

Abstract
We present the high quality, complete genome assembly of Neisseria lactamica Y92–1009 used to manufacture an outer membrane vesicle (OMV)-based vaccine, and a member of the Neisseria genus. The strain is available on request from the Public Health England Meningococcal Reference Unit. This Gram negative, dipplococcoid bacterium is an organism of worldwide clinical interest because human nasopharyngeal carriage is related inversely to the incidence of meningococcal disease, caused by Neisseria meningitidis. The organism sequenced was isolated during a school carriage survey in Northern Ