

Abstract
It is well known that excessive production of reactive oxygen and nitrogen species is linked to the development of oxidative stress-driven disorders. In particular, nitric oxide (NO) and superoxide (O2−•) play critical roles in many physiological and pathological processes. This paper reports the use of 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM DA) and MitoSOX Red (or Mito-HE) in conjunction with microchip electrophoresis and laser-induced fluorescence detection for the simultaneous detection of NO and O2−• in RAW 264.7 macrophage cell lysates following different stimulation procedures. Cell stimulations were performed in the presence and absence of cytosolic (diethyldithiocarbamate) and mitochondrial (2-methoxyestradiol) superoxide dismutase (SOD) inhibitors. The NO/O2−• ratios in macrophage cell lysates under physiological and pro-inflammatory conditions were determined. The ratios of NO to O2−• were 0.60 ± 0.07 for unstimulated cells pre-treated with SOD inhibitors, 1.08 ± 0.06 for unstimulated cells in the absence of SOD inhibitors, and 3.14 ± 0.13 for stimulated cells. The effect of carnosine (antioxidant) or Ca2+ (intracellular messenger) on the NO/O2−• ratio was also investigated.
Keywords: Bioanalytical methods, Inflammation, Macrophages, Microchip electrophoresis, Nitric oxide, Superoxide
Graphical abstract
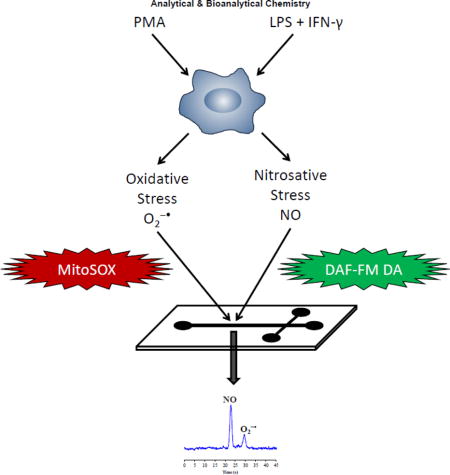
Introduction
Reactive nitrogen and oxygen species (RNOS) play a variety of roles in biological systems [1,2]. In mammals, specialized enzymes, such as NADPH-oxidase and nitric oxide synthase (NOS), are involved in the production of RNOS as part of the immune system. Excessive production of these reactive species can lead to oxidative and nitrosative stress, resulting in damage to important biomolecules [3]. Moreover, this damage has been linked to neurodegenerative diseases, cardiovascular disorders, and cancer [4–6].
Nitric oxide (NO) is a water-soluble, free radical gas that acts as an intracellular and intercellular messenger in all vertebrates [7]. NO is able to modulate cytokine and chemokine release [8] during the immune response [9] and plays important roles in both the cardiovascular [10] and nervous systems [11]. NO has a half-life of 3 to 6 s in vivo and is synthesized by a complex family of NOS enzymes through the conversion of L-arginine to L-citrulline [7].
The superoxide anion (O2−•) is a reactive oxygen species (ROS) naturally produced in the human body when oxygen (O2) gains an excess electron during various enzymatic reactions in mitochondria; it is involved in many physiological and pathological signaling processes [12]. An overproduction of O2−• can lead to cell death due to oxidative damage to DNA, lipids, carbohydrates, and proteins [13]. In living organisms, the intracellular enzyme superoxide dismutase (SOD) protects the cell from the deleterious effects of O2−• by catalyzing the conversion of O2−• to O2 and hydrogen peroxide [14].
Simultaneous production of intracellular NO and O2−• can lead to the formation of peroxynitrite [15]. This dangerous molecule has the ability to nitrate, nitrosylate, and oxidize proteins, DNA, and lipids, inhibiting their functions and causing cytotoxicity within the cell [16]. Additionally, peroxynitrite has been linked to neurodegenerative disorders, cardiovascular disease, and cancer [7]. Therefore, the simultaneous detection of NO and O2−• is necessary in order to obtain a thorough understanding of intracellular nitrosative and oxidative stress.
Macrophages are cells involved in the primary immune defense mechanism that, when activated in vivo under pro-inflammatory conditions, can lead to a higher expression of inducible NOS (iNOS) coupled to the production of a large amount of NO and, thus, RNOS [17,18]. It is well known that a combination of lipopolysaccharides (LPS) and interferon-gamma (IFN-γ) results in the stimulation of macrophages to produce a large amount of NO via iNOS [19]. In addition, high amounts of intracellular O2−• can be generated by incubating macrophages with phorbol 12-myristate 13-acetate (PMA) [20].
A method for the simultaneous detection of O2−• and hydrogen peroxide in stimulated macrophages using microchip electrophoresis with laser-induced fluorescence detection (ME-LIF) was previously reported by Li et al. [21]. Our group has also employed ME-LIF for the determination of the intracellular production of NO in lymphocytes [22], as well as detection of O2−• in macrophage cells [23]. ME has several advantages over conventional methods for the analysis of cultured cells, especially for the detection of RNOS. These advantages include fast separation times, isolation of the intended product from interfering substances, high throughput, and the ability to easily integrate multiple detection platforms. Additionally, ME systems are ideal for single cell analysis because they can be automated and permit on-chip cell manipulation and lysis [24–27].
In the present work, the use of ME-LIF for the simultaneous detection of intracellular NO and O2−• in RAW 264.7 macrophage cells is reported. These RNOS are detected by ME-LIF in the cell lysates of macrophages following incubation of the cells with both 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM DA) and MitoSOX Red. Changes in the NO/O2−• ratio were then investigated under physiological and pro-inflammatory conditions. The resulting method provides an additional tool for understanding the physiopathological role of RNOS in oxidative stress-driven disorders.
