

Abstract
Acute myeloid leukaemia (AML) is a heterogeneous disease that is, in general, associated with a very poor prognosis. Multiple cytogenetic and molecular abnormalities that characterize different forms of AML have been used to better prognosticate patients and inform treatment decisions. Indeed, risk status in patients with this disease has classically been based on cytogenetic findings; however, additional molecular characteristics have been shown to inform risk assessment, including FLT3, NPM1, KIT, and CEBPA mutation status. Advances in sequencing technology have led to the discovery of novel somatic mutations in tissue samples from patients with AML, providing deeper insight into the mutational landscape of the disease. The majority of patients with AML (>97%) are found to have a clonal somatic abnormality on mutational profiling. Nevertheless, our understanding of the utility of mutation profiling in clinical practice remains incomplete and is continually evolving, and evidence-based approaches to application of these data are needed. In this Review, we discuss the evidence-base for integrating mutational data into treatment decisions for patients with AML, and propose novel therapeutic algorithms in the era of molecular medicine.
Acute myeloid leukaemia (AML) is the most-common acute leukaemia in adults, and is primarily a disease of older adults (defined in this Review as those aged ≥60 years, unless otherwise stated), with a median age at diagnosis of 67 years1,2. The survival rates for younger adults with AML (aged <60 years) have improved, to some extent, over time, owing mostly to the development of intensive consolidation chemotherapy regimens, and improvements in supportive care and allogeneic haematopoietic-stem-cell transplantation (allo-HSCT) — the standard induction chemotherapy regimens have not changed substantially over the past 40 years3. In older patients, however, limited or no improvement in survival rates has been achieved, especially in patients aged >75 years, for whom no improvement in outcome has been demonstrated over the past three decades4.
The use of different therapies in the postremission treatment of AML is largely determined by prognostic risk stratification, which has classically been based on cytogenetic findings. The largest subset of patients with AML falls into the ‘intermediate-risk’ group, which includes those with normal-karyotype AML (NK-AML), as well as patients without either adverse or favourable cytogenetic abnormalities5,6. Considerable efforts have been made to refine risk stratification among this large and diverse subset of patients, with the emergence of multiple molecular abnormalities, including FLT3-ITD (FLT3 internal tandem duplication), and mutations in NPM1, KIT, and/or CEBPA, which have been shown to predict outcome and have been incorporated into modern prognostic and treatment guidelines7,8, although, in addition to cytogenetic and molecular findings, clinical factors can also predict outcomes9. Advances in next-generation sequencing (NGS) technology have led to the discovery of novel somatic mutations in patients with AML, providing greater insight into the mutational landscape of the disease. The majority of patients with AML (>97%) demonstrate at least one clonal somatic abnormality on mutational profiling10–12. Through genomic analysis of samples from 200 patients with de novo AML, The Cancer Genome Atlas (TCGA) investigators demonstrated that the distribution of genomic aberrations includes mutations in signalling-pathway genes in 59%, DNA-methylation-related genes in 44%, chromatin-modifying genes in 30%, NPM1 in 27%, myeloid-transcription-factor genes in 22%, transcription-factor-gene fusions in 18%, tumour-suppressor genes in 16%, spliceosome-complex genes in 14%, and cohesin-complex genes in 13% of patients12 (TABLE 1), with findings from several other studies corroborating a similar distribution of mutations13–15. The use of mutation profiling in the clinic continues to evolve, and a substantial degree of variance remains among oncologists on how to apply genomic findings to clinical practice16. Consequently, evidence-based approaches are urgently required for the application of genetic data in molecular medicine for patients with AML.
Table 1.
Frequency of mutations in relevant AML-associated genes*
| Gene | Overall frequency | Frequency in patients aged <60 years | Frequency in patients aged ≥60 years |
|---|---|---|---|
| FLT3 | 19–28% (FLT3-ITD)145 and 5–10% (FLT3-TKD)12,146 | 30% (FLT3-ITD) and 7% (FLT3-TKD)10 NK-AML only: 35% (FLT3-ITD) and 8% (FLT3-TKD)147 |
17–21% (FLT3-ITD)27,148‡ and 5% (FLT3-TKD)27 NK-AML only: 23% (FLT3-ITD) and 5% (FLT3-TKD)147 |
| NPM1 | 27–35%12,13 | 29%10 NK-AML only: 57%147 |
24–34%27,148‡ NK-AML only: 42%147 |
| DNMT3A | 26%12 | 18–23%10,149–151 | NA |
| NRAS | 8–9%12,152,153 | 10%10 | NA |
| ASXL1 | 17–19%154,155§ | 3–6%10,156‖,157 NK-AML only: 3%157 |
NA NK-AML only: 16%157 |
| CEBPA (biallelic) | 4–6%12,153,158,159 | 8–9%10,159 NK-AML only: 10%147 |
NA NK-AML only: 9–10%27¶,147 |
| TET2 | 8–27%12,160,161 | 8%10 | NA |
| WT1 | 6–7%12,162 | 8–11%10,163 | NA |
| IDH2 | 8–9%12,164 | 8–9%10,165 | NA |
| IDH1 | 9%12,152,164 | 7–8%10,165 | NA |
| KIT | 2–4%12,153 | 6%10 | NA |
| RUNX1 | 5–10%12,153 | 5%10 NK-AML only: 8%166 |
NA NK-AML only: 16%166 |
| MLL-PTD | 5%145 | 5%10 NK-AML only: 4%147 |
4%27 NK-AML only: 11%147 |
| NRAS | 8–9%12,152,153 | 10%10 | NA |
| PHF6 | 3%167 | 3%10 | NA |
| KRAS | 2–4%12,153 | 2%10 | NA |
| TP53 | 2–8%12,153,168 | 2%10 | NA |
| EZH2 | 2%169 | 0%10 | NA |
| JAK2 | 1–3%153,170 | NA | NA |
AML, acute myeloid leukaemia; FLT3-ITD, FLT3 internal tandem duplication mutation; FLT3-TKD, FLT3 tyrosine-kinase-domain mutation; MLL-PTD, MLL (KMT2A) partial tandem duplication; NA, not available; NK‑AML, normal‑karyotype acute myeloid leukaemia.
Inclusive of all karyotypes, except when noted; discrepancies between the mutational frequencies in younger (age <60 years) and elderly (age ≥60 years) patients with NK‑AML have been reported, when available.
Ostronoff et al.148 used the age of >65 years as the cut‑point definition for ‘elderly’ patients.
The study by Schnittger et al.155 included only patients with intermediate-risk AML.
Paschka et al.156 defined younger adult patients as those aged 18–61 years.
Schlenk et al.27 analyzed CEBPA mutations in elderly patients with normal karyotypes only.
In this Review, we propose that the lack of progress in improving the outcomes of patients with AML over the past four decades calls for consideration of a major paradigm shift regarding the present clinical treatment algorithms; for example, consideration of the use of upfront targeted therapy in elderly or unfit patients who are not candidates for standard intensive induction regimens, and addition of targeted agents to induction chemotherapy for younger patients, all in the setting of clinical trials (FIGS 1,2). We hypothesize that the integration of mutational profiling into the care of patients with AML will improve the outcomes associated with this characteristically devastating disorder. To accommodate the integration of mutational profiling into daily practice, more-high-throughput and more-comprehensive sequencing assays need to be used, in order to inform not only the approach to therapy for relapsed or refractory disease, but also decisions on upfront treatments.
Figure 1. Proposed treatment algorithm for patients aged >60 years with newly diagnosed AML.
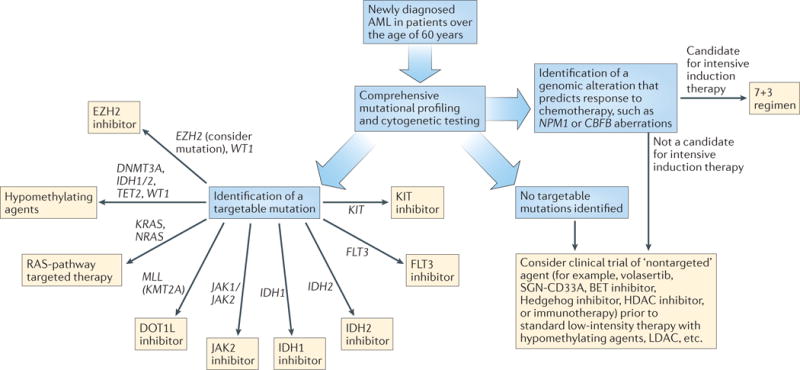
All novel agents, both targeted and ‘non-targeted’, should be administered in the setting of a clinical trial. HDAC, histone deacetylase; LDAC, low‑dose cytarabine.
Figure 2. Proposed treatment algorithm for patients aged ≤60 years with newly diagnosed AML.
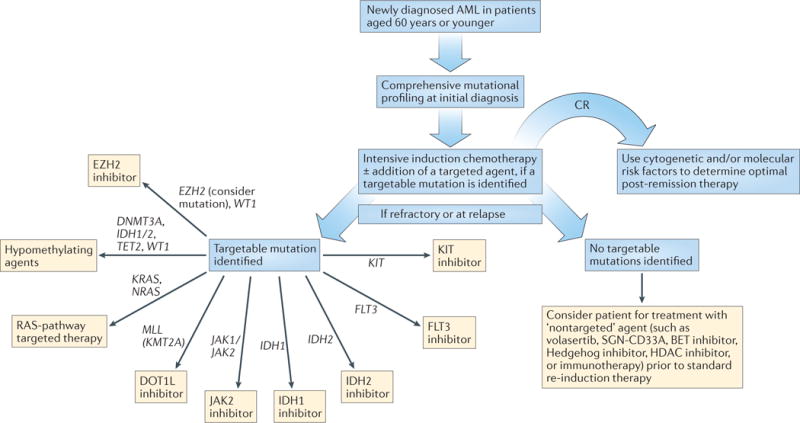
All novel agents, both targeted and ‘non‑targeted’, should be administered in the setting of a clinical trial. CR, complete response; HDAC, histone deacetylase.
Genetic profiling and induction therapy
Karyotype and molecular alterations are powerful prognostic markers in AML; however, at present, these data are often unavailable at the initiation of induction therapy, particularly outside of a clinical trial. As such, patients with AML generally receive induction therapy independent of their subsequent risk stratification; the choice of induction regimen is based on the patient’s age and comorbidities, with ‘fit’ younger patients typically receiving standard induction therapy with cytarabine and an anthracycline7. Daunorubicin and idarubicin are the anthracyclines used most commonly in induction regimens, and neither agent is clearly superior to the other17,18. The findings of studies performed in the past decade, which are outlined in the following sections, indicate that molecular subtypes of AML correlate with improved responses to induction therapy in various subsets of patients, stressing the importance of integrating upfront comprehensive mutational profiling into initial treatment decisions.
