

Abstract
This communication describes a method for the nucleophilic radiofluorination of electron-rich arenes. The reaction involves the initial C(sp2)–H functionalization of an electron-rich arene with MesI(OH)OTs to form a (mesityl)(aryl)iodonium salt. This salt is then used in situ in a Cu-mediated radiofluorination with [18F]KF. This approach leverages the stability and availability of electron-rich arene starting materials to enable mild late-stage radiofluorination of toluene, anisole, aniline, pyrrole, and thiophene derivatives. The radiofluorination has been automated to access a 41 mCi dose of an 18F-labeled nimesulide derivative in high (2800 ± 700 Ci/mmol) specific activity.
The development of methods for the nucleophilic radiofluorination of electron-rich (hetero)arene precursors represents a long-standing challenge for the radiochemistry community.1−3 Most existing routes to 18F-labeled electron-rich aromatics are limited by the requirement for prefunctionalization of the labeling site with, for example, a boron, tin, hypervalent iodine, or transition-metal substituent.3,4 These prefunctionalized precursors are often not commercially available and must be accessed via multistep synthesis and purification sequences.3a−3c,3e−3k Furthermore, a number of radiofluorination precursors (e.g., hypervalent iodine reagents and Ni/Pd complexes) have limited shelf lives, which precludes widespread clinical applications.5 Overall, methods for the direct nucleophilic radiofluorination of arene C–H bonds would help to address these challenges.6
Several methods have been reported for the electrophilic C–H radiofluorination of electron-rich arenes (Scheme 1A).1c,1f These involve the initial generation of an 18F+ source (e.g., [18F]F2 or [18F]Selectfluor) and subsequent electrophilic aromatic substitution (SEAr) between the 18F+ reagent and the arene substrate. This approach eliminates the need for prefunctionalization. However, the requirement for 18F+ reagents leads to three key limitations. First, 18F+ reagents afford radiofluorinated products with low specific activity due to the need to use carrier [19F]F2 gas for their synthesis.7 Second, the production of 18F+ sources requires specialized facilities.8 Third, the high reactivity of electrophilic fluorinating reagents often results in poor site and chemoselectivity.9
Scheme 1. Strategies for the C–H Radiofluorination of Electron-Rich Arenes.
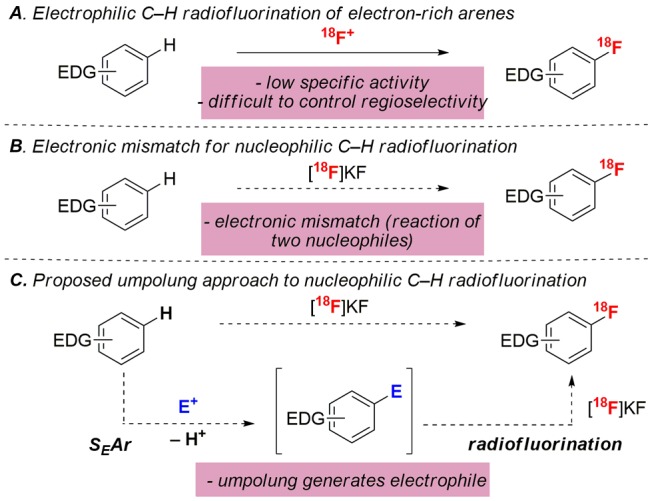
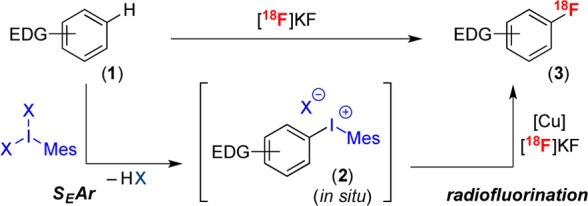
These limitations could be addressed through the development of a nucleophilic radiofluorination of (hetero)aryl C–H bonds (Scheme 1B), as this would enable the use of easily accessible bench-stable precursors in combination with high specific activity [18F]KF. The key challenge for this approach is the electronic mismatch between nucleophilic [18F]KF and the nucleophilic electron-rich arene substrate (Ar–H). We hypothesized that this could be overcome via an umpolung strategy that would invert the polarity of the arene substrate (Scheme 1C). Specifically, an initial SEAr reaction with an electrophile (E+) would functionalize the C–H bond and generate an electrophilic coupling partner (Ar–E).10 Ar–E could then undergo in situ nucleophilic radiofluorination with [18F]KF. We demonstrate herein the realization of this approach using IIII-based electrophiles. This enables the selective radiofluorination of electron-rich aromatics, including toluene, anisole, aniline, pyrrole, and thiophene derivatives. Furthermore, it can be scaled to automated radiosynthesis modules and used to access high specific activity 18F-labeled products.
The approach in Scheme 1C is predicated on the identification of an electrophile (E+) that (i) undergoes site-selective SEAr with diverse electron-rich (hetero)arenes and (ii) generates an intermediate (Ar–E) that can participate in a nucleophilic radiofluorination. Notably, it is not critical that Ar–E be stable; indeed, a key advantage of this approach is that Ar–E will be used in situ, thus precluding the need for isolation/storage of this intermediate. On the basis of these considerations, we selected IIII reagents as E+. As shown in Scheme 2, ArIIIIX2 salts are known to participate in site-selective SEAr reactions with electron-rich (hetero)arenes.11−13 Furthermore, these transformations yield diaryliodonium salts, which we have shown to undergo Cu-catalyzed radiofluorination with [18F]KF.3a,14 While electron-rich diaryliodonium salts are notoriously unstable,5a,15 this approach offers the advantage that they are generated in situ from stable and readily available C–H starting materials.
Scheme 2. Proposed Use of Hypervalent Iodine Reagents as E+.
Our initial investigations focused on the reaction of anisole (1a) with MesI(OH)OTs (Table 1). These partners were selected because they are well-known to undergo SEAr in the presence of Lewis or Brønsted acid activators16 (MY or HY, respectively, in eq 1) to form a single isomeric product, [4-MeOC6H4-I-Mes]+ (2a). Furthermore, 2a is known to undergo Cu-catalyzed radiofluorination with [18F]KF.3a,13,14,17 There were two key considerations for our choice of aryliodonium electrophile and activator for this one-pot radiofluorination. First, these reagents should not contain nucleophilic counterions (Y) (e.g., carboxylates), as these are susceptible to undesired coupling reactions with the diaryliodonium intermediate 2 (eq 1).18 Second, their byproducts should be compatible with the Cu-mediated radiofluorination step.
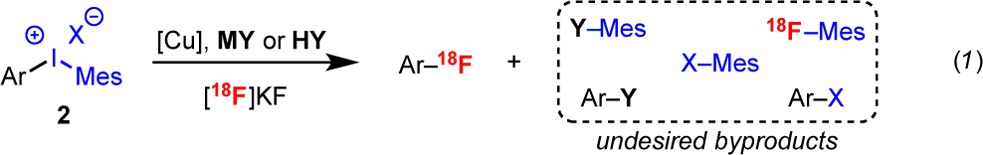 |
1 |
Table 1. Optimization of the Radiofluorination of Anisolea.
| entry | activator | base | RCC 3ab (%) |
|---|---|---|---|
| 1 | TsOH·H2O | 27 ± 4 (n = 4) | |
| 2 | TsOH·H2O | iPr2NEt (20 μmol) | 61 ± 8 (n = 9) |
| 3 | TMSOTf | 5 ± 2 (n = 5) | |
| 4 | TMSOTf | iPr2NEt (10 μmol) | 58 ± 0.4 (n = 3) |
| 5 | TMSOTf | iPr2NEt (20 μmol) | 78 ± 4 (n = 3) |
| 6c,d | TMSOTf | iPr2NEt (20 μmol) | 87 ± 4 (n = 13) |
Conditions: (1) 1a (10 μmol), MesI(OH)OTs (10 μmol), activator (10 μmol), CH2Cl2 (40 μL); (2) (MeCN)4CuOTf (10 μmol), [18F]KF·18-crown-6·K2CO3 complex in DMF (100 μL, 80–1200 μCi), total volume 1.0 mL.
RCC was determined by radio-TLC (average of n runs). The identity of 3a was confirmed by HPLC.
Quinaldic acid (10 μmol) was included in step 2.
98:2 selectivity (3a/[18F]fluoromesitylene) detected by radio-HPLC.
With these criteria in mind, we first examined the use of TsOH as the activator. The two-step procedure involved (1) stirring 1a, MesI(OH)OTs, and TsOH (10 μmol) in CH2Cl2 for 18 h at 25 °C and then (2) adding the reaction mixture to a solution of (MeCN)4CuOTf and [18F]KF·18-crown-6 in DMF and heating at 85 °C for 20 min. The reactions were assayed using radio-TLC and radio-HPLC, and these initial conditions afforded a 27% radiochemical conversion (RCC) to 4-[18F]fluoroanisole (3a) (Table 1, entry 1).3a We hypothesized that the addition of a base in step 2 would quench the residual acid from the diaryliodonium formation step. This was expected to enhance the radiofluorination yield, as Cu-catalyzed nucleophilic fluorinations are known to be acid-sensitive.19 Indeed, a screen of bases revealed that iPr2NEt (20 μmol) increased the RCC to 61 ± 8% (n = 9, entry 2).20,21
Trimethylsilyl trifluoromethanesulfonate (TMSOTf) also proved to be an effective activator for the SEAr reaction. The use of this activator in step 1 coupled with iPr2NEt (20 μmol) in step 2 delivered 4-[18F]fluoroanisole (3a) in 78 ± 4% RCC (n = 3, entry 5). Finally, the evaluation of different additives revealed that the addition of quinaldic acid (10 μmol) to the Cu-mediated radiofluorination under otherwise identical conditions resulted in a RCC of 87 ± 4% (n = 13, entry 6).22 Notably, the RCC of this transformation of 1a to 3a (87%) is comparable to that reported using isolated [4-MeOC6H4-I-Mes]+ as the substrate for Cu-catalyzed radiofluorination (79%).3a
The site-selectivity of the reaction of anisole was determined by radio-HPLC analysis23 via comparison to authentic standards of the three possible isomers (see the Supporting Information for complete details). The 18F-fluorination proceeded to afford the para-isomer with >99:1 selectivity.12,13a−13d This high selectivity is fully consistent with that reported for SEAr reactions between hypervalent iodine reagents and related electron-rich arene substrates.12 Importantly, <2% of [18F]fluoromesitylene was detected under any of the conditions examined.
We next evaluated the scope of this C–H radiofluorination (Figure 1).24 A series of anisoles (3a–f), protected anilines (3g–i), and toluene (3j,k) derivatives reacted under the standard conditions to form 18F-labeled arenes in >80% RCC. The transformation was tolerant of aryl halides (3c and 3o), esters and tertiary amides (3d, 3g, and 3m), sulfonamides (3h, 3o, and 3q), and nitro groups (3q) as well as substitution adjacent to the site of fluorination (3e). Protic functional groups (e.g., alcohols, amines, and amides) were not compatible and were thus protected prior to the reaction.27 The radiofluorination of heterocycles, including thiophene, pyrrole, and uracil, required elevated temperature (105 °C) to afford 3l–n. Notably, the radiofluorination of these heterocycles proceeded with lower selectivity, and significant quantities of [18F]fluoromesitylene were observed in these systems. However, in all cases, this byproduct was readily separable by HPLC.25,26 The formation of 2-[18F]fluoro-3-methylthiophene (3l) in 25% RCC is particularly noteworthy, as thiophenes are particularly challenging to radiofluorinate using other methods.1d−1f,28 Indeed, thiophene is typically used as the inert ligand on diaryliodonium salts to direct radiofluorination to the other arene ligand.1d−1f,29
Figure 1.

Cu-mediated C(sp2)–H radiofluorination of electron-rich arenes. Conditions: (1) 1 (10 μmol), MesI(OH)OTs (10 μmol), TMSOTf (10 μmol), CH2Cl2 (40 μL); (2) (MeCN)4CuOTf (10 μmol), quinaldic acid (10 μmol), iPr2NEt (20 μmol), [18F]KF·18-crown-6·K2CO3 complex in DMF (100 μL, 80–1200 μCi), total volume 1.0 mL. Reported radiochemical conversions were measured by radio-TLC (average of n runs) and were corrected for presence of 4 (ratio of 3/4 determined by radio-HPLC). The identity of each product was confirmed by radio-HPLC. Key: (a) 105 °C; (b) TMSOTf (5 μmol).25,26
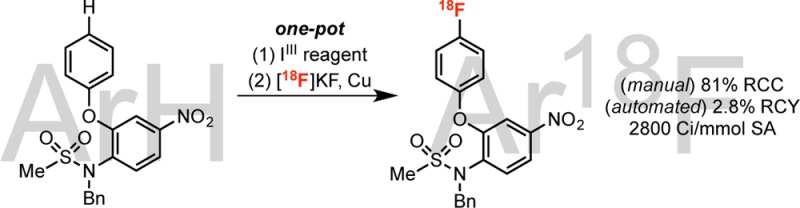
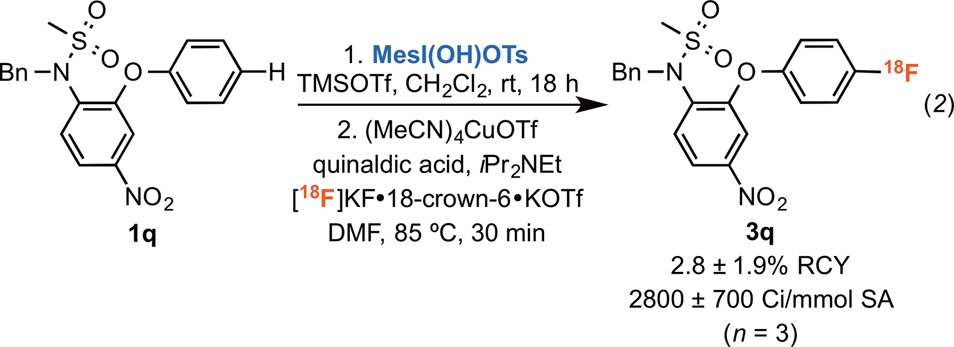
We next examined the late-stage C–H radiofluorination of fragments that appear in pharmaceuticals. In one example, the dibenzothiazepine fragment of tianeptine (Coaxil), underwent radiofluorination to yield 3o in 18% RCC. Additionally, both O-Bn-protected propofol (an anesthetic and sedative) and N-Bn-protected nimesulide (a nonsteroidal anti-inflammatory) reacted under our standard conditions to yield 18F-analogues 3p and 3q in 87% and 81% RCC, respectively.
 |
2 |
A final set of experiments focused on automation of this reaction on a TRACERLab FXFN radiosynthesis module used for the preparation of clinical radiotracers.2b Initial automated studies were conducted for anisole (1a) using ∼1.5 Ci of [18F]fluoride ([18F]KF·18-crown-6·KOTf) and afforded 4-[18F]fluoroanisole (3a) in 56 ± 4% RCC and a specific activity of 2700 ± 1900 Ci/mmol (n = 4).20,30 We also automated the radiofluorination of N-Bn-protected nimesulide without any further optimization. Radiolabeling was conducted using ∼1.5 Ci of [18F]fluoride, and the product was purified via semipreparative HPLC to yield a formulated 41 ± 31 mCi dose of 3q in 2.8 ± 1.9% non-decay-corrected radiochemical yield (RCY) with 2800 ± 700 Ci/mmol specific activity (n = 3, eq 2). This proof-of-concept demonstration provides a foundation for the application of this method to the synthesis of other radiotracer targets.
In summary, this report describes a method for the nucleophilic radiofluorination of electron-rich arene C–H bonds that enables the late-stage C–H functionalization of (hetero)aromatic scaffolds. This approach eliminates the need to isolate/store prefunctionalized starting materials, instead leveraging the bench stability and availability of electron-rich aromatic substrates. Current efforts are focused on the application of this C(sp2)–H to C–18F radiofluorination chemistry to clinically relevant targets.
Acknowledgments
This work was supported by Merck Sharp & Dohme, NIH (R01EB021155) and DOE (DE-SC0012484). We thank Drs. Idriss Bennacef, Thomas Graham, Eric Hostetler, Wenping Li, James Mulhearn, and Petr Vachal from Merck for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.7b01902.
Experimental and spectral data for all new compounds and all reactions (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Ametamey S. M.; Honer M.; Schubiger P. A. Chem. Rev. 2008, 108, 1501. 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]; b Cai L.; Lu S.; Pike V. W. Eur. J. Org. Chem. 2008, 2008, 2853. 10.1002/ejoc.200800114. [DOI] [Google Scholar]; c Miller P. W.; Long N. J.; Vilar R.; Gee A. D. Angew. Chem., Int. Ed. 2008, 47, 8998. 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]; d Brooks A. F.; Topczewski J. J.; Ichiishi N.; Sanford M. S.; Scott P. J. H. Chem. Sci. 2014, 5, 4545. 10.1039/C4SC02099E. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Campbell M. G.; Ritter T. Chem. Rev. 2015, 115, 612. 10.1021/cr500366b. [DOI] [PubMed] [Google Scholar]; f Preshlock S.; Tredwell M.; Gouverneur V. Chem. Rev. 2016, 116, 719. 10.1021/acs.chemrev.5b00493. [DOI] [PubMed] [Google Scholar]
- a Sanford M. S.; Scott P. J. H. ACS Cent. Sci. 2016, 2, 128. 10.1021/acscentsci.6b00061. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Campbell M. G.; Mercier J.; Genicot C.; Gouverneur V.; Hooker J. M.; Ritter T. Nat. Chem. 2017, 9, 1. 10.1038/nchem.2693. [DOI] [PubMed] [Google Scholar]
- Recent examples of the nucleophilic radiofluorination of electron-rich arenes:; a Ichiishi N.; Brooks A. F.; Topczewski J. J.; Rodnick M. E.; Sanford M. S.; Scott P. J. H. Org. Lett. 2014, 16, 3224. 10.1021/ol501243g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Makaravage K. J.; Brooks A. F.; Mossine A. V.; Sanford M. S.; Scott P. J. H. Org. Lett. 2016, 18, 5440. 10.1021/acs.orglett.6b02911. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lee E.; Kamlet A. S.; Powers D. C.; Neumann C. N.; Boursalian G. B.; Furuya T.; Choi D. C.; Hooker J. M.; Ritter T. Science 2011, 334, 639. 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lee E.; Hooker J. M.; Ritter T. J. Am. Chem. Soc. 2012, 134, 17456. 10.1021/ja3084797. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hoover A. J.; Lazari M.; Ren H.; Narayanam M. K.; Murphy J. M.; van Dam R. M.; Hooker J. M.; Ritter T. Organometallics 2016, 35, 1008. 10.1021/acs.organomet.6b00059. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Tredwell M.; Preshlock S. M.; Taylor N. J.; Gruber S.; Huiban M.; Passchier J.; Mercier J.; Génicot C.; Gouverneur V. Angew. Chem., Int. Ed. 2014, 53, 7751. 10.1002/anie.201404436. [DOI] [PubMed] [Google Scholar]; g Preshlock S.; Calderwood S.; Verhoog S.; Tredwell M.; Huiban M.; Hienzsch A.; Gruber S.; Wilson T. C.; Taylor N. J.; Cailly T.; Schedler M.; Collier T. L.; Passchier J.; Smits R.; Mollitor J.; Hoepping A.; Mueller M.; Genicot C.; Mercier J.; Gouverneur V. Chem. Commun. 2016, 52, 8361. 10.1039/C6CC03295H. [DOI] [PubMed] [Google Scholar]; h Rotstein B. H.; Stephenson N. A.; Vasdev N.; Liang S. H. Nat. Commun. 2014, 5, 4365. 10.1038/ncomms5365. [DOI] [PubMed] [Google Scholar]; i Rotstein B. H.; Wang L.; Liu R. Y.; Patteson J.; Kwan E. E.; Vasdev N.; Liang S. H. Chem. Sci. 2016, 7, 4407. 10.1039/C6SC00197A. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Zlatopolskiy B. D.; Zischler J.; Krapf P.; Zarrad F.; Urusova E. A.; Kordys E.; Endepols H.; Neumaier B. Chem. - Eur. J. 2015, 21, 5972. 10.1002/chem.201405586. [DOI] [PubMed] [Google Scholar]; k Haskali M. B.; Telu S.; Lee Y.-S.; Morse C. L.; Lu S.; Pike V. W. J. Org. Chem. 2016, 81, 297. 10.1021/acs.joc.5b02332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Phelps M. E. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 9226. 10.1073/pnas.97.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Müller K.; Faeh C.; Diederich F. Science 2007, 317, 1881. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; c Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Chem. Soc. Rev. 2008, 37, 320. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- a Seidl T. L.; Sundalam S. K.; McCullough B.; Stuart D. R. J. Org. Chem. 2016, 81, 1998. 10.1021/acs.joc.5b02833. [DOI] [PubMed] [Google Scholar]; b Moon B. S.; Kil H. S.; Park J. H.; Kim J. S.; Park J.; Chi D. Y.; Lee B. C.; Kim S. E. Org. Biomol. Chem. 2011, 9, 8346. 10.1039/c1ob06277h. [DOI] [PubMed] [Google Scholar]
- Recent examples of C–H radiofluorination:; a Huang X.; Liu W.; Ren H.; Neelamegam R.; Hooker J. M.; Groves J. T. J. Am. Chem. Soc. 2014, 136, 6842. 10.1021/ja5039819. [DOI] [PubMed] [Google Scholar]; b Nodwell M. B.; Yang H.; Čolović M.; Yuan Z.; Merkens H.; Martin R. E.; Bénard F.; Schaffer P.; Britton R. J. Am. Chem. Soc. 2017, 139, 3595. 10.1021/jacs.6b11533. [DOI] [PubMed] [Google Scholar]
- Bergman J.; Solin O. Nucl. Med. Biol. 1997, 24, 677. 10.1016/S0969-8051(97)00078-4. [DOI] [PubMed] [Google Scholar]
- Channing M. A.; Musachio J. L.; Kusmierz J. J.. Synthesis of 6-[18F]Fluorodopamine (6-[18F]FDA). In Radiochemical Syntheses; Scott P. J. H., Hockley B. G., Eds.; John Wiley & Sons: Hoboken, 2012; Vol. 1, pp 125–138. [Google Scholar]
- Coenen H. H.Fluorine-18 labeling methods: features and possibilities of basic reactions. In PET Chemistry. Ernst Schering Research Foundation Workshop; Schubiger P. A., Lehmann L., Friebe M., Eds; Springer: Berlin, 2007; Vol. 64, pp 15–50. [DOI] [PubMed] [Google Scholar]
- Taylor R.Electrophilic Aromatic Substitution; John Wiley & Sons: Chichester, 1990. [Google Scholar]
- Reviews on the synthesis of diaryliodonium salts:; a Zhdankin V. V.; Stang P. J. Chem. Rev. 2008, 108, 5299. 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Merritt E. A.; Olofsson B. Angew. Chem., Int. Ed. 2009, 48, 9052. 10.1002/anie.200904689. [DOI] [PubMed] [Google Scholar]; c Dohi T.; Yamaoka N.; Kita Y. Tetrahedron 2010, 66, 5775. 10.1016/j.tet.2010.04.116. [DOI] [Google Scholar]; d Yusubov M. S.; Svitich D. Y.; Larkina M. S.; Zhdankin V. V. ARKIVOC 2013, 364. [Google Scholar]
- Examples of the synthesis of diaryliodonium salts via SEAr:; a Kitamura T.; Matsuyuki J.-i.; Nagata K.; Furuki R.; Taniguchi H. Synthesis 1992, 1992, 945. 10.1055/s-1992-26272. [DOI] [Google Scholar]; b Kitamura T.; Matsuyuki J.-i.; Taniguchi H. Synthesis 1994, 1994, 147. 10.1055/s-1994-25423. [DOI] [Google Scholar]; c Chun J.-H.; Pike V. W. J. Org. Chem. 2012, 77, 1931. 10.1021/jo202517v. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bielawski M.; Malmgren J.; Pardo L. M.; Wikmark Y.; Olofsson B. ChemistryOpen 2014, 3, 19. 10.1002/open.201300042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Examples of the in situ generation and subsequent nucleophilic functionalization of diaryliodonium reagents:; a Lubriks D.; Sokolovs I.; Suna E. J. Am. Chem. Soc. 2012, 134, 15436. 10.1021/ja305574k. [DOI] [PubMed] [Google Scholar]; b Sokolovs I.; Lubriks D.; Suna E. J. Am. Chem. Soc. 2014, 136, 6920. 10.1021/ja502174d. [DOI] [PubMed] [Google Scholar]; c Berzina B.; Sokolovs I.; Suna E. ACS Catal. 2015, 5, 7008. 10.1021/acscatal.5b01992. [DOI] [Google Scholar]; d Sokolovs I.; Suna E. J. Org. Chem. 2016, 81, 371. 10.1021/acs.joc.5b02728. [DOI] [PubMed] [Google Scholar]; e Reitti M.; Villo P.; Olofsson B. Angew. Chem. 2016, 128, 9074. 10.1002/ange.201603175. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Kita Y.; Morimoto K.; Ito M.; Ogawa C.; Goto A.; Dohi T. J. Am. Chem. Soc. 2009, 131, 1668. 10.1021/ja808940n. [DOI] [PubMed] [Google Scholar]; g Morimoto K.; Yamaoka N.; Ogawa C.; Nakae T.; Fujioka H.; Dohi T.; Kita Y. Org. Lett. 2010, 12, 3804. 10.1021/ol101498r. [DOI] [PubMed] [Google Scholar]
- a Ichiishi N.; Canty A. J.; Yates B. F.; Sanford M. S. Org. Lett. 2013, 15, 5134. 10.1021/ol4025716. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ichiishi N.; Canty A. J.; Yates B. F.; Sanford M. S. Organometallics 2014, 33, 5525. 10.1021/om5007903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crivello J. V.; Lam J. H. W. Macromolecules 1977, 10, 1307. 10.1021/ma60060a028. [DOI] [Google Scholar]
- Izquierdo S.; Essafi S.; del Rosal I.; Vidossich P.; Pleixats R.; Vallribera A.; Ujaque G.; Lledós A.; Shafir A. J. Am. Chem. Soc. 2016, 138, 12747. 10.1021/jacs.6b07999. [DOI] [PubMed] [Google Scholar]
- a Phipps R. J.; Grimster N. P.; Gaunt M. J. J. Am. Chem. Soc. 2008, 130, 8172. 10.1021/ja801767s. [DOI] [PubMed] [Google Scholar]; b Phipps R. J.; Gaunt M. J. Science 2009, 323, 1593. 10.1126/science.1169975. [DOI] [PubMed] [Google Scholar]; c Phipps R. J.; McMurray L.; Ritter S.; Duong H. A.; Gaunt M. J. J. Am. Chem. Soc. 2012, 134, 10773. 10.1021/ja3039807. [DOI] [PubMed] [Google Scholar]
- Schimler S. D.; Sanford M. S. Synlett 2016, 27, 2279. 10.1055/s-0035-1562529. [DOI] [Google Scholar]
- Fier P. S.; Hartwig J. F. J. Am. Chem. Soc. 2012, 134, 10795. 10.1021/ja304410x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For experimental details, see the Supporting Information.
- These conditions and others employing trifluoromethanesulfonic acid or trifluoroacetic anhydride for iodonium formation afforded low RCCs and/or did not prove to be general.
- The RCC of isolated [4-MeOC6H4-I-Mes]OTf (2a) was also improved to 85% with the addition of quinalidic acid (10 μmol) and iPr2NEt (10 μmol). At this time, the role of quinaldic acid in the radiofluorination is unknown.
- The regioselectivity is set in the SEAr reaction and was confirmed by 1H NMR spectroscopic analysis of the iodonium salts generated under these conditions. See the SI for details.
- For consistency, step 1 was allowed to run for 18 h. However, in many cases, the SEAr reaction was complete after a shorter time.
- In select examples, decreasing the amount of TMSOTf in the first step helped to eliminate minor 18F-labeled byproducts believed to originate from residual TMSOTf.
- The RCCs of 3l–o were lower due to formation of larger amounts of [18F]fluoromesitylene and other unidentified [18F]byproducts. See the Supporting Information for details.
- A summary of unreactive or noncompatible arene substrates is available in the Supporting Information.
- Kilbourn M. R. Nucl. Med. Biol. 1989, 16, 681. 10.1016/0883-2897(89)90138-4. [DOI] [Google Scholar]
- In general, selectivity for 3 over 4 was greater than 95:5 in favor of 3. The only exceptions were 3d, 3l, 3m, 3n, and 3o. See the Supporting Information for details.
- [18F]KF·18-crown-6·K2CO3 performed poorly as a [18F]fluoride source for the automated reactions. Improved reactivity was obtained using [18F]KF·18-crown-6·KOTf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.