Materials and methods
Materials and reagents
Murine RAW 264.7 cells (ATCC® TIB71™), Dulbecco’s Modified Eagle’s Medium (DMEM), phenol red-free DMEM, fetal bovine serum (FBS), and penicillin/streptomycin antibiotic solution were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). L-carnosine, diethyldithiocarbamate (DDC), 2-methoxyestradiol (2-ME), phorbol 12-myristate 13-acetate (PMA), anhydrous dimethyl sulfoxide (DMSO), phosphate-buffered saline (PBS), Trypan blue solution, lipopolysaccharides (LPS), sodium dodecyl sulfate (SDS), calcium chloride, and bovine serum albumin (BSA) were all supplied by Sigma-Aldrich (St. Louis, MO, USA). Sodium hydroxide (NaOH), hydrochloric acid (HCl), 25 mL polystyrene culture flasks, boric acid, ethanol (95%), and C-Chip disposable hemocytometers were obtained from Fisher Scientific (Pittsburgh, PA, USA). Interferon-γ (IFN-γ) was supplied by Calbiochem (Gibbstown, NJ, USA). 4-Amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM DA) and MitoSOX Red were purchased from Life Technologies (Carlsbad, CA, USA). Polyethersulfone (PES) membrane (3 kDa) centrifuge filters were purchased from VWR International (West Chester, PA, USA). Polydimethylsiloxane (PDMS) microdevices were prepared from the Sylgard 184 Elastomer Kit (Ellsworth Adhesives, Germantown, WI, USA). All water used was ultrapure (18.3 MΩ cm) (Milli-Q Synthesis A10, Millipore, Burlington, MA, USA).
Cell culture and preparation
RAW 264.7 macrophage cells were cultured in DMEM containing 10% (v/v) FBS, L- glutamine (2 mM), penicillin (50 IU mL−1), and streptomycin (0.3 mg mL−1). The cells were cultured in 25 cm2 polystyrene culture flasks at a density of 5 × 106 cells/flask, maintained in a humidified environment at 37 °C, 5% CO2, and 95% air, and passaged every 2 or 3 days depending on the cell confluence to avoid overgrowth.
Stimulation protocol for the simultaneous detection of NO and O2−•
The protocol used for cell sample preparation is shown in Figure 1. The day of the experiment, cells were harvested using a cell scraper, counted with a C-Chip disposable hemocytometer, and plated at a density of 1.2 × 107 cells/flask. Stock solutions of 1 mg/mL LPS and 200,000 U/mL IFN-γ were prepared in 10 mM PBS and 10 mM PBS with 0.1% BSA, respectively. Once the cells adhered to the flask surface, they were stimulated to increase the production of NO by diluting the stock solutions to 100 ng/mL LPS and 600 U/mL INF-γ in 5 mL of cell culture medium. Immediately after the stimulation, the macrophages were incubated for 20 h in a humidified environment at 37 °C, 5% CO2, and 95% air. A stock solution of 5 mM DAF-FM DA was prepared in 99% sterile DMSO. After the 20-h incubation with LPS + INF-γ, the medium was replaced with 5 mL of phenol red-free DMEM containing 10 μL of DAF-FM DA for 60 min (10 μM DAF-FM DA final concentration) [22]. A 5 mM MitoSOX Red stock solution was prepared in 99% sterile DMSO [23]. Additionally, a stock solution of 100 mM DDC was prepared in 10 mM PBS and stock solutions of 16.5 mM 2-ME and 1 mg/mL PMA were prepared in DMSO. Each flask of cells containing 5 mL of culture medium was then incubated for 1 h with a combination of the cytosolic SOD inhibitor DDC (1 mM final concentration), mitochondrial SOD inhibitor 2-ME (50 μM final concentration), and MitoSOX Red (10 μM MitoSOX final concentration). Finally, the cells were stimulated with PMA (1 μg/mL final concentration) for 30 min. During the incubation of the macrophages with DAF-FM DA and MitoSOX Red, the flasks were covered with aluminum foil to minimize any photo-bleaching of the dyes.
Fig. 1.
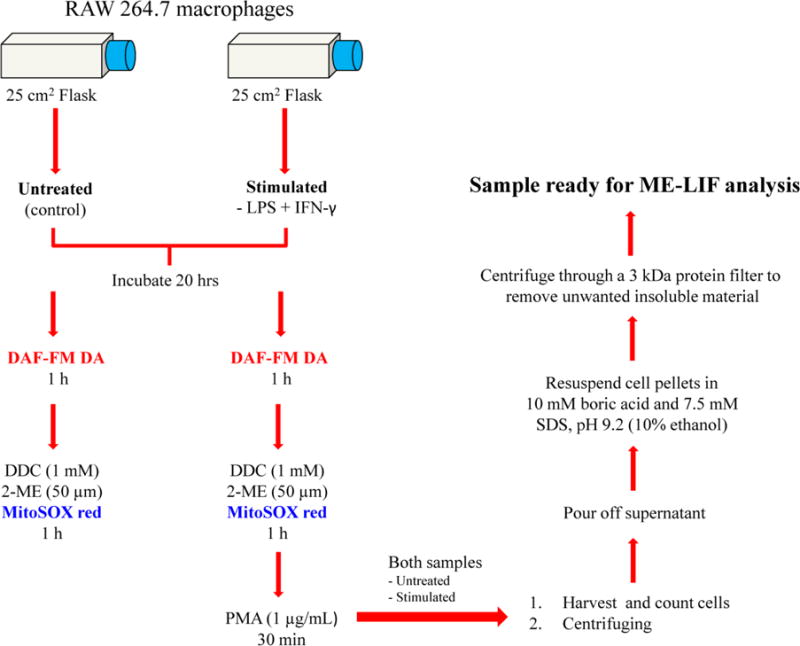
Flowchart of the protocol used for preparation of RAW 264.7 cell lysates
Native untreated cells from the same population were used as a control. These were incubated under the same conditions as the cells above, except that no stimulants were added. Another set of untreated cells was incubated in the presence of both SOD inhibitors (to increase the detectable concentration of O2−•).
At the end of the stimulation process, the cells were harvested, and 100 μL of the cell suspension was removed for cell counting. The suspension was then centrifuged at 1,137 × g for 4 min. The supernatant was removed, and the cell pellet was washed twice with 1 mL of cold 10 mM PBS at pH 7.4. Cells were lysed with 50 μL of pure ethanol. The lysate solution was filtered using a 3 kDa molecular weight cut-off filter with centrifugation at 18,690 × g for 10 min. Then 10 μL of the filtered cell lysates were added to a 90 μL solution of 10 mM boric acid and 7.5 mM SDS at pH 9.2 (10% ethanol final concentration) and immediately analyzed with the microfluidic device. Peak identification of NO or O2−• was accomplished by using the same stimulation protocol, except that the cells were incubated in the presence of DAF-FM DA or MitoSOX Red only.
Alternative stimulation protocol for measuring changes in NO/O2−• ratio
The effect of specific NO or O2−• stimulation procedures was evaluated through the analysis of changes in the NO/O2−• ratio in the macrophage cells. The protocol used for this analysis is the same as that described for the simultaneous detection of both analytes, except that the cells were stimulated with LPS + INF-γ (NO stimulation) or PMA (O2−• stimulation) only.
Effect of pre-treatment with carnosine or calcium on NO/O2−• ratio changes
To investigate changes in the NO/O2−• ratio due to the presence of carnosine or calcium ions, cells were incubated with either carnosine (10 μL of 500 mM stock solution in 10 mM PBS; 1 mM final concentration) 1 h prior to the stimulation with LPS + INF-γ or calcium chloride (25 μL of 200 mM stock solution in DI water; 1 mM final concetration) 1 h prior to the stimulation with PMA. Native untreated (nonstimulated) cells from the same population were incubated under identical conditions, in the presence of carnosine or calcium chloride, and used as controls.
Cell density and viability
Cell density and viability were measured using a C-Chip disposable hemocytometer and Trypan blue exclusion assay, respectively. The cell suspension was diluted either 1:3 or 1:5 (stimulated and untreated, respectively) with 0.4% Trypan blue solution.
Microchip fabrication and instrumental setup
The fabrication of hybrid PDMS-glass microfluidic devices has been described previously [22]. Briefly, a silicon master containing the design of the microchip was fabricated with SU-8 photoresist and soft lithography. A 10:1 (w/w) PDMS pre-polymer to curing agent mixture was degassed in a vacuum desiccator and poured onto the master. The PDMS was cured overnight in an oven at 70 °C. Then the cured PDMS was peeled off the master and 3 mm reservoirs were punched in the substrate with a biopsy punch (Harris Uni-core, Ted Pella, Redding, CA, USA). To make the final microfluidic device, the PDMS substrate was reversibly bonded to a borosilicate glass substrate (Precision Glass and Optics, Santa Ana, CA, USA). For these experiments, a microchip with a simple-T design was employed with a 5 cm separation channel, 0.75 cm side arms, and 40 μm by 15 μm channels throughout.
Prior to operation, the microchip was conditioned with 0.1 M NaOH and run buffer. The run buffer utilized in these experiments consisted of 10 mM boric acid and 7.5 mM SDS at pH 9.2. A separation field was generated with a high voltage power supply (Ultravolt Inc., Ronkonkoma, NY, USA). A 1s gated injection was used for sample introduction. A gate was established by applying +2400 V and +2200 V to the buffer and sample reservoirs, respectively.
For LIF detection, the microchip was placed on the stage of an Eclipse Ti-U inverted microscope (Nikon Instruments Inc., Melville, NY, USA). A 488 nm diode laser (Spectra-Physics, Irvine, CA, USA) was aligned 3.75 cm below the sample/buffer intersection as the excitation source. Fluorescence signals were collected with a photon multiplier tube (Hamamatsu Corporation, Bridgewater, NJ, USA) and amplified with a SR570 low noise preamplifier at 1 μA V−1 (Stanford Research Systems, Sunnyvale, CA). Data were acquired with a D/A converter (National Instruments, Austin, TX, USA) and a homemade LabView program (National Instruments, Austin, TX, USA). Data analysis was accomplished with Origin 8.6 software (OriginLab, Northhampton, MA, USA).
Comparison of the sensitivity of DAF-FM DA and MitoSOX Red probes
The fluorescence quantum yield of DAF-FM is ~0.005, but increases about 160-fold to ~0.81 after reacting with NO [28]. In the case of MitoSOX Red, the literature does not present a uniform view of the fluorescence quantum yield before and after reaction with O2−•. To ensure that the ratio accurately depicts the relative concentration of NO to O2−• in the cell, the response factors for the products of the two probes were determined as described previously in [22] and [23]. The ratio of the slope of the response curve for DAF-FM T to that of OH-MitoE+ was determined to be 1.2. All NO/O2−• ratios were corrected for the difference in sensitivity (quantum yield) between the two products. This provided a more accurate assessment of the relative amounts that are produced. (more details can be found in Fig. S1 in the Electronic Supplementary Material, ESM).
Results and discussion
Optimization of stimulation protocol and electrophoretic separation
Before monitoring the NO/O2−• ratio, it was crucial to ensure that the method would be able to generate separable, measurable, and reproducible signals for both NO and O2−• in complex matrices such as RAW 264.7 macrophage cell lysates. Several factors needed to be considered because of their possible influence on the ability to detect both analytes by ME-LIF. Initially, the cell stimulation protocol was optimized for generation of NO and O2−•. In the first studies, cells were stimulated for 20 h with LPS + IFN-γ, followed by the addition of PMA (500 ng/mL final concentration) and incubation for an additional 60 min. However, this protocol was not optimal in terms of cell viability and NO and O2−• production. The PMA-stimulation time was then decreased from 60 to 30 min and the concentration was doubled to 1 μg/mL. The MitoSOX probe was incubated with cells for 1 h prior to stimulation with PMA. This new protocol was found to be the best for cell viability and NO and O2−• production.
Once the optimal cell stimulation protocol was established, attention was focused on the separation and detection conditions. Our group previously reported the detection of NO using DAF-FM and ME-LIF using a run buffer consisting of 10 mM boric acid and 7.5 mM SDS at pH 9.2 [22]. A separate method was developed for O2−• that employed a similar run buffer but with a lower SDS concentration (3.5 mM) [23]. In order to determine the optimal concentration of SDS needed for the simultaneous detection of NO and O2−•, several SDS concentrations (3.5, 5.5, and 7.5 mM) in combination with 10 mM boric acid at pH 9.2 were investigated as background electrolytes. It was found that 7.5 mM SDS provided the best resolution for DAF-FM-T, 2-OH-MitoE+, and potential interferences.
The previous ME-LIF methods mentioned above used a detection distance of 4.5 cm from the T intersection of the simple T microchip. Therefore, intial experiments in these studies used the same detection distance. However, it was found in these experiments that a distance of 3.5 cm provided a faster separation and better resolution, so it was used for all further studies. The migration times for DAF-FM T and 2-OH-MitoE+ under the different experimental conditions are reported in Table 1. The RSD for migration times was below 5% for both DAF-FM T and 2-OH-MitoE+ within sample type. However, there was a drift to longer migration times with the stimulated samples, which could be due to changes in the sample matrix effects and fouling of the PDMS substrate. The final optimized method including sample preparation is described in detail in the experimental section.
Table 1.
Migration times for DAF-FM T and 2-OH-MitoE+ for untreated and stimulated cells
| Migration time | ||
|---|---|---|
| Cell treatment | DAF-FM T | 2-OH-MitoE+ |
| Untreated | 21.82 ± 0.11 | 27.87 ± 0.15 |
| Unstimulated + SOD inhibitors | 22.81 ± 0.59 | 29.41 ± 0.45 |
| LPS + IFN-γ + PMA + SOD inhibitors | 23.64 ± 1.13 | 30.90 ± 1.42 |
All the migration time values are represented as numbers ± standard deviations
Simultaneous detection of NO and O2−• in macrophage cell lysates: determination of changes in the ratio of NO to O2−• as a function of stimulation protocol
Prior to the investigation of the effect of inflammation on the NO/O2−• ratio in macrophage cell lysates, the identity of the NO- and O2−•-product-specific fluorescence peaks had to be verified. Figure 2a shows representative electropherograms of the simultaneous detection of NO and O2−• in RAW 264.7 macrophage cell lysates. The identification of NO and O2−• peaks was performed by incubating stimulated cells in the presence of one probe (DAF-FM DA or MitoSOX Red for NO and O2−•, respectively). DAF-FM T and 2-OH-MitoE+ represent the fluorescence reaction products of DAF-FM DA with NO and MitoSOX with O2−• [22,23], respectively. Although DAF-FM is very selective for NO, it has been shown that it can react with dehydroascorbate (DHA), giving DAF-FM DHA [29–31]. In earlier studies, the DAF-FM DHA peak was effectively separated from the NO-specific peak in cell lysates [22]. In these studies, a peak for DAF-FM DHA was observed only in the electropherogram of a stimulated cell sample (Figure 2c).
Fig. 2.
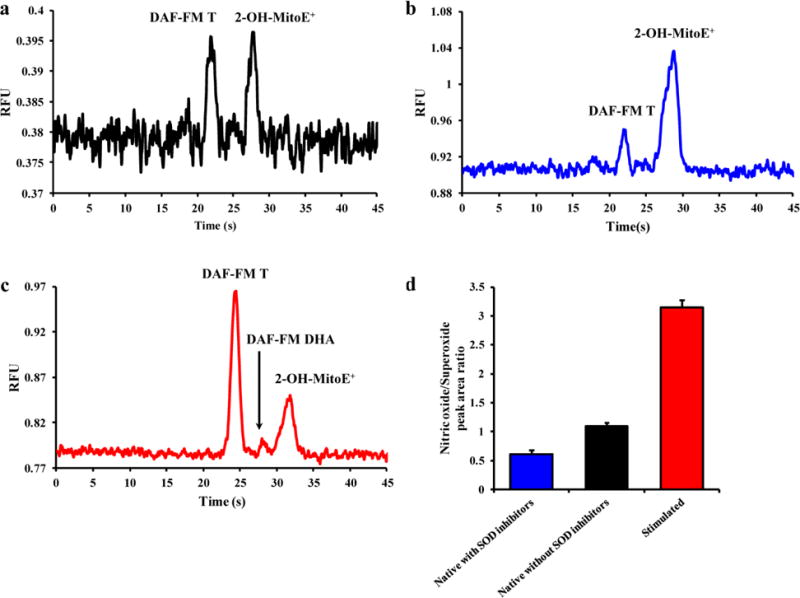
Representative electropherograms of (a) native macrophage cell lysate, (b) macrophage cell lysate treated with DDC and 2-ME, and (c) cell lysate stimulated with LPS + IFN-γ + PMA in the presence of SOD inhibitors. (d) A histogram comparing the NO/O2−• peak area ratios between unstimulated cells in the presence or absence of SOD inhibitors, and cells stimulated with LPS + IFN-γ + PMA in the presence of SOD inhibitors. Standard deviations are represented by vertical bars
Figure 2a shows a representative electropherogram for untreated macrophage cells. As can be seen by this electropherogram, the amount of NO and O2−• produced by native macrophages is very small due to the natural occurrence of endogenous intracellular scavenging molecules, such as SOD. Cytosolic and mitochondrial SOD regulates the intracellular concentration of O2−•, which can make O2−• difficult to detect in the cell lysate samples [32]. Therefore, to reduce the degradation of intracellular O2−• by SOD, two different SOD inhibitors, 2-ME and DDC, were introduced into the cells, along with MitoSOX Red, and incubated for an hour prior to analysis (Figure 1). Figure 2b shows a representative electropherogram obtained for unstimulated macrophages pre-treated with 2-ME and DDC, with a corresponding increase in the 2-OH-MitoE+ peak in relation to the DAF-FM T peak. An electropherogram of the cell lysate obtained for cells stimulated with LPS, IFN-γ, and then PMA in the presence of SOD inhibitors is shown in Figure 2c. In this case, the products for both NO and O2−• signals were increased, indicating an expected enhancement in intracellular production of both species, with a greater amount of NO being produced. This yielded a higher NO/O2−• ratio than for the SOD inhibitors alone.
A bar graph showing the comparison of the NO/O2−• ratios obtained under the different experimental conditions is provided in Figure 2d. The ratios obtained for unstimulated cells pre-treated with SOD inhibitors (0.60 ± 0.07), unstimulated cells without SOD inhibitors (1.08 ± 0.06), and stimulated (3.14 ± 0.13) macrophages show that, along with peak areas, the ratio of NO to O2−• changes as a function of the stimulation conditions. In these experiments, the number of viable cells was determined prior to analysis because the stimulation process can reduce the amount of cell division [33], increase cell differentiation [34], and also cause cell death [35]. Fig. S2 in the ESM shows the variation in cell numbers as a function of the different stimulation protocols used in the present study.
Changes in the NO/O2−• ratio for LPS + IFN-γ-stimulated versus PMA-stimulated macrophages
NO [36] and O2−• [37] have been implicated in the development of several neurodegenerative disorders and cardiovascular disease. Therefore, a series of experiments was performed to investigate the effect of two different stimulation protocols on the intracellular NO/O2−• ratio. Figure 3a shows the comparison between NO and O2−• peak areas obtained for cells stimulated with LPS + INF-γ (NO stimulation) or PMA (O2−• stimulation) only. As expected, the NO peak area was higher than that of O2−• for the samples stimulated with LPS + INF-γ, while an opposite situation was observed for samples treated with only PMA. Interestingly, the average O2−• peak area was more than 3 times higher in the case of iNOS-activated cells (LPS + INF-γ) than with cells stimulated by PMA alone. These data imply that increased NO production or iNOS activation may catalyze O2−• production, which agrees with previous reports in the literature [38–41].
Fig. 3.
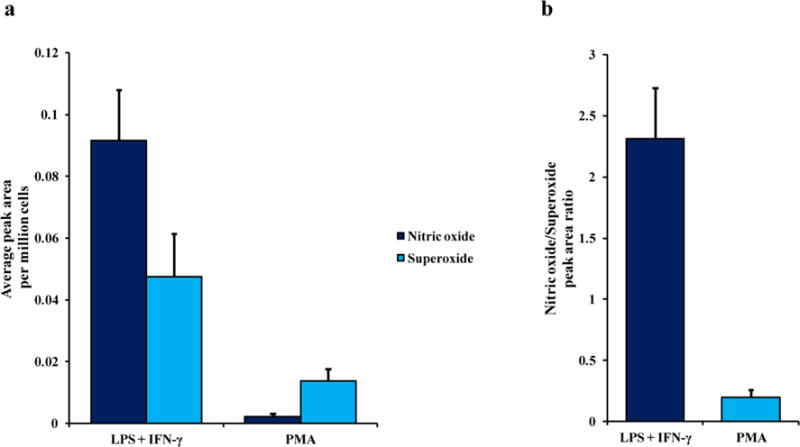
(a) The average peak area per million cells of NO and O2−• in cell lysate samples only stimulated with LPS + INF-γ (NO stimulation) or PMA (O2−• stimulation). (b) A comparison of the NO/O2−• peak area ratios between the two different stimulation protocols. Standard deviations are represented by vertical bars
A bar graph showing the effect of stimulation protocol on the NO/O2−• ratio is shown in Figure 3b. The NO/O2−• value for cells stimulated with LPS + INF-γ was 2.31 ± 0.41 while the value for cells treated with PMA alone was 0.19 ± 0.06.
Effect of carnosine or calcium on the NO/O2−• ratio in native and stimulated macrophages
Carnosine is an endogenous dipeptide that exhibits antioxidant properties and protects cells against free radicals. It has been clearly demonstrated that carnosine is able to scavenge RNOS [42]. Recently, Caruso et al. have reported that carnosine can catalyze the conversion of NO into nitrite, thereby causing a decrease in the apparent intracellular NO concentration [43]. We have also shown that significant amounts of carnosine are taken up by macrophages when it is incorporated in the cell culture medium [44]. In these studies, the effect of Ca+2 on the NO/O2−• ratio was also investigated. Ca2+ is an intracellular second messenger involved in signal transduction and many pathological processes [45]. Calcium, along with RNOS, participates in the regulation and integration of many cellular functions [46]. Increases in cytoplasmic Ca2+ have been correlated with increased O2−• [46–48].
In this study, the effect of pretreatment of the cells with either carnosine or Ca2+ on the NO/O2−• ratio in macrophage cell lysates was investigated under native and pro-inflammatory conditions. Figure 4 depicts the change in NO/O2−• ratio due to pretreatment of the cells with carnosine or Ca2+ in unstimulated (Figure 4a) and stimulated (Figure 4b) cells. When compared to the control (unstimulated cells), the samples pre-treated with carnosine showed a decrease in the NO/O2−• ratio (from 0.60 ± 0.07 to 0.35 ± 0.04). This ratio decrease was even more prominent for cells pre-treated with Ca2+ (from 0.60 ± 0.07 to 0.29 ± 0.05). A comparable trend was observed for stimulated samples, where the difference in the NO/O2−• ratio between each sample was slightly lower (from 3.14 ± 0.13 for cells stimulated with LPS + INF-γ + PMA to 2.35 ± 0.16 and 1.91 ± 0.10 for cells challenged with the same stimuli and pre-treated with carnosine or Ca2+, respectively).
Fig. 4.
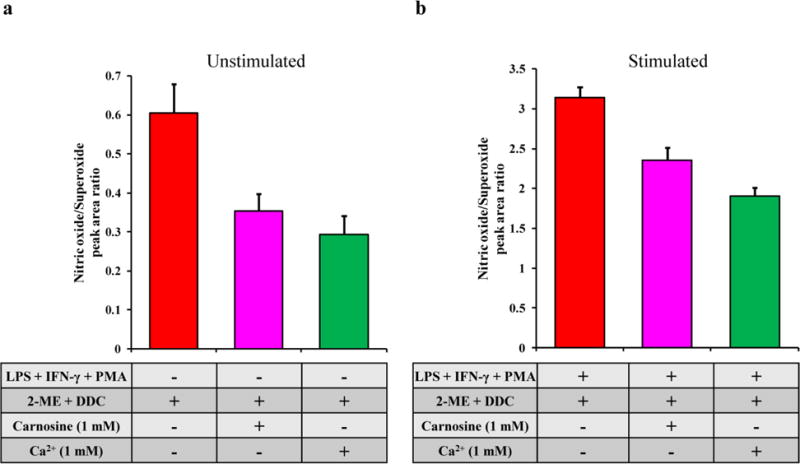
Changes in the NO/O2−• ratio due to pre-treatment with carnosine or Ca2+ in (a) unstimulated and (b) LPS + IFN-γ + PMA-stimulated cells. Standard deviations are represented by vertical bars
We believe that the decrease in the NO/O2−• ratio produced by pre-treatment with carnosine is due to a decrease in NO production (with little or no change in O2−• production). Previous studies by our group and others have shown that carnosine reduces iNOS-facilitated NO production in cells [43,49–50]. In contrast, it is proposed that Ca2+ pre-treatment caused a decrease in the NO/O2−• ratio due to an increase in intracellular O2−• (and not a decrease in NO). High intracellular Ca2+ concentrations have been shown to enhance the production of O2−• in the both the cytoplasm and mitochondria [46–48].
Conclusions
In this investigation, ME-LIF was employed for the simultaneous detection of NO and O2−• using the fluorescent probes DAF-FM DA and MitoSOX. This method was also employed to study the variations of the NO/O2−• ratio in RAW 264.7 macrophage cell lysates under physiological and pro-inflammatory conditions. Additionally, the effect of the natural antioxidant carnosine and the second messenger Ca2+ in modulating this ratio was investigated. These results highlight the roles played by different stimulation protocols in influencing the release and bioavailability of NO with respect to O2−•. It is well known that NO and O2−• oxidative stress-driven disorders; thus, the development of new cell stimulation protocols along with the application of this method in single cell analysis formats will provide new perspectives that can be used for a better understanding of the role of RNOS in neurodegenerative and cardiovascular disease.
Supplementary Material
Acknowledgments
This study was funded by National Science Foundation (CHE-1411993) and National Institutes of Health (COBRE P20GM103638). GC gratefully acknowledges the support of an American Heart Association-Midwest Affiliate Postdoctoral Research Fellowship (NFP0075515). JMS acknowledges the Madison and Lila Self Graduate Fellowship for support. We would also like to thank Dr. Richard Piffer and Nancy Harmony for their helpful comments and editorial support.
Biographies
Giuseppe Caruso, American Heart postdoctoral fellow at the University of Kansas, USA. He has been working for several years on the development of methods for the detection of reactive nitrogen and oxygen species in immune and cancer cells using microfluidic-based systems. Giuseppe’s research also includes the study of antioxidants on cell activation and differentiation. He recently discovered that the antioxidant carnosine modulates the energy metabolism as well as the differentiation and activation of macrophages.

Claudia G. Fresta, research assistant at the University of Kansas, USA. She has been working on the development of stimulation protocols for the concurrent determination of multiple intracellular reactive nitrogen and oxygen species to study the activity of inducible nitric oxide synthase in macrophages using microfluidic devices coupled to fluorescence. Her research also involves the study of the uptake of antioxidants under physiological and pro-inflammatory conditions in immune cells.

Joseph Siegel is a Madison and Lila Self graduate fellow and a Ph.D. candidate in the Department of Chemistry at the University of Kansas. His research focuses on developing separation-based microfluidic platforms to study nitrosative and oxidative stress and their relationships to neurological disorders.

Susan Lunte is the Ralph N. Adams Professor of Chemistry and Pharmaceutical Chemistry at the University of Kansas, USA. Dr. Lunte has over 35 years of experience in bioanalytical chemistry and pharmaceutical analysis. Her research interests include the development of separation-based methods for the determination of biological markers of oxidative stress and neurodegenerative disease. Most recently her group has focused on the development of microfluidic devices for single cell analysis and on-line monitoring of neurotransmitters using microdialysis sampling coupled to microchip electrophoresis.

Manjula Wijesinghe is a 4th year Ph.D. candidate in the Department of Chemistry at the University of Kansas. His research concerns the development of a unique detection system for microchip electrophoresis based on electrochemically generated fluorescence.

Footnotes
Compliance with ethical standards
Conflict of Interest: The authors declare that they have no conflict of interest.
Research involving human participants and/or animals: This article does not contain any studies with human participants performed by any of the authors.
Informed consent: Not applicable. This is an in vitro study.
References
- 1.Aaltoma SH, Lipponen PK, Kosma VM. Inducible nitric oxide synthase (iNOS) expression and its prognostic value in prostate cancer. Anticancer Res. 2001;21:3101–6. [PubMed] [Google Scholar]
- 2.Patel RP, McAndrew J, Sellak H, White CR, Jo H, Freeman BA, Darley-Usmar VM. Biological aspects of reactive nitrogen species. Biochim Biophys Acta. 1999;1411:385–400. doi: 10.1016/s0005-2728(99)00028-6. [DOI] [PubMed] [Google Scholar]
- 3.Weidinger A, Kozlov AV. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules. 2015;5:472–84. doi: 10.3390/biom5020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, O W, Li W, Jiang ZG, Ghanbari HA. Oxidative stress and neurodegenerative disorders. Int J Mol Sci. 2013;14:24438–75. doi: 10.3390/ijms141224438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fearon IM, Faux SP. Oxidative stress and cardiovascular disease: novel tools give (free) radical insight. J Mol Cell Cardiol. 2009;47:372–81. doi: 10.1016/j.yjmcc.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 6.Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H, LLeonart ME. Ageing Res Rev. 2013;12:376–90. doi: 10.1016/j.arr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevanin TM, Laver JR, Poole RK, Moir JW, Read RC. Metabolism of nitric oxide by Neisseria meningitidis modifies release of NO-regulated cytokines and chemokines by human macrophages. Microbes Infect. 2007;9:981–7. doi: 10.1016/j.micinf.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–16. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 10.Cooke JP. The pivotal role of nitric oxide for vascular health. Can J Cardiol. 2004;20(Suppl B):7B–15B. [PubMed] [Google Scholar]
- 11.Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–70. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- 12.Ma ZA, Zhao Z, Turk J. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Exp Diabetes Res. 2012;2012:703538. doi: 10.1155/2012/703538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Estevez AG, Jordan J. Nitric oxide and superoxide, a deadly cocktail. Ann N Y Acad Sci. 2002;962:207–11. doi: 10.1111/j.1749-6632.2002.tb04069.x. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer-Sueta G, Radi R. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol. 2009;4:161–77. doi: 10.1021/cb800279q. [DOI] [PubMed] [Google Scholar]
- 15.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–80. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 16.Espey MG, Miranda KM, Feelisch M, Fukuto J, Grisham MB, Vitek MP, Wink DA. Mechanisms of cell death governed by the balance between nitrosative and oxidative stress. Ann N Y Acad Sci. 2000;899:209–21. doi: 10.1111/j.1749-6632.2000.tb06188.x. [DOI] [PubMed] [Google Scholar]
- 17.Chi DS, Qui M, Krishnaswamy G, Li C, Stone W. Regulation of nitric oxide production from macrophages by lipopolysaccharide and catecholamines. Nitric Oxide. 2003;8:127–32. doi: 10.1016/s1089-8603(02)00148-9. [DOI] [PubMed] [Google Scholar]
- 18.Panaro MA, Brandonisio O, Acquafredda A, Sisto M, Mitolo V. Evidences for iNOS expression and nitric oxide production in the human macrophages. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3:210–21. doi: 10.2174/1568008033340216. [DOI] [PubMed] [Google Scholar]
- 19.Seminara AR, Ruvolo PP, Murad F. LPS/IFNgamma-induced RAW 264.7 apoptosis is regulated by both nitric oxide-dependent and -independent pathways involving JNK and the Bcl-2 family. Cell Cycle. 2007;6:1772–8. doi: 10.4161/cc.6.14.4438. [DOI] [PubMed] [Google Scholar]
- 20.Abbas K, Hardy M, Poulhes F, Karoui H, Tordo P, Ouari O, Peyrot F. Detection of superoxide production in stimulated and unstimulated living cells using new cyclic nitrone spin traps. Free Radic Biol Med. 2014;71:281–90. doi: 10.1016/j.freeradbiomed.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Li Q, Wang X, Xu K, Chen Z, Gong X, Liu X, Tong L, Tang B. Simultaneous Determination of Superoxide and Hydrogen Peroxide in Macrophage RAW 264.7 Cell Extracts by Microchip Electrophoresis with Laser-Induced Fluorescence Detection. Anal Chem. 2009;81:2193–98. doi: 10.1021/ac801777c. [DOI] [PubMed] [Google Scholar]
- 22.Mainz ER, Gunasekara DB, Caruso G, Jensen DT, Hulvey MK, da Silva JAF, Metto EC, Culbertson AH, Culbertson CT, Lunte SM. Monitoring Intracellular Nitric Oxide Production Using Microchip Electrophoresis and Laser-Induced Fluorescence Detection. Anal Methods. 2012;4:414–20. [Google Scholar]
- 23.de Campos RP, Siegel JM, Fresta CG, Caruso G, da Silva JA, Lunte SM. Indirect detection of superoxide in RAW 264.7 macrophage cells using microchip electrophoresis coupled to laser-induced fluorescence. Anal Bioanal Chem. 2015;407:7003–12. doi: 10.1007/s00216-015-8865-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zare RN, Kim S. Microfluidic platforms for single-cell analysis. Annu Rev Biomed Eng. 2010;12:187–201. doi: 10.1146/annurev-bioeng-070909-105238. [DOI] [PubMed] [Google Scholar]
- 25.Culbertson CT. Single cell analysis on microfluidic devices. Methods Mol Biol. 2006;339:203–16. doi: 10.1385/1-59745-076-6:203. [DOI] [PubMed] [Google Scholar]
- 26.Price AK, Culbertson CT. Chemical analysis of single mammalian cells with microfluidics. Strategies for culturing, sorting, trapping, and lysing cells and separating their contents on chips. Anal Chem. 2007;79:2614–21. doi: 10.1021/ac071891x. [DOI] [PubMed] [Google Scholar]
