose tolerance or type 2 diabetes. Am. J. Clin. Nutr. 2002;76:1000–1006. doi: 10.1093/ajcn/76.5.1000. [DOI] [PubMed] [Google Scholar]
- 59.Vessby B, et al. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: The KANWU Study. Diabetologia. 2001;44:312–319. doi: 10.1007/s001250051620. [DOI] [PubMed] [Google Scholar]
- 60.Ragogna F, Lattuada G, Ruotolo G, Luzi L, Perseghin G. Lack of association of apoE ε4 allele with insulin resistance. Acta Diabetol. 2012;49:25–32. doi: 10.1007/s00592-011-0255-3. [DOI] [PubMed] [Google Scholar]
- 61.Meigs JB, et al. Apolipoprotein E isoform polymorphisms are not associated with insulin resistance: the Framingham Offspring Study. Diabetes Care. 2000;23:669–674. doi: 10.2337/diacare.23.5.669. [DOI] [PubMed] [Google Scholar]
- 62.Elosua R, et al. Obesity modulates the association among APOE genotype, insulin, and glucose in men. Obes. Res. 2003;11:1502–1508. doi: 10.1038/oby.2003.201. [DOI] [PubMed] [Google Scholar]
- 63.Hodson L, Skeaff CM, Fielding BA. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog. Lipid Res. 2008;47:348–380. doi: 10.1016/j.plipres.2008.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information - APOE genotype in the Metabolic Syndrome