Candidates for intensive induction therapy
In the large, randomized, phase III Eastern Cooperative Oncology Group (ECOG) E1900 trial in which patients <60 years of age with AML were enrolled19, investigators observed an increased complete response (CR) rate and improved overall survival in patients who received induction therapy with high-dose daunorubicin (90 mg/m2 daily for 3 days), compared with those given the ‘standard’ dose of 45 mg/m2 daily for 3 days (CR rate 70.6% versus 57.3%, P <0.001; median overall survival 23.7 months versus 15.7 months, P = 0.003). The improved CR rate with 90 mg/m2 versus 45 mg/m2 daunorubicin was replicated in a second trial conducted in Korea by the Cooperative Study Group A for Haematology (COSAH)20. By contrast, the UK National Cancer Research Institute (NCRI) AML17 trial compared 90 mg/m2 daunorubicin with the more-conventionally used daunorubicin dose of 60 mg/m2, and the investigators found no differences in CR or overall survival between the treatment groups21. Notably, this trial was closed before the planned patient accrual target was met, after a signal of increased mortality within 60 days of treatment was noted in the 90 mg/m2 daunorubicin arm21. In addition to the use of a higher dose of daunorubicin in the comparator arm, the AML17 trial design also incorporated a second course of induction therapy for all patients and included additional randomizations, such as the potential addition of lestaurtinib, gemtuzumab, or everolimus depending on FLT3 status and cytogenetics21, thus limiting direct comparison of these findings with those of prior studies.
On subgroup analysis of the E1900 trial19, the benefit of high-dose daunorubicin was found to be limited to patients with favourable-risk and intermediate-risk disease, according to the definition proposed by Slovak et al.22, without a statistically significant benefit in patients with high-risk AML (median overall survival in the pooled favourable-risk and intermediate-risk subgroups was 20.7 months with standard-dose daunorubicin versus 34.3 months with high-dose daunorubicin; P = 0.004)19. Patel et al.11 performed a mutational analysis of 18 genes in samples from 398 of the 657 patients enrolled in this trial to characterize whether unique mutational profiles predicted response to high-dose daunorubicin. The authors found that DNMT3A and NPM1 mutations, and MLL translocations predicted better outcomes after receipt of high-dose daunorubicin, compared with the standard dose11. More recently, Sehgal et al.23 confirmed the benefit of anthracycline-dose intensification for patients with AML and DNMT3A mutations in a retrospective cohort study. By contrast, cytogenetic findings can inform decisions on de-escalation of induction therapy, such as in the case of PML–RARA fusions seen in patients with acute promyelocytic leukaemia (APL), in whom excellent outcomes have been demonstrated with the use of a chemotherapy-free regimen comprising all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) for the treatment of low-risk disease, defined by a white-blood-cell count <10,000/μL24. Collectively, these findings suggest that mutational profiling might be useful in determination of the upfront induction treatment regimen in patients with AML.
A randomized study conducted by the European Organisation for Research and Treatment of Cancer (EORTC) and the Gruppo Italiano Malattie Ematologiche dell’ Adulto (GIMEMA) Leukaemia Groups compared the efficacy of remission-induction therapy with daunorubicin, etoposide, and either standard-dose cytarabine (100 mg/m2 per day by continuous infusion for 10 days) or high-dose cytarabine (3,000 mg/m2 every 12 h by 3-hour infusion on days 1, 3, 5, and 7)25. In a planned subgroup analysis of this trial25, the investigators found that patients of all ages with FLT3-ITD mutations (n = 263) benefited from high-dose cytarabine, compared with the standard-dose regimen; however, the benefit was statistically significant in patients aged <46 years (hazard ratio (HR) 0.70, P = 0.02), but not in patients aged 46–60 years (HR 0.80, P = 0.14)25. Additional molecular abnormalities were not examined in this study, limiting the application of the findings to broader mutational profiling results. Nevertheless, the results of this trial suggest that certain genetically-defined subsets of patients with AML might benefit from dose-intensification, and that it might be possible to tailor chemotherapy regimens based on genetic profiles.
The randomized AML HD98B trial26, performed by the German–Austrian AML Study Group (AMLSG), demonstrated an improvement in the outcome of older patients (aged ≥61 years) with non-APL AML who received ATRA during and after intensive induction chemotherapy, as compared with those who received intensive induction chemotherapy alone. A subsequent correlative study demonstrated that NPM1 mutations in the absence of FLT3-ITD mutation are a predictive marker of response to ATRA27. This correlative study was limited, however, by the relatively small numbers of patients in each subgroup (n = 60 with NPM1 mutation and n = 51 with FLT3-ITD mutation), owing to incomplete profiling of the study cohort. Similar results were observed in the larger prospective AMLSG 07–04 trial28, in which younger adults were randomly assigned to receive intensive induction chemotherapy with and without ATRA. This trial included 289 patients with NPM1 mutations28. The CR rate was significantly increased by addition of ATRA to the treatment regimen in only the patients with NPM1-mutated AML (OR 2.20; P = 0.05), although the overall survival of all patients treated with ATRA (n = 549) was significantly improved (P = 0.02), compared with that of the patients who received chemotherapy only (n = 562)28. In a UK Medical Research Council (MRC trial), however, Burnett and co-investigators29 found no difference in outcomes with the addition of ATRA to induction chemotherapy with daunorubicin, standard-dose or high-dose cytarabine, and thioguanine in any of the molecular subgroups examined (NPM1, FLT3-ITD, and CEPBA mutated). Cross-trial comparisons limit the conclusions that can be drawn regarding whether ATRA improves outcomes among certain molecular subgroups of patients with AML, as the individual induction regimens used, and the dose and schedule of ATRA administration differed in these studies. For example, lower cumulative ATRA doses were used in the AMLSG trials26,28 compared with the MRC trial29, in which ATRA was administered at the full 45 mg/m2 dose on days 1–60. Taken together, the role of ATRA in any specific subset of patients with non-APL AML remains unclear. Preclinical data have demonstrated that ATRA and ATO induce proteasome-mediated degradation of mutant NPM1 protein, and apoptosis of NPM1-mutant AML cell-lines and primary AML-cell samples from patients with such mutations30,31, suggesting another potential therapeutic approach that merits exploration.
On the basis of data showing valproic acid (VPA) acts as a potent histone deacetylase (HDAC) inhibitor, Tassara and colleagues32 examined the therapeutic potential of the addition of VPA to standard induction therapy plus ATRA in patients with AML. This trial was stopped early owing to a lack of efficacy of the investigational VPA arm, the emergence of a trend towards a lower CR rate (40% versus 52%; P = 0.14) and higher early mortality (26% versus 14%; P = 0.06) in the VPA group32. Nevertheless, in an explorative subset analysis, it was noted that patients carrying NPM1 mutations might be more sensitive to the addition of VPA, based on an improvement in 5-year overall survival on univariate analysis (52% in patients who received VPA versus 37% in patients treated with standard therapy; P = 0.03)32. The absolute number of patients with NPM1 mutations enrolled in this trial was, however, low (n = 40), and as such these data are hypothesis generating only and cannot be used to guide clinical practice.
Lower-intensity therapy
Molecular data can also influence therapeutic decisions among patients who are not candidates for standard induction regimens. Data from TCGA Research Network indicates that 44% of patients with de novo AML carry mutations in genes encoding proteins that regulate DNA modifications, which have been demonstrated to have therapeutic implications12. In a small cohort of elderly patients with AML (median age of 74 years; range 32–85 years, with only one patient <60 years of age), Metzeler et al.33 reported a significantly greater CR rate to the hypomethylating agent (HMA) decitabine in patients with DNMT3A mutations (n = 8) versus those without such mutations (n = 38): 75% versus 34% (P = 0.05). The mechanistic link between mutations in DNMT3A — which reduce DNA methyltransferase activity — and increased response to HMAs has not, however, been elucidated. In addition, Itzykson and colleagues34 have reported an increased response rate to HMA therapy with azacitidine in 13 patients with TET2-mutant myelodysplastic syndrome (MDS) or low-blast-count AML, compared with that observed in 73 patients without TET2 mutations (82% versus 45%; P = 0.007). Furthermore, in a small retrospective analysis of 42 patients with AML who received either decitabine or azacitidine, those with IDH mutations (n = 7) had a higher response rate compared with those without mutations in the IDH1/2 genes (71% versus 23%; P = 0.01)35. Finally, preclinical evidence has demonstrated that WT1 mutations lead to a loss of TET2 function, suggesting that WT1-mutated AML might also be sensitive to HMA therapy36,37. These findings indicate that unified epigenetic pathway regulation by mutations in TET2, IDH1/2, WT1 and/or DNMT3A might confer increased sensitivity to HMAs; although promising, validation of the findings in prospective trials is needed.
In contrast to these data, Quintás-Cardama et al.38 noted a similar CR rate in patients with and those without FLT3-ITD mutations when treated with HMAs (51% versus 45%; P = 0.74); however, they demostrated an inferior CR rate in patients with NPM1 mutations who received HMAs as opposed to intensive chemotherapy (0% versus 70%; P = 0.02), although only three patients with NPM1-mutations received HMAs38. By contrast, in a randomized, phase II trial that examined responses to low-dose cytarabine with and without the polo-like kinase 1 (PLK1) inhibitor volasertib (a mitotic regulator that has been shown to inhibit the proliferation of leukaemia cells in preclinical studies39), the response rate among NPM1-mutant patients was 50% (7 of 14 patients)40. Low absolute numbers of NPM1-mutant patients in both studies prevent firm conclusions, although chemotherapy, in suitable candidates, seems to lead to more favourable responses than lower-intensity therapy with HMAs and volasertib in such patients.
Addition of targeted agents
A series of small-molecule FLT3 tyrosine kinase inhibitors (TKIs) have been developed and tested in patients with AML, including midostaurin, lestaurtinib, lefitinib, sorafenib, quizartinib, and crenolanib. The preclinical and clinical experience with FLT3 inhibition has been the subject of numerous dedicated review articles41–44, and a comprehensive analysis of these agents is outside the scope of this Review. These agents were among the first targeted agents to be added to standard induction chemotherapy for patients with AML, and current evidence for this approach will be briefly discussed in the following paragraph.
Sorafenib inhibits a broad spectrum of kinases, including FLT3, PDGFR, VEGFR, KIT, and the RAF proteins, although an active metabolite, sorafenib N-oxide, is a more potent inhibitor of FLT3 than the parent compound45. Serve et al.46 reported the results of a randomized, placebo-controlled trial investigating sorafenib in combination with intensive induction chemotherapy, using the 7 + 3 regimen comprising cytarabine and daunorubicin dosed at 60 mg/m2, in older patients with AML, and found that treatment in the sorafenib arm did not improve either event-free or overall survival. Moreover, use of sorafenib was associated with greater treatment-related mortality and lower CR rates, compared with the intensive chemotherapy regimen only46. This study was not restricted to patients carrying FLT3-ITD mutations; however, on subgroup analysis, the patients with FLT3-ITD-mutated AML also had poorer event-free and overall survival with sorafenib versus placebo treatment46. In the randomized, placebo-controlled SORAML trial in patients of ≤60 years of age with AML47, investigators evaluated the efficacy of addition of sorafenib to standard induction chemotherapy, followed by high-dose cytarabine consolidation therapy; patient enrolment was not restricted by genotype, and the findings of the study demonstrated an improvement in 3-year relapse-free survival (RFS) in the patients who received sorafenib versus the placebo group (56% versus 38%; P = 0.017)47. This benefit did not translate into an improvement in overall survival (63% with sorafenib versus 56% with placebo; P = 0.382), and a higher incidence of some toxicities (fever, bleeding, and hand–foot syndrome) was observed in the sorafenib arm, calling into question the overall clinical benefit of this approach47. In a phase I/II study48, sorafenib was combined with low-dose cytarabine therapy for elderly patients (median age of 77 years (range 63–83 years)) who were ineligible for intensive induction therapy, independent of tumour genotype, and the results demonstrated a favourable safety profile; however, efficacy was limited, with an overall response rate (ORR) of 10%48. Finally, sorafenib has been combined with azacitidine in a phase II trial restricted to patients with FLT3-ITD mutations49, in which investigators demonstrated a 46% ORR rate and a 27% CR rate. The favourable results of this trial, although not directly comparable to the permissive trials (that is, those that did not select patients based on genotype), nonetheless are suggestive of the importance of genotype-selected approaches in this patient population. The lack of specificity of sorafenib, coupled with a limited dataset in FLT3-ITD-positive patients, limits the ability to use these data to assess the importance of FLT3-ITD as a therapeutic target in newly diagnosed, and in relapsed or refractory, AML. Additional FLT3 inhibitors have been combined with standard intensive induction therapy, or used as monotherapies in patients deemed unfit for intensive induction, including midostaurin (in the phase Ib setting50; a phase III trial is currently ongoing51) and lestaurtinib52,53. In the phase III setting, lestaurtinib treatment did not result in an improved CR rate compared with standard first-line chemotherapy52. Studies with newer agents, including crenolanib54, AC220 (REF. 55), and ASP2215 (REF. 56) are all ongoing.
Future directions
As discussed, the presence of somatic mutations can clearly influence the response of patients with newly diagnosed AML to induction therapy, highlighting the underlying heterogeneity of this disease. The turnaround time required for high-throughput, comprehensive DNA sequencing, in addition to standard cytogenetic testing, to deliver clinically actionable data is the major barrier to clinical implementation of mutational data at time of diagnosis. The addition of targeted agents to induction therapy in a small set of AML trials demonstrates that rapid testing for alterations in a single gene is possible, but needs to be coordinated across cooperative sites in order to allow for adequate accrual of patients with rare mutations. Thus, future directions for research should include the continued development of faster DNA-sequencing platforms to allow for a reasonable turnaround time from sample receipt to reporting of results, enabling integration of these data to inform decisions on induction therapy57–60. To obtain statistically meaningful results from clinical trials, multicentre studies with upfront application of mutational data, acquired through a central lab or via a platform that can be performed at the different centres, will be required to investigate which molecularly targeted therapies are most effective in newly diagnosed patients with AML. A reasonable approach to the evaluation of novel agents would be to first examine their use as single agents in older patients who are not candidates for standard induction regimens, and subsequently apply those agents that show promise to the treatment of younger patients, in combination with standard induction therapy (FIGS 1,2).
Application to postremission therapy
Mutational profiling has been demonstrated to facilitate decision-making regarding postremission therapy, such as the decision to proceed with allo-HSCT in patients with high-risk AML versus proceeding with chemotherapy-based consolidation alone in lower-risk groups; the data supporting this approach are reviewed in the following sections. As further mutational data emerge, determining whether unique mutational patterns can predict outcome after allo-HSCT will be important, as such patterns might inform the development of investigational agents in patients whose genomic profile is predictive of an adverse outcome with allo-HSCT.
Postremission allografting
Studies assessing the effect of allo-HSCT on outcome are inherently limited by selection bias, in that often patients who do not undergo transplantation are ‘sicker’ than those who do61,62. Prospective studies that compare patients with available donors to ‘no-donor’ subsets limit this bias, but have other important limitations, including exclusion of patients without siblings and patients who never underwent HLA-typing63. Overall, the results of analyses have demonstrated a benefit of allo-HSCT for patients with intermediate-risk and high-risk cytogenetics64–67. Another approach to establish the benefit of allo-HSCT is the use of matched-pairs analyses, which incorporate patients with both sibling and unrelated donors to limit selection bias68. Many ‘donor versus no-donor’ studies have, however, been conducted using traditional cytogenetic-risk categories, without incorporating comprehensive mutational data.
The influence of NPM1 and FLT-ITD mutations
Schlenk et al.69 evaluated the prognostic and therapeutic relevance of NPM1, FLT3, CEBPA, MLL, and NRAS mutations in 872 adults <60 years of age with NK-AML who were enrolled in one of four multicentre prospective AMLSG trials; 663 patients received postremission therapy, with 150 undergoing allo-HSCT from an HLA-matched related donor. Among the patients without a donor, who received either chemotherapy or autologous HSCT (auto-HSCT), no statistically significant differences were observed in patient outcome according to mutational status of the five genes analysed69. The authors performed Cox regression analyses of the RFS data to explore the role of allo-HSCT according to genotype; they concluded that patients with FLT3-ITD mutations, and patients with wild-type NPM1 and CEBPA without the FLT3-ITD allele benefited from transplantation, whereas other subgroups either did not benefit or the numbers of patients were too small to conduct meaningful statistical analyses69. In 2015, Röllig et al.70 reported the findings of another donor versus no-donor analysis regarding the role of allo-HSCT in patients with NPM1-mutant AML, which indicated that allo-HSCT led to improved RFS in this group; however, no improvement in overall survival was demonstrated, probably owing to the excellent response to salvage therapy in this patient subset70.
Extensive analyses regarding the effect of FLT3-ITD mutation status on outcomes of allo-HSCT have been performed, both in regard to the benefit of allo-HSCT and to the risk of relapse post-transplantation in this high-risk patient population. Firstly, in a prospective study in patients with intermediate-risk AML (n = 555; 175 with FLT3-ITD mutations) by Bornhauser et al.71, patients with FLT3-ITD mutations had a significantly higher risk of relapse compared with those without FLT3-ITD mutations after consolidation chemo therapy (94% versus 59%, HR 4.0; P <0.001) or allo-HSCT (35% versus 19%, HR 2.7; P = 0.03), but not auto-HSCT; however, the absolute rates of relapse for all patients undergoing allo-HSCT were lower than those for other treatments, and overall survival was significantly worse for the FLT3-ITD-mutant patients after consolidation with chemotherapy only (21% versus 46% in those without FLT3-ITD mutations, HR 2.2; P = 0.001)71. The difference in the cumulative incidence of relapse (CIR) in recipients of allo-HSCT compared with patients not undergoing allo-HSCT has been confirmed in a smaller retrospective analysis of 66 patients, 24 of whom had FLT3-ITD mutations72. Results have also indicated that the benefit of allo-HSCT might be restricted to patients with a high FLT3-ITD allelic ratio ≥0.51 (P = 0.02 for RFS and P = 0.03 for overall survival)73. In a retrospective analysis of 206 patients who underwent allo-HSCT, 42% of whom had FLT3-ITD mutations, Brunet et al.74 found that FLT3-ITD-mutant patients had a higher incidence of relapse and worse leukaemia-free survival compared with patients without FLT3-ITD mutations. In a study of data from 511 patients in the Center for International Blood and Marrow Transplant Research (CIBMTR) database, 31% of whom were FLT3-ITD positive, Deol and colleagues75 found that the presence of a FLT3-ITD mutation increased the CIR after allo-HSCT, but did not have an effect on overall survival. Given the high risk of post-transplantation relapse in patients with FLT3-ITD-mutant AML, several investigators have examined incorporating FLT3-inhibitors as maintenance therapy after allo-HSCT. For example, phase I trials that examine the use of sorafenib76 and quizartinib77 in this context were reported at the 2014 American Society of Haematology (ASH) meeting, and the organizers of a midostaurin trial are currently recruiting patients78. Conclusions regarding these trials are restricted to the safety of the agents, and subsequent trials are needed to determine treatment efficacy.
The influence of MLL rearrangements
Wang et al.79 have prospectively evaluated the effect of MLL rearrangements on the outcomes of 56 consecutive patients with acute leukaemias who underwent allo-HSCT; during the study period, an additional 29 patients with MLL-rearranged leukaemias did not undergo allo-HSCT for various reasons, including disease relapse while awaiting transplant, disease resistance, patient preference, donor availability, and death during induction therapy79. In total, 26 of the 56 patients who underwent allo-HSCT were adults with AML79. Of the patients who underwent allo-HSCT, 12 relapsed (11 died from relapsed disease and one was subsequently lost to follow up), seven died from other causes, and 37 were alive without disease recurrence (median follow-up duration of 742 days)79. Of note, a lower relapse rate was noted among the patients transplanted in first CR (CR1) compared with patients transplanted beyond CR1 (17.9% versus 48.1%; P = 0.03)79. By contrast, patients who did not undergo transplantation had a median overall survival of 145 days79. These findings, although preliminary and from a heterogeneous population of adult and paediatric patients with AML or ALL, suggest that allo-HSCT might be a viable option for patients with MLL-rearranged leukaemias. A particularly high-risk subset of MLL-rearranged AMLs has been identified by Gröschel et al.80, who demonstrated that patients with EVI1 overexpression had inferior overall survival versus those with other MLL-rearranged AMLs; however, the outcomes of this patient group improved with the use of consolidation therapy with allo-HSCT, compared with other consolidation approaches80.
The influence of RUNX1 mutations
The effect of RUNX1 mutations on AML-treatment outcomes has been examined in 945 patients treated in two consecutive AMLSG multicentre trials (AML HD98A and AMLSG 07–04); 53 patients in these trials had a total of 59 different RUNX1 mutations81. Of the 53 patients with RUNX1 mutations, 32 attained a CR after induction chemotherapy (60%)81. In an exploratory analysis, the investigators examined the role of allo-HSCT, which was performed in 14 of the 32 patients with RUNX1-mutations who achieved a CR. In the patients undergoing allo-HSCT, 4-year RFS was 52%, compared with 0% among patients who received postremission therapy with high-dose cytarabine or auto-HSCT (P <0.0001)81. Moreover, Chou et al.82 have reported improved overall survival of patients with RUNX1 mutations, compared with those without such mutations, after allo-HSCT (HR 0.33; P = 0.04), and poor outcomes in patients with RUNX1 mutations, compared with patients who were RUNX1 wild type, among those who did not receive allo-HSCT (HR 1.74, P = 0.04). Caution is required not to overinterpret these findings, however, given that the nontransplanted patients might have been sicker than the patients who underwent allo-HSCT.
The influence of TP53 mutations
Patients with complex-karyotype AMLs (CK-AML) have been demonstrated to have dismal outcomes with chemotherapy alone, and the current recommendation is to consider these patients for allo-HSCT in CR1 (REF. 7). Rucker et al.83 examined a group of 234 patients with CK-AML to assess the frequency of TP53 alterations and the effect of these genetic aberrations on clinical outcomes. In total, 70% of patients had TP53 alter ations (60% of whom had TP53 mutations and 40% had TP53 losses)83. Patients with TP53 alterations had substantially lower CR rates after induction chemotherapy and inferior overall survival; this effect persisted after multivariable analysis83. 30 patients in this series underwent allo-HSCT for consolidation therapy, 15 of whom had TP53 alterations; 14 of the 15 patients with TP53 mutations relapsed and died, compared with 9 of the 15 patients who were TP53 wild type (P = 0.04)83. The absolute number of patients who underwent allo-HSCT in this study was low; however, these findings suggest extremely poor post-transplant outcomes in patients with TP53 alterations, and investigational therapies should be considered in lieu of, or in addition to, allografting.
The influence of CEBPA mutation
Schlenk et al.84 examined the clinical outcomes of patients with CEBPA mutations, who were treated using various consolidation approaches, among a large cohort of patients with AML (n = 2,983) who were enrolled in four Dutch–Belgian Haemato-Oncology Cooperative Group and Swiss Group for Clinical Cancer Research (HOVON/SAKK) trials and three AMLSG trials. 124 of the patients had AML with biallelic CEBPA mutations and achieved CR1 (REF. 84). In CR1, 32 of these patients underwent allo-HSCT, with 29 having matched related donors and three with matched unrelated donors; 20 underwent auto-HSCT, and 72 received chemotherapy84. The authors of this analysis reported that the patients who underwent allo-HSCT or auto-HSCT had improved RFS compared with those who received chemotherapy, but did not have better overall survival84. Furthermore, in the patients who relapsed (n = 45; one after allo-HSCT, five after auto-HSCT, and 39 after chemotherapy), re-induction chemotherapy followed by allo-HSCT was associated with favourable outcomes: 83% (35 of 42) of patients undergoing reinduction therapy achieved a second CR84.
Nontransplant postremission therapies
In a study of 185 patients who achieved a CR in the Cancer and Leukaemia Group B (CALGB) study 8525, in which investigators randomly assigned patients to postremission treatment with one of three cytarabine doses, Neubauer and colleagues85 examined the influence of RAS mutations on clinical outcomes — as in vitro data provided evidence of cytarabine sensitivity in RAS-mutant cells86. RAS mutations were present in 34 of the 185 patients85. The authors found that patients with RAS mutations who received high-dose cytarabine consolidation (3 g/m2 every 12 h on days 1, 3, and 5; or 400 mg/m2 per day for 5 days) had the lowest 10-year CIR (45% compared with 68% for patients with wild-type RAS)85. In patients with wild-type RAS, those who received high-dose cytarabine had a lower relapse risk than those who received low-dose cytarabine (HR 0.67; P = 0.04); this reduction in relapse risk with high-dose cytarabine was magnified in patients with RAS mutations (HR 0.28; P = 0.002), after adjusting for confounding variables such as presence of core-binding factor (CBF) cytogenetics85. Findings of in vitro studies have suggested that the presence of RAS mutations leads to altered cellular responses, ranging from cytostatic to cytotoxic, in the presence of cytarabine, potentially explaining these differences86,87.
Future directions
Data are limited regarding the relationships between other mutations, including those in ASXL1, DNMT3A, TET2, IDH1/2, WT1, EZH2, and PHF6, and postremission outcomes of patients with AML. Furthermore, the existing data are often from small cohorts of patients, thus limiting the power of statistical analysis, especially with regard to infrequent mutations, such as those in EZH2, WT1, and PHF6. A meta-analysis of published trials with available biospecimens and/or comprehensive mutational profiling results would increase the number of patients with data available for meaningful statistical analysis of the effects of different mutations, or combinations of mutations. Alternatively, prospective comprehensive sequencing of well-annotated, homogeneously treated patient cohorts would assist in understanding the clinical implications of integrated mutational profiling in AML. Questions for future research include the role of mutation-directed inhibitors for maintenance therapy in the post-transplantation setting, or for maintenance therapy in patients harbouring targetable mutations in genes, such as IDH1/2 and FLT3, who achieve CR, but are not candidates for transplantation. In nontransplant candidates with mutations in genes affecting DNA methylation, such as TET2, DNMT3A, IDH1/2, and WT1, the role of postinduction therapy with HMAs merits further investigation.
Novel therapies for patients with AML
Over the past decade, novel molecular findings obtained using NGS technology have resulted in a rapid expansion of the armamentarium of targeted agents, which is expected to continue over time. At present, new therapies are typically offered to patients in the relapsed/refractory disease setting, although, as we have proposed, these agents should be strongly considered (in the setting of a clinical trial) for the treatment of newly diagnosed elderly or unfit patients who are not candidates for intensive induction therapy (FIGS 1,2). Some of the novel molecular medicine approaches that are under investigation in patients with AML are discussed in the following sections.
IDH1/IDH2
Preclinical data demonstrated the efficacy of IDH2 inhibition in models of AML88,89, leading to the development of a multicentre, open-label, phase I dose-escalation study examining AG-221, a first-in-class, potent, reversible, selective inhibitor of the mutant form of IDH2 (REF. 90). Data from this phase I trial were first presented at the ASH meeting 2014 (REF. 91), and were updated at the European Haematology Association (EHA) meeting in 2015 (REF. 92). Eligibility criteria included the presence of an IDH2 mutation in patients with advanced-stage haematological malignancies, with 75% of patients having relapsed/refractory AML91. The data from this trial indicate that AG-221 is well tolerated, and the maximum-tolerated dose has not yet been reached91,92. Of the 11 deaths reported to date, most were disease-related, with only two deaths being reported as ‘possibly’ related to effects of the study drug91,92.
The updated findings presented at the EHA meeting revealed that among 45 efficacy-evaluable patients, objective responses were observed in 25 patients (ORR of 56%): six CRs, four bone-marrow CRs, five CRs with incomplete count recovery (CRi), and 10 partial responses (PRs)92. Five patients who achieved a CR proceeded to allo-HSCT91,92. Ultimately, the final, mature results from this trial must be awaited before we can draw any firm conclusions and decide whether these findings warrant any further investigation in the phase II setting. Additional considerations for the future include whether IDH2 inhibition can be moved into the front-line setting, alone and/or in combination with conventional induction therapy, and whether IDH2 blockade should be continued post-transplantation as maintenance therapy.
Early results from a phase I trial of the IDH1 inhibitor AG-120 were also presented at the EHA meeting in 2015 (REF. 93), and demonstrated that seven of 14 efficacy-evaluable patients had objective responses (four CR, two bone-marrow CRs, and one PR). Development of a phase I trial to evaluate AG-881, a dual IDH1–IDH2 inhibitor, is currently underway94.
Kinase inhibitors
FLT3 inhibitors
Numerous FLT3 inhibitors have been developed for the treatment of FLT3-mutant AML; typically, these agents have been first used in the relapsed or refractory disease setting, with some advancing to phase III trials, as discussed previously. Use of the early FLT3 inhibitors as single agents generally failed to produce robust or sustainable responses in phase I/II trials in this setting44. Newer-generation FLT3 inhibitors have been demonstrated to have higher potency and selectively for FLT3 (REF. 42). In a phase I clinical trial95, the second-generation FLT3 inhibitor quizartinib was assessed as a single agent, and responses were reported in patients with FLT3-mutant, FLT3-indeterminate, and FLT3-wild-type disease (response rates of 53%, 41%, and 14%, respectively). Quizartinib progressed to phase II clinical trials, with the results showing a high degree of activity as a single agent in both patients with mutant and wild-type FLT3 (composite CR (CRc) and PR rates of 68% and 47%, respectively)96. Crenolanib is a newer FLT3 inhibitor, which has demonstrated activity against both FLT3-ITD and FLT3-TKD mutations, with high selectivity for FLT3 relative to the closely-related protein KIT97,98. In the phase II setting99, crenolanib was associated with an ORR of 47%, with greater responses rate and longer overall survival seen in the FLT3-inhibitor-naive arm, compared with patients who had previously received a FLT3 inhibitor, suggesting that on-target FLT3 inhibition is responsible for the efficacy of this agent.
KIT inhibitors
A phase I/II study was performed to examine the safety and efficacy of imatinib (an inhibitor of KIT, as well as BCR–ABL1-kinase fusion proteins) combined with mitoxantrone, etoposide, and cytarabine in patients with KIT-positive relapsed/refractory AML100. Of 21 patients treated with imatinib at a dose of 400 mg per day, 62% achieved a CR100. Investigators determined that the patients who responded to therapy had a higher degree of phospho-AKT inhibition compared with nonresponders, indicating that agents that more-effectively inhibit AKT might have greater clinically utility100. Furthermore, imatinib has been examined as a single agent at higher doses, but with no responses observed101. In the phase I setting in a population of patients with relapsed/refractory AML, imatinib in combination with cytarabine and daunorubicin was associated with a frequency of CR or CR with incomplete platelet recovery (CRp) of 57%102, and in combination with low-dose cytarabine, an objective haematological response rate of 11% was reported in older patients who were not candidates for intensive induction therapy103. In all of these studies, KIT-positive patients were defined as those with AML blasts showing positivity for CD117 (KIT receptor), most commonly by flow cytometry, without evaluation of KIT-mutation status. Prolonged therapy with KIT TKIs can lead to a resistant phenotype via acquired secondary KIT mutations104. In preclinical models of gastrointestinal stromal tumours, which also harbour KIT mutations, the heat-shock-protein inhibitor AUY922 demonstrated growth inhibition in both imatinib-sensitive and imatinib-resistant cell-lines, implicating a resistance pathway that demands further investigation105. Owing to the high incidence of CD117 expression and KIT mutations in CBF AMLs, dasatinib has been examined as a maintenance therapy in CR1 for patients with high-risk disease, with a low 2-year disease-free survival rate of 25.7% reported106. Of the four patients with CBF AML harbouring KIT mutations at initial diagnosis who received dasatinib maintenance therapy, 75% no longer had KIT mutations detected at the time of relapse, suggesting that clonal evolution contributed to relapse106.
JAK2 inhibitors
JAK2 mutations are rarely found in patients with AML, and are observed most often in the setting of an antecedent myeloproliferative neoplasm (MPN)107. Nevertheless, JAK2 inhibition with ruxolitinib has been tested in the phase II setting in patients with refractory leukaemias, including post-MPN AML — 12 of the 38 patients treated harboured the JAK2V617F mutation108. Three of the 18 patients with post-MPN AML had a response to therapy in this study, with two achieving a CR and one a CRi108; of the three patients who achieved a CR, two harboured JAK2V617F mutations108. Despite these promising initial findings, a subsequent study was terminated early owing to a lack of satisfactory clinical benefit, with only one CRp observed among 13 evaluable patients with AML — the patient with the CRp was negative for the JAK2V617F mutation109. JAK2 inhibitors are currently under continued development in combination with HMAs for the treatment of post-MPN AML110.
Targeting the RAS pathway
Farnesyltransferase inhibition
RAS activity is dependent on post-translational farnesylation; therefore, farnesyltransferase inhibitors have been tested in clinical trials involving patients with AML111. In a trial examining the farnesyltransferase inhibitor tipifarnib, however, no correlation between treatment response and RAS-mutation status or inhibition of protein farnesylation was found112, thus calling the precise mechanism of action of this agent into question. Overall, responses to farnesyltransferase inhibitors have been disappointing; for example, in a trial in which investigators examined 348 elderly patients with AML (aged ≥70 years) who received tipifarnib, the highest ORR, 20%, was observed in patients treated at a dose of 300 mg twice daily113.
MEK–AKT-pathway inhibition
Given the disappointing results obtained with farnesyltransferase inhibition, further avenues of targeting the RAS-signalling pathway have been explored, predominantly inhibition of downstream mediators, such as MEK and AKT — with a preclinical rationale for dual-pathway inhibition114. The efficacy of combination therapy targeting MEK and AKT has been established in patients with BRAF-mutant melanoma, and is currently being investigated in clinical trials in patients with AML and RAS mutations115,116.
Chromatin modulators
DOT1L inhibition in MLL-rearranged AML
DOT1L is a histone methyltransferase that is required for the development and maintenance of MLL-rearranged leukaemias, and preclinical data have supported the potential clinical utility of DOT1L inhibition in this setting117,118. Subsequently, a phase I clinical trial was initiated to examine the safety of EPZ-5676, a small-molecule inhibitor of DOT1L, with preliminary results reported at the ASH meeting, 2014 (REF. 119). At the time of reporting, 37 patients had been enrolled in the study, 31 of whom had AML, with 36 patients evaluable for safety outcomes (having received at least one dose) and 28 evaluable for antileukaemic activity (having completed one or more post-baseline bone-marrow biopsy)119. Median time on therapy was 29 days, and EPZ-5676 was generally found to be well tolerated119. Best responses included one morphological CR, one cytogenetic CR, and two patients had resolution of leukaemia cutis; six patients had a treatment-related increase in neutrophils and/or monocytes119. These data support ongoing clinical investigation of EPZ-5676 and further exploration of DOT1L as a therapeutic target in patients with AML.
EZH2 inhibitors
EZH2 is a member of the polycomb group complex, which has histone methyltransferase activity. EZH2 is critical for haematopoietic-stem-cell development, influencing the balance between cell self-renewal and differentiation120. EZH2 inhibitors are currently under clinical development, with most clinical trials of such agents enrolling patients with diffuse large-B-cell lymphoma121, although inactivating mutations in EZH2 are also associated with AML122. Preclinical data suggest that the treatment of WT1-mutant AML cells with EZH2 inhibitors promotes myeloid differentiation, highlighting another potential application of this class of drugs123. The observation that loss-of-function EZH2 mutations are observed in myeloid malignancies, however, suggests a complex role of EZH2 in AML, and indicates the need for carefully designed preclinical studies and judicious patient selection when evaluating the use of EZH2 inhibitors in AML.
BET inhibitors
Inhibitors of the bromodomain and extraterminal (BET) family proteins act by targeting the epigenetic regulators that maintain aberrant chromatin states commonly associated with AML124. The BET-bromodomain-containing protein 4 (BRD4) was identified using an advanced RNA-interference screening method as a critical factor for maintenance of the AML-cell phenotype125, and murine models have supported the clinical use of BET inhibition across multiple cytogenetic and molecular subtypes of this disease126. Data from a phase I trial of a BET inhibitor, OTX015, were recently reported at the ASH meeting 2014 (REF. 127), and demonstrated single-agent antileukaemic efficacy at a wide range of doses (evidence of activity reported in five of 28 patients (18%)), with a reasonable toxicity profile. These data necessitate further investigation of OTX015 in the phase II setting.
HDAC inhibitors
Histone deacetylases (HDACs) are a class of enzymes that influence gene expression by altering the acetylation status of nucleosomal histones and other, nonhistone, proteins in chromatin128. HDAC inhibitors promote cell-cycle arrest, growth inhibition, and apoptosis in multiple cell types, including leukaemia cell-lines129. In a phase II study130, the HDAC inhibitor vorinostat had minimal single-agent activity in patients with untreated or relapsed AML, leading to the initiation of combination studies this agent. In particular, the use of vorinostat in combination with gemtuzumab ozogamicin in patients >60 years of age was associated with CR and CRp rates of 19% and 3%, respectively, with better responses observed in patients with normal or favourable AML karyotypes, compared with patients of the same age and performance status with other cytogenetic profiles (CR plus CRp rate of 46% versus 0%)131. Further complicating investigation of this combination, gemtuzumab ozogamicin was voluntarily withdrawn by Pfizer in 2010, at the request of the FDA, owing to concerns regarding an initial lack of clinical benefit and liver toxicity132. In 2014, however, the results of a meta-analysis of randomized studies demonstrated that addition of gemtuzumab ozogamicin to induction chemotherapy was associated with a survival benefit; thus, this drug could conceivably return to the market in the future133,134.
A trial in which investigators are combining vorinostat with azacitidine for the treatment of patients with newly diagnosed AML who are not eligible for intensive induction is currently ongoing135. Panobinostat, a pan-HDAC inhibitor, has also been combined with azacitidine in a phase Ib/II study involving patients with AML and high-risk MDS136, the results of which demonstrated a 31% ORR (CR, CRi, and PR) in the subgroup of patients with AML. The combination of the HDAC inhibitor pracinostat with azacitidine is also under investigation in an ongoing trial137. Of note, data from a preclinical model of AML indicate a synergism between the HDAC inhibitor pracinostat and the JAK2 inhibitor pacritinib; further studies are needed to determine if this approach has any clinical utility138.
Future directions
At present, targeted agents are typically used in the setting of relapsed and/or refractory AML, at a time when patients are generally sicker and when the disease is more resistant to therapy. To facilitate the future development of targeted therapies, we propose a paradigm shift in the general approach to therapy for newly diagnosed AML, in which mutational profiling should be performed upfront in all patients. If a targetable mutation is identified in older patients who are not candidates for intensive induction, they should be considered for frontline treatment with the relevant targeted agent in the context of a clinical trial — or an alternate investigational agent if no molecular targets are identified (FIG. 1). For younger patients, the addition of a targeted agent to standard induction therapy should be considered if an appropriate trial is available (FIG. 2). At first relapse, targeted agents should be considered, again, in the setting of a clinical trial.
To achieve these goals, the current mechanism for mutational profiling needs to be adjusted in order to enable quicker access to mutational data and to prevent unnecessary delays in administering upfront therapy. Admittedly, concerns exist regarding the timing of therapy relative to the window when informative mutational profiling is available. In a study of 599 patients with newly diagnosed AML, Bertoli et al.139 determined that time from diagnosis to treatment (TDT) had no effect on survival, CR, or early death rates. By contrast, Sekeres et al.140 found that a TDT >5 days negatively affected the CR rate and overall survival of patients aged <60 years, but not older patients. Both of these studies are limited by their nonrandomized nature, as the TDT among patients with more-favourable disease could conceivably have been longer than in patients who seemed to have more-aggressive disease — who might have been treated quicker. A randomized trial is unlikely to be performed to address this question, however, and the findings of both studies indicate that reasonable delays in the initiation of induction therapy might be safe in older patients with AML.
On the basis of the seminal work regarding clonal evolution in AML141–144, additional concerns regarding the use of targeted therapies are bound to arise over time: in a clonally heterogeneous disorder, does the mutant-allele frequency influence responsiveness to targeted agents? Are patients with more clonally heterogeneous AML less responsive to a single targeted agent than those with more-homogenous disease? Are combinations of targeted agents safe and effective? The design of clinical trials involving targeted agents should include correlative studies with well-organized biospecimen collection to help answer these questions.
Given the rarity of certain mutations in AML, accrual of patients for trials is often difficult. To examine the role of targeted agents in the treatment of patients with rare mutations, performing multicentre and/or cooperative-group trials could help to increase accrual by providing geographically diverse access to patients, enabling sufficient statistical power for meaningful comparisons. Finally, standardized, high-throughput, and rapid DNA-sequencing techniques are imperative for the clinical application of genomic data to patient care.
Conclusions
We are currently at a crossroads where our knowledge of AML biology is rapidly expanding, and we must endeavour to apply this knowledge to the clinical context as soon as possible in order to improve the outcomes of our patients. To enhance the clinical care of patients with AML, especially older patients for whom clinical outcomes have improved little over the past several decades, we advocate for a paradigm shift in the way that novel agents are introduced into the clinic. Instead of delaying introduction of novel agents to the setting of relapsed/refractory disease, we propose consideration of frontline treatment with targeted agents either alone or in combination with chemotherapy, in the context of multicentre and/or cooperative-group clinical trials, when available.
Key points.
More than 97% of patients with acute myeloid leukaemia (AML) demonstrate at least one clonal somatic abnormality on comprehensive mutational profiling, which is increasingly being performed in the clinic
Unique mutational profiles can be used to predict a response to both standard and investigational therapies in patients with newly diagnosed or relapsed and/or refractory AML
Molecular abnormalities associated with AML are also predictors of outcome following allogeneic haematopoietic-stem-cell transplantation
Comprehensive mutational profiling should be performed in all newly diagnosed patients with AML using standardized high-throughput assays
Comprehensive mutational profiling will enable consideration of novel targeted agents in the upfront setting, either alone or in combination with chemotherapy, and we hypothesize that this approach will improve outcomes of patients with this disease
Footnotes
Author contributions
All of the authors made substantial contributions to researching data for article, discussion of content, and review/editing of manuscript before submission. C.C.C. wrote the manuscript.
Competing interests statement
The authors declare no competing interests.
References
- 1.Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.National Cancer Institute. SEER Stat Fact Sheets: Acute Myeloid Leukemia (AML) NIH [online] 2015 http://seer.cancer.gov/statfacts/html/amyl.html.
- 3.Pulte D, Gondos A, Brenner H. Improvements in survival of adults diagnosed with acute myeloblastic leukemia in the early 21st century. Haematologica. 2008;93:594–600. doi: 10.3324/haematol.12304. [DOI] [PubMed] [Google Scholar]
- 4.Thein MS, Ershler WB, Jemal A, Yates JW, Baer MR. Outcome of older patients with acute myeloid leukemia: an analysis of SEER data over 3 decades. Cancer. 2013;119:2720–2727. doi: 10.1002/cncr.28129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rockova V, et al. Risk stratification of intermediate-risk acute myeloid leukemia: integrative analysis of a multitude of gene mutation and gene expression markers. Blood. 2011;118:1069–1076. doi: 10.1182/blood-2011-02-334748. [DOI] [PubMed] [Google Scholar]
- 6.Dohner K, Paschka P. Intermediate-risk acute myeloid leukemia therapy: current and future. Hematology Am Soc Hematol Ed Program. 2014;2014:34–43. doi: 10.1182/asheducation-2014.1.34. [DOI] [PubMed] [Google Scholar]
- 7.National Comprehensive Cancer Network. NCCN Guidelines for Treatment of Cancer by Site: Acute Myeloid Leukemia (Version 1.2015) [online] 2015 http://www.nccn.org/professionals/physician_gls/pdf/aml.pdf.
- 8.Dohner H, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 9.Krug U, et al. Complete remission and early death after intensive chemotherapy in patients aged 60 years or older with acute myeloid leukaemia: a web-based application for prediction of outcomes. Lancet. 2010;376:2000–2008. doi: 10.1016/S0140-6736(10)62105-8. [DOI] [PubMed] [Google Scholar]
- 10.Patel JP, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel JP, Levine RL. How do novel molecular genetic markers influence treatment decisions in acute myeloid leukemia? Hematology Am Soc Hematol Ed Program. 2012;2012:28–34. doi: 10.1182/asheducation-2012.1.28. [DOI] [PubMed] [Google Scholar]
- 12.The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falini B, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 14.Cho YU, et al. Preferential occurrence of spliceosome mutations in acute myeloid leukemia with preceding myelodysplastic syndrome and/or myelodysplasia morphology. Leuk Lymphoma. 2015;56:2301–2308. doi: 10.3109/10428194.2014.995648. [DOI] [PubMed] [Google Scholar]
- 15.Aslanyan MG, et al. Clinical and biological impact of TET2 mutations and expression in younger adult AML patients treated within the EORTC/GIMEMA AML-12 clinical trial. Ann Hematol. 2014;93:1401–1412. doi: 10.1007/s00277-014-2055-7. [DOI] [PubMed] [Google Scholar]
- 16.Gray SW, Hicks-Courant K, Cronin A, Rollins BJ, Weeks JC. Physicians’ attitudes about multiplex tumor genomic testing. J Clin Oncol. 2014;32:1317–1323. doi: 10.1200/JCO.2013.52.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohtake S, et al. Randomized study of induction therapy comparing standard-dose idarubicin with high-dose daunorubicin in adult patients with previously untreated acute myeloid leukemia: the JALSG AML201 Study. Blood. 2011;117:2358–2365. doi: 10.1182/blood-2010-03-273243. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Xu S, Tan Y, Chen J. The effects of idarubicin versus other anthracyclines for induction therapy of patients with newly diagnosed leukaemia. Cochrane Database of Systematic Reviews. 2015;(6) doi: 10.1002/14651858.CD010432.pub2. Art. No.: CD010432 http://dx.doi.org/10.1002/14651858.CD010432.pub2. [DOI] [PMC free article] [PubMed]
- 19.Fernandez HF, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361:1249–1259. doi: 10.1056/NEJMoa0904544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JH, et al. A randomized trial comparing standard versus high-dose daunorubicin induction in patients with acute myeloid leukemia. Blood. 2011;118:3832–3841. doi: 10.1182/blood-2011-06-361410. [DOI] [PubMed] [Google Scholar]
- 21.Burnett AK, et al. A randomized comparison of daunorubicin 90 mg/m2 versus 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood. 2015;125:3878–3885. doi: 10.1182/blood-2015-01-623447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slovak ML, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–4083. [PubMed] [Google Scholar]
- 23.Sehgal AR, et al. DNMT3A mutational status affects the results of dose-escalated induction therapy in acute myelogenous leukemia. Clin Cancer Res. 2015;21:1614–1620. doi: 10.1158/1078-0432.CCR-14-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo-Coco F, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–121. doi: 10.1056/NEJMoa1300874. [DOI] [PubMed] [Google Scholar]
- 25.Willemze R, et al. High-dose cytarabine in induction treatment improves the outcome of adult patients younger than age 46 years with acute myeloid leukemia: results of the EORTC-GIMEMA AML-12 trial. J Clin Oncol. 2014;32:219–228. doi: 10.1200/JCO.2013.51.8571. [DOI] [PubMed] [Google Scholar]
- 26.Schlenk RF, et al. Phase III study of all-trans retinoic acid in previously untreated patients 61 years or older with acute myeloid leukemia. Leukemia. 2004;18:1798–1803. doi: 10.1038/sj.leu.2403528. [DOI] [PubMed] [Google Scholar]
- 27.Schlenk RF, et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from the AMLSG Trial AML HD98B. Haematologica. 2009;94:54–60. doi: 10.3324/haematol.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlenk RF, et al. All-trans retinoic acid improves outcome in younger adult patients with nucleophosmin-1 mutated acute myeloid leukemia — results of the AMLSG 07–04 Randomized Treatment Trial [online] 2011 https://ash.confex.com/ash/2011/webprogram/Paper37138.html.
- 29.Burnett AK, et al. The impact on outcome of the addition of all-trans retinoic acid to intensive chemotherapy in younger patients with nonacute promyelocytic acute myeloid leukemia: overall results and results in genotypic subgroups defined by mutations in NPM1, FLT3, and CEBPA. Blood. 2010;115:948–956. doi: 10.1182/blood-2009-08-236588. [DOI] [PubMed] [Google Scholar]
- 30.El Hajj H, et al. Retinoic acid and arsenic trioxide trigger degradation of mutated NPM1, resulting in apoptosis of AML cells. Blood. 2015;125:3447–3454. doi: 10.1182/blood-2014-11-612416. [DOI] [PubMed] [Google Scholar]
- 31.Martelli MP, et al. Arsenic trioxide and all-trans retinoic acid target NPM1 mutant oncoprotein levels and induce apoptosis in NPM1-mutated AML cells. Blood. 2015;125:3455–3465. doi: 10.1182/blood-2014-11-611459. [DOI] [PubMed] [Google Scholar]
- 32.Tassara M, et al. Valproic acid in combination with all-trans retinoic acid and intensive therapy for acute myeloid leukemia in older patients. 2014;123:4027–4036. doi: 10.1182/blood-2013-12-546283. [DOI] [PubMed] [Google Scholar]
- 33.Metzeler KH, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26:1106–1107. doi: 10.1038/leu.2011.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itzykson R, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25:1147–1152. doi: 10.1038/leu.2011.71. [DOI] [PubMed] [Google Scholar]
- 35.Emadi A, et al. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am J Hematol. 2015;90:E77–E79. doi: 10.1002/ajh.23965. [DOI] [PubMed] [Google Scholar]
- 36.Rampal R, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9:1841–1855. doi: 10.1016/j.celrep.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. 2015;57:662–673. doi: 10.1016/j.molcel.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quintas-Cardama A, et al. Epigenetic therapy is associated with similar survival compared with intensive chemotherapy in older patients with newly diagnosed acute myeloid leukemia. Blood. 2012;120:4840–4845. doi: 10.1182/blood-2012-06-436055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Renner AG, et al. Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood. 2009;114:659–662. doi: 10.1182/blood-2008-12-195867. [DOI] [PubMed] [Google Scholar]
- 40.Dohner H, et al. Randomized, phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy. Blood. 2014;124:1426–1433. doi: 10.1182/blood-2014-03-560557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knapper S. The clinical development of FLT3 inhibitors in acute myeloid leukemia. Expert Opin Investig Drugs. 2011;20:1377–1395. doi: 10.1517/13543784.2011.611802. [DOI] [PubMed] [Google Scholar]
- 42.Kayser S, Levis MJ. FLT3 tyrosine kinase inhibitors in acute myeloid leukemia: clinical implications and limitations. Leuk Lymphoma. 2014;55:243–255. doi: 10.3109/10428194.2013.800198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiernik PH. FLT3 inhibitors for the treatment of acute myeloid leukemia. Clin Adv Hematol Oncol. 2010;8:429–436. [PubMed] [Google Scholar]
- 44.Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5:65–77. doi: 10.1177/2040620714532123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inaba H, et al. Phase I pharmacokinetic and pharmacodynamic study of the multikinase inhibitor sorafenib in combination with clofarabine and cytarabine in pediatric relapsed/refractory leukemia. J Clin Oncol. 2011;29:3293–3300. doi: 10.1200/JCO.2011.34.7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Serve H, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31:3110–3118. doi: 10.1200/JCO.2012.46.4990. [DOI] [PubMed] [Google Scholar]
- 47.Röllig C, et al. Sorafenib versus placebo in addition to standard therapy in younger patients with newly diagnosed acute myeloid leukemia: results from 267 patients treated in the randomized placebo-controlled SAL-soraml trial [online] 2014 https://ash.confex.com/ash/2014/webprogram/Paper75091.html.
- 48.Macdonald DA, et al. A Phase I/II study of sorafenib in combination with low dose cytarabine in elderly patients with acute myeloid leukemia or high-risk myelodysplastic syndrome from the National Cancer Institute of Canada Clinical Trials Group: trial IND. 186. Leuk Lymphoma. 2013;54:760–766. doi: 10.3109/10428194.2012.737917. [DOI] [PubMed] [Google Scholar]
- 49.Ravandi F, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121:4655–4662. doi: 10.1182/blood-2013-01-480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stone RM, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26:2061–2068. doi: 10.1038/leu.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.US National Library of Medicine. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT00651261.
- 52.Knapper S, et al. A randomised comparison of the sequential addition of the FLT3 inhibitor lestaurtinib (CEP701) to standard first line chemotherapy for FLT3-Mutated acute myeloid leukemia: the UK experience [online] 2014 https://ash.confex.com/ash/2014/webprogram/Paper71906.html.
- 53.Knapper S, et al. A Phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood. 2006;108:3262–3270. doi: 10.1182/blood-2006-04-015560. [DOI] [PubMed] [Google Scholar]
- 54.US National Library of Medicine. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT02283177?term=NCT02283177&rank=1.
- 55.US National Library of Medicine. ClinicalTrials.gov [online] 2014 https://clinicaltrials.gov/ct2/show/NCT02272478?term=NCT02272478&rank=1.
- 56.US National Library of Medicine. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT02236013?term=NCT02236013&rank=1.
- 57.Duncavage EJ, Tandon B. The utility of next-generation sequencing in diagnosis and monitoring of acute myeloid leukemia and myelodysplastic syndromes. Int J Lab Hematol. 2015;37(Suppl. 1):115–121. doi: 10.1111/ijlh.12361. [DOI] [PubMed] [Google Scholar]
- 58.Ibanez M, et al. Rapid screening of ASXL1, IDH1, IDH2, and c-CBL mutations in de novo acute myeloid leukemia by high-resolution melting. J Mol Diagn. 2012;14:594–601. doi: 10.1016/j.jmoldx.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 59.Cheng DT, et al. Detection of mutations in myeloid malignancies through paired-sample analysis of microdroplet-PCR deep sequencing data. J Mol Diagn. 2014;16:504–518. doi: 10.1016/j.jmoldx.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luthra R, et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring. Haematologica. 2014;99:465–473. doi: 10.3324/haematol.2013.093765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schlenk RF, et al. Prospective evaluation of allogeneic hematopoietic stem-cell transplantation from matched related and matched unrelated donors in younger adults with high-risk acute myeloid leukemia: German–Austrian trial AMLHD98A. J Clin Oncol. 2010;28:4642–4648. doi: 10.1200/JCO.2010.28.6856. [DOI] [PubMed] [Google Scholar]
- 62.Anderson JR, Cain KC, Gelber RD. Analysis of survival by tumor response. J Clin Oncol. 1983;1:710–719. doi: 10.1200/JCO.1983.1.11.710. [DOI] [PubMed] [Google Scholar]
- 63.Buchner T, Berdel WE, Kienast J. Cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;359:651. doi: 10.1056/NEJMc081230. [DOI] [PubMed] [Google Scholar]
- 64.Cornelissen JJ, et al. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in first remission acute myeloid leukemia in young and middle-aged adults: benefits for whom? Blood. 2007;109:3658–3666. doi: 10.1182/blood-2006-06-025627. [DOI] [PubMed] [Google Scholar]
- 65.Stelljes M, et al. Allogeneic transplantation as post-remission therapy for cytogenetically high-risk acute myeloid leukemia: landmark analysis from a single prospective multicenter trial. Haematologica. 2011;96:972–979. doi: 10.3324/haematol.2011.041004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Estey E, et al. Prospective feasibility analysis of reduced-intensity conditioning (RIC) regimens for hematopoietic stem cell transplantation (HSCT) in elderly patients with acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (MDS) Blood. 2007;109:1395–1400. doi: 10.1182/blood-2006-05-021907. [DOI] [PubMed] [Google Scholar]
- 67.Koreth J, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA. 2009;301:2349–2361. doi: 10.1001/jama.2009.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stelljes M, et al. Allogeneic transplantation versus chemotherapy as postremission therapy for acute myeloid leukemia: a prospective matched pairs analysis. J Clin Oncol. 2014;32:288–296. doi: 10.1200/JCO.2013.50.5768. [DOI] [PubMed] [Google Scholar]
- 69.Schlenk RF, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 70.Röllig C, et al. Allogeneic stem-cell transplantation in patients with NPM1-mutated acute myeloid leukemia: results from a prospective donor versus no-donor analysis of patients after upfront HLA typing within the SAL-AML 2003 trial. J Clin Oncol. 2015;33:403–410. doi: 10.1200/JCO.2013.54.4973. [DOI] [PubMed] [Google Scholar]
- 71.Bornhauser M, et al. Improved outcome after stem-cell transplantation in FLT3/ITD-positive AML. Blood. 2007;109:2264–2265. doi: 10.1182/blood-2006-09-047225. [DOI] [PubMed] [Google Scholar]
- 72.Laboure G, et al. Potent graft-versus-leukemia effect after reduced-intensity allogeneic SCT for intermediate-risk AML with FLT3-ITD or wild-type NPM1 and CEBPA without FLT3-ITD. Biol Blood Marrow Transplant. 2012;18:1845–1850. doi: 10.1016/j.bbmt.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 73.Schlenk RF, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014;124:3441–3449. doi: 10.1182/blood-2014-05-578070. [DOI] [PubMed] [Google Scholar]
- 74.Brunet S, et al. Impact of FLT3 internal tandem duplication on the outcome of related and unrelated hematopoietic transplantation for adult acute myeloid leukemia in first remission: a retrospective analysis. J Clin Oncol. 2012;30:735–741. doi: 10.1200/JCO.2011.36.9868. [DOI] [PubMed] [Google Scholar]
- 75.Deol A, et al. FLT3 mutation increases relapse risk after allogeneic hematopoietic cell transplant for acute myeloid leukemia in first or second complete remission: a center for international blood and marrow transplant research (CIBMTR) analysis [online] 2014 https://ash.confex.com/ash/2014/webprogram/Paper68629.html.
- 76.Chen YB, et al. Phase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for patients with FLT3-ITD AML [online] 2014 doi: 10.1016/j.bbmt.2014.09.007. https://ash.confex.com/ash/2014/webprogram/Paper67358.html. [DOI] [PMC free article] [PubMed]
- 77.Sandmaier BM, et al. Results of a phase 1 study of quizartinib (AC220) as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic cell transplantation [online] 2014 doi: 10.1002/ajh.24959. https://ash.confex.com/ash/2014/webprogram/Paper73502.html. [DOI] [PMC free article] [PubMed]
- 78.University of Ulm. Protocol in acute myeloid leukemia with FLT3-ITD. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT01477606?term=NCT01477606&rank=1.
- 79.Wang Y, et al. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of patients with mixed-lineage-leukemia-rearranged acute leukemia: results from a prospective, multi-center study. Am J Hematol. 2014;89:130–136. doi: 10.1002/ajh.23595. [DOI] [PubMed] [Google Scholar]
- 80.Groschel S, et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: a study of the German–Austrian Acute Myeloid Leukemia Study Group and the Dutch–Belgian–Swiss HOVON/SAKK Cooperative Group. J Clin Oncol. 2013;31:95–103. doi: 10.1200/JCO.2011.41.5505. [DOI] [PubMed] [Google Scholar]
- 81.Gaidzik VI, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29:1364–1372. doi: 10.1200/JCO.2010.30.7926. [DOI] [PubMed] [Google Scholar]
- 82.Chou SC, et al. Prognostic implication of gene mutations on overall survival in the adult acute myeloid leukemia patients receiving or not receiving allogeneic hematopoietic stem cell transplantations. Leuk Res. 2014;38:1278–1284. doi: 10.1016/j.leukres.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 83.Rucker FG, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–2121. doi: 10.1182/blood-2011-08-375758. [DOI] [PubMed] [Google Scholar]
- 84.Schlenk RF, et al. The value of allogeneic and autologous hematopoietic stem cell transplantation in prognostically favorable acute myeloid leukemia with double mutant CEBPA. Blood. 2013;122:1576–1582. doi: 10.1182/blood-2013-05-503847. [DOI] [PubMed] [Google Scholar]
- 85.Neubauer A, et al. Patients with acute myeloid leukemia and RAS mutations benefit most from postremission high-dose cytarabine: a Cancer and Leukemia Group B study. J Clin Oncol. 2008;26:4603–4609. doi: 10.1200/JCO.2007.14.0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koo HM, et al. Enhanced sensitivity to 1-β-D-arabinofuranosylcytosine and topoisomerase II inhibitors in tumor cell lines harboring activated ras oncogenes. Cancer Res. 1996;56:5211–5216. [PubMed] [Google Scholar]
- 87.Koo HM, McWilliams MJ, Alvord WG, Vande Woude GF. Ras oncogene-induced sensitization to 1-β-D-arabinofuranosylcytosine. Cancer Res. 1999;59:6057–6062. [PubMed] [Google Scholar]
- 88.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang F, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 90.US National Library of Medicine. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT01915498?term=NCT01915498&rank=1.
- 91.Stein EM, et al. AG-221, an oral, selective, first-in-class, potent inhibitor of the IDH2 mutant metabolic enzyme, induces durable remissions in a phase I study in patients with IDH2 mutation positive advanced hematologic malignancies [online] 2014 https://ash.confex.com/ash/2014/webprogram/Paper70721.html.
- 92.DiNardo CS, et al. AG-221, An oral, selective, first-in-class, potent inhibitor of the IDH2 mutant enzyme, induced durable responses in a phase 1 study of IDH2 mutation-positive advanced hematologic malignancies [online] 2015 http://learningcenter.ehaweb.org/eha/2015/20th/100710/eyal.attar.ag-221.an.oral.selective.first-in-class.potent.inhibitor.of.the.html?f=14269p16m3.
- 93.de Botton S, et al. Clinical safety and activity of AG-120, a first-in-class, potent inhibitor of the IDH1-mutant protein, in a phase 1 study of patients with advanced IDH1-mutant hematologic malignancies [online] 2015 http://learningcenter.ehaweb.org/eha/2015/20th/100704/stphane.debotton.clinical.safety.and.activity.of.ag-120.a.first-in-class.html?f=p3m3.
- 94.US National Library of Medicine. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT02492737?term=NCT02492737&rank=1.
- 95.Cortes JE, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31:3681–3687. doi: 10.1200/JCO.2013.48.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Levis MJ, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia after second-line chemotherapy or hematopoietic stem cell transplantation [online] 2012 https://ash.confex.com/ash/2012/webprogram/Paper54037.html.
- 97.Galanis A, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014;123:94–100. doi: 10.1182/blood-2013-10-529313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith CC, et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci USA. 2014;111:5319–5324. doi: 10.1073/pnas.1320661111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Randhawa JK, et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (pts) with activating FLT3 mutations [online] 2014 https://ash.confex.com/ash/2014/webprogram/Paper74499.html.
- 100.Brandwein JM, et al. A phase I/II study of imatinib plus reinduction therapy for c-kit-positive relapsed/refractory acute myeloid leukemia: inhibition of Akt activation correlates with complete response. Leukemia. 2011;25:945–952. doi: 10.1038/leu.2011.34. [DOI] [PubMed] [Google Scholar]
- 101.Chevallier P, et al. A phase II trial of high-dose imatinib mesylate for relapsed or refractory c-kit positive and Bcr-Abl negative acute myeloid leukaemia: the AFR-15 trial. Leuk Res. 2009;33:1124–1126. doi: 10.1016/j.leukres.2008.09.030. [DOI] [PubMed] [Google Scholar]
- 102.Advani AS, et al. A phase 1 study of imatinib mesylate in combination with cytarabine and daunorubicin for c-kit positive relapsed acute myeloid leukemia. Leuk Res. 2010;34:1622–1626. doi: 10.1016/j.leukres.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 103.Heidel F, et al. Results of a multicenter Phase II trial for older patients with c-Kit-positive acute myeloid leukemia (AML) and high-risk myelodysplastic syndrome (HR-MDS) using low-dose Ara-C and Imatinib. Cancer. 2007;109:907–914. doi: 10.1002/cncr.22471. [DOI] [PubMed] [Google Scholar]
- 104.Wardelmann E, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12:1743–1749. doi: 10.1158/1078-0432.CCR-05-1211. [DOI] [PubMed] [Google Scholar]
- 105.Hsueh YS, et al. Autophagy is involved in endogenous and NVP-AUY922-induced KIT degradation in gastrointestinal stromal tumors. Autophagy. 2013;9:220–233. doi: 10.4161/auto.22802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Boissel N, et al. Dasatinib in high-risk core binding factor acute myeloid leukemia in first complete remission: a French Acute Myeloid Leukemia Intergroup trial. Haematologica. 2015;100:780–785. doi: 10.3324/haematol.2014.114884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Levine RL, et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106:3377–3379. doi: 10.1182/blood-2005-05-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eghtedar A, et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood. 2012;119:4614–4618. doi: 10.1182/blood-2011-12-400051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pemmaraju N, et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2015;15:171–176. doi: 10.1016/j.clml.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.US National Library of Medicine. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT02076191?term=NCT02076191&rank=1.
- 111.Rowinsky EK, Windle JJ, Von Hoff DD. Ras protein farnesyltransferase: a strategic target for anticancer therapeutic development. J Clin Oncol. 1999;17:3631–3652. doi: 10.1200/JCO.1999.17.11.3631. [DOI] [PubMed] [Google Scholar]
- 112.Lancet JE, et al. A phase 2 study of the farnesyltransferase inhibitor tipifarnib in poor-risk and elderly patients with previously untreated acute myelogenous leukemia. Blood. 2007;109:1387–1394. doi: 10.1182/blood-2006-04-014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Erba HP, et al. Four different regimens of farnesyltransferase inhibitor tipifarnib in older, untreated acute myeloid leukemia patients: North American Intergroup Phase II study SWOG S0432. Leukemia Res. 2014;38:329–333. doi: 10.1016/j.leukres.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Posch C, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci USA. 2013;110:4015–4020. doi: 10.1073/pnas.1216013110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Johnson DB, Smalley KS, Sosman JA. Molecular pathways: targeting NRAS in melanoma and acute myelogenous leukemia. Clin Cancer Res. 2014;20:4186–4192. doi: 10.1158/1078-0432.CCR-13-3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.US National Library of Medicine. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT01907815?term=NCT01907815&rank=1.
- 117.Daigle SR, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–1025. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Daigle SR, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stein EM, et al. The DOT1L inhibitor EPZ-5676: safety and activity in relapsed/refractory patients with MLL-rearranged leukemia [online] 2014 https://ash.confex.com/ash/2014/webprogram/Paper70025.html.
- 120.Lund K, Adams PD, Copland M. EZH2 in normal and malignant hematopoiesis. Leukemia. 2014;28:44–49. doi: 10.1038/leu.2013.288. [DOI] [PubMed] [Google Scholar]
- 121.McCabe MT, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 122.Ohgami RS, et al. Next-generation sequencing of acute myeloid leukemia identifies the significance of TP53, U2AF1, ASXL1, and TET2 mutations. Mod Pathol. 2015;28:706–714. doi: 10.1038/modpathol.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sinha S, et al. Mutant WT1 is associated with DNA hypermethylation of PRC2 targets in AML and responds to EZH2 inhibition. Blood. 2015;125:316–326. doi: 10.1182/blood-2014-03-566018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Valent P, Zuber J. BRD4: a BET(ter) target for the treatment of AML? Cell Cycle. 2014;13:689–690. doi: 10.4161/cc.27859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zuber J, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dawson MA, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. 2014;28:311–320. doi: 10.1038/leu.2013.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dombret H, et al. A phase 1 study of the BET-bromodomain inhibitor OTX015 in patients with advanced acute leukemia [online] 2014 https://ash.confex.com/ash/2014/webprogram/Paper70684.html.
- 128.Grassadonia A, et al. Role of hydroxamate-based histone deacetylase inhibitors (Hb-HDACIs) in the treatment of solid malignancies. Cancers. 2013;5:919–942. doi: 10.3390/cancers5030919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Silva G, Cardoso BA, Belo H, Almeida AM. Vorinostat induces apoptosis and differentiation in myeloid malignancies: genetic and molecular mechanisms. PLoS ONE. 2013;8:e53766. doi: 10.1371/journal.pone.0053766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schaefer EW, et al. A phase 2 study of vorinostat in acute myeloid leukemia. Haematologica. 2009;94:1375–1382. doi: 10.3324/haematol.2009.009217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Walter RB, et al. Phase II trial of vorinostat and gemtuzumab ozogamicin as induction and post-remission therapy in older adults with previously untreated acute myeloid leukemia. Haematologica. 2012;97:739–742. doi: 10.3324/haematol.2011.055822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.U.S. Food and Drug Administration. Mylotarg (gemtuzumab ozogamicin): market withdrawal [online] 2010 http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm216458.htm.
- 133.Ravandi F, et al. Gemtuzumab ozogamicin: time to resurrect? J Clin Oncol. 2012;30:3921–3923. doi: 10.1200/JCO.2012.43.0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hills RK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15:986–996. doi: 10.1016/S1470-2045(14)70281-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.US National Library of Medicine. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT00948064?term=NCT00948064&rank=1.
- 136.Tan P, et al. Dual epigenetic targeting with panobinostat and azacitidine in acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood Cancer J. 2014;4:e170. doi: 10.1038/bcj.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.US National Library of Medicine. ClinicalTrials.gov [online] 2015 https://clinicaltrials.gov/ct2/show/NCT01912274?term=NCT01912274&rank=1.
- 138.Novotny-Diermayr V, et al. The oral HDAC inhibitor pracinostat (SB939) is efficacious and synergistic with the JAK2 inhibitor pacritinib (SB1518) in preclinical models of AML. Blood Cancer J. 2012;2:e69. doi: 10.1038/bcj.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bertoli S, et al. Time from diagnosis to intensive chemotherapy initiation does not adversely impact the outcome of patients with acute myeloid leukemia. Blood. 2013;121:2618–2626. doi: 10.1182/blood-2012-09-454553. [DOI] [PubMed] [Google Scholar]
- 140.Sekeres MA, et al. Time from diagnosis to treatment initiation predicts survival in younger, but not older, acute myeloid leukemia patients. Blood. 2009;113:28–36. doi: 10.1182/blood-2008-05-157065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hughes AE, et al. Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing. PLoS Genet. 2014;10:e1004462. doi: 10.1371/journal.pgen.1004462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Walter MJ, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Steudel C, et al. Comparative analysis of MLL partial tandem duplication and FLT3 internal tandem duplication mutations in 956 adult patients with acute myeloid leukemia. Genes Chromosomes Cancer. 2003;37:237–251. doi: 10.1002/gcc.10219. [DOI] [PubMed] [Google Scholar]
- 146.Marcucci G, Haferlach T, Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–486. doi: 10.1200/JCO.2010.30.2554. [DOI] [PubMed] [Google Scholar]
- 147.Schneider F, et al. Age-dependent frequencies of NPM1 mutations and FLT3-ITD in patients with normal karyotype AML (NK-AML) Ann Hematol. 2012;91:9–18. doi: 10.1007/s00277-011-1280-6. [DOI] [PubMed] [Google Scholar]
- 148.Ostronoff F, et al. Prognostic significance of NPM1 mutations in the absence of FLT3-internal tandem duplication in older patients with acute myeloid leukemia: a SWOG and UK National Cancer Research Institute/Medical Research Council report. J Clin Oncol. 2015;33:1157–1164. doi: 10.1200/JCO.2014.58.0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gaidzik VI, et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML Study Group (AMLSG) Blood. 2013;121:4769–4777. doi: 10.1182/blood-2012-10-461624. [DOI] [PubMed] [Google Scholar]
- 150.Thol F, et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol. 2011;29:2889–2896. doi: 10.1200/JCO.2011.35.4894. [DOI] [PubMed] [Google Scholar]
- 151.Ribeiro AF, et al. Mutant DNMT3A: a marker of poor prognosis in acute myeloid leukemia. Blood. 2012;119:5824–5831. doi: 10.1182/blood-2011-07-367961. [DOI] [PubMed] [Google Scholar]
- 152.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Link DC, et al. Distinct patterns of mutations occurring in de novo AML versus AML arising in the setting of severe congenital neutropenia. Blood. 2007;110:1648–1655. doi: 10.1182/blood-2007-03-081216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Carbuccia N, et al. Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia. 2010;24:469–473. doi: 10.1038/leu.2009.218. [DOI] [PubMed] [Google Scholar]
- 155.Schnittger S, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 2013;27:82–91. doi: 10.1038/leu.2012.262. [DOI] [PubMed] [Google Scholar]
- 156.Paschka P, et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: a study by the German–Austrian Acute Myeloid Leukemia Study Group. Haematologica. 2015;100:324–330. doi: 10.3324/haematol.2014.114157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Metzeler KH, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN favorable genetic category. Blood. 2011;118:6920–6929. doi: 10.1182/blood-2011-08-368225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dufour A, et al. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol. 2010;28:570–577. doi: 10.1200/JCO.2008.21.6010. [DOI] [PubMed] [Google Scholar]
- 159.Taskesen E, et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood. 2011;117:2469–2475. doi: 10.1182/blood-2010-09-307280. [DOI] [PubMed] [Google Scholar]
- 160.Metzeler KH, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Weissmann S, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26:934–942. doi: 10.1038/leu.2011.326. [DOI] [PubMed] [Google Scholar]
- 162.Hou HA, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood. 2010;115:5222–5231. doi: 10.1182/blood-2009-12-259390. [DOI] [PubMed] [Google Scholar]
- 163.Paschka P, et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2008;26:4595–4602. doi: 10.1200/JCO.2007.15.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yamaguchi S, et al. IDH1 and IDH2 mutations confer an adverse effect in patients with acute myeloid leukemia lacking the NPM1 mutation. Eur J Haematol. 2014;92:471–477. doi: 10.1111/ejh.12271. [DOI] [PubMed] [Google Scholar]
- 165.Paschka P, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–3643. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 166.Mendler JH, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J Clin Oncol. 2012;30:3109–3118. doi: 10.1200/JCO.2011.40.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Van Vlierberghe P, et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia. 2011;25:130–134. doi: 10.1038/leu.2010.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hou HA, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015;5:e331. doi: 10.1038/bcj.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang X, et al. EZH2 mutations are related to low blast percentage in bone marrow and-7/del(7q) in de novo acute myeloid leukemia. PLoS ONE. 2013;8:e61341. doi: 10.1371/journal.pone.0061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lee JW, et al. The JAK2 V617F mutation in de novo acute myelogenous leukemias. Oncogene. 2006;25:1434–1436. doi: 10.1038/sj.onc.1209163. [DOI] [PubMed] [Google Scholar]