- 27.Lin Y, Trouillon R, Safina G, Ewing AG. Chemical analysis of single cells. Anal Chem. 2011;83:4369–92. doi: 10.1021/ac2009838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kojima H, Urano Y, Kikuchi K, Higuchi T, Hirata Y, Nagano T. Fluorescent Indicators for Imaging Nitric Oxide Production. Angew Chem Int Ed Engl. 1999;38:3209–12. doi: 10.1002/(sici)1521-3773(19991102)38:21<3209::aid-anie3209>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 29.Kim WS, Ye X, Rubakhin SS, Sweedler JV. Measuring nitric oxide in single neurons by capillary electrophoresis with laser-induced fluorescence: use of ascorbate oxidase in diaminofluorescein measurements. Anal Chem. 2006;78:1859–65. doi: 10.1021/ac051877p. [DOI] [PubMed] [Google Scholar]
- 30.Balcerczyk A, Soszynski M, Bartosz G. On the specificity of 4-amino-5-methylamino-2′,7′-difluorofluorescein as a probe for nitric oxide. Free Radic Biol Med. 2005;39:327–35. doi: 10.1016/j.freeradbiomed.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Kim WS, Hatcher N, Potgieter K, Moroz LL, Gillette R, Sweedler JV. Interfering with nitric oxide measurements. 4,5-diaminofluorescein reacts with dehydroascorbic acid and ascorbic acid. J Biol Chem. 2002;277:48472–8. doi: 10.1074/jbc.M209130200. [DOI] [PubMed] [Google Scholar]
- 32.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vadiveloo PK, Keramidaris E, Morrison WA, Stewart AG. Lipopolysaccharide-induced cell cycle arrest in macrophages occurs independently of nitric oxide synthase II induction. Biochim Biophys Acta. 2001;1539:140–6. doi: 10.1016/s0167-4889(01)00102-1. [DOI] [PubMed] [Google Scholar]
- 34.Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One. 2010;5:e8668. doi: 10.1371/journal.pone.0008668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishio K, Horie M, Akazawa Y, Shichiri M, Iwahashi H, Hagihara Y, Yoshida Y, Niki E. Attenuation of lipopolysaccharide (LPS)-induced cytotoxicity by tocopherols and tocotrienols. Redox Biol. 2013;1:97–103. doi: 10.1016/j.redox.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Togo T, Katsuse O, Iseki E. Nitric oxide pathways in Alzheimer’s disease and other neurodegenerative dementias. Neurol Res. 2004;26:563–6. doi: 10.1179/016164104225016236. [DOI] [PubMed] [Google Scholar]
- 37.Wu Z, Zhao Y, Zhao B. Superoxide anion, uncoupling proteins and Alzheimer’s disease. J Clin Biochem Nutr. 2010;46:187–94. doi: 10.3164/jcbn.09-104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia Y, Roman LJ, Masters BS, Zweier JL. Inducible nitric-oxide synthase generates superoxide from the reductase domain. J Biol Chem. 1998;273:22635–9. doi: 10.1074/jbc.273.35.22635. [DOI] [PubMed] [Google Scholar]
- 39.Heinzel B, John M, Klatt P, Bohme E, Mayer B. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem J. 1992;281:627–30. doi: 10.1042/bj2810627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pou S, Pou WS, Bredt DS, Snyder SH, Rosen GM. Generation of superoxide by purified brain nitric oxide synthase. J Biol Chem. 1992;267:24173–6. [PubMed] [Google Scholar]
- 41.Pou S, Keaton L, Surichamorn W, Rosen GM. Mechanism of superoxide generation by neuronal nitric-oxide synthase. J Biol Chem. 1999;274:9573–80. doi: 10.1074/jbc.274.14.9573. [DOI] [PubMed] [Google Scholar]
- 42.Hipkiss AR. Carnosine and its possible roles in nutrition and health. Adv Food Nutr Res. 2009;57:87–154. doi: 10.1016/S1043-4526(09)57003-9. [DOI] [PubMed] [Google Scholar]
- 43.Caruso G, Fresta CG, Martinez-Becerra F, Antonio L, Johnson RT, de Campos RPS, Siegel JM, Wijesinghe MB, Lazzarino G, Lunte SM. Carnosine modulates nitric oxide in stimulated RAW 264.7 macrophages. Mol Cell Biochem. 2017 doi: 10.1007/s11010-017-2991-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fresta CG, Hogard HL, Caruso G, Costa EEM, Lazzarino G, Lunte SM. Monitoring carnosine uptake by RAW 264.7 macrophage cells using microchip electrophoresis with fluorescence detection. Anal Methods. 2017;9:402–8. doi: 10.1039/C6AY03009B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophys Acta. 2009;1787:1416–24. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan Y, Wei CL, Zhang WR, Cheng HP, Liu J. Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol Sin. 2006;27:821–6. doi: 10.1111/j.1745-7254.2006.00390.x. [DOI] [PubMed] [Google Scholar]
- 47.Scully SP, Segel GB, Lichtman MA. Relationship of superoxide production to cytoplasmic free calcium in human monocytes. J Clin Invest. 1986;77:1349–56. doi: 10.1172/JCI112440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valentin F, Bueb J, Capdeville-Atkinson C, Tschirhart E. Rac-1-mediated O2- secretion requires Ca2+ influx in neutrophil-like HL-60 cells. Cell Calcium. 2001;29:409–15. doi: 10.1054/ceca.2001.0203. [DOI] [PubMed] [Google Scholar]
- 49.Fleisher-Berkovich S, Abramovitch-Dahan C, Ben-Shabat S, Apte R, Beit-Yannai E. Inhibitory effect of carnosine and N-acetyl carnosine on LPS-induced microglial oxidative stress and inflammation. Peptides. 2009;30:1306–12. doi: 10.1016/j.peptides.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 50.Nicoletti VG, Santoro AM, Grasso G, Vagliasindi LI, Giuffrida ML, Cuppari C, Purrello VS, Stella AM, Rizzarelli E. Carnosine interaction with nitric oxide and astroglial cell protection. J Neurosci Res. 2007;85:2239–45. doi: 10.1002/jnr.21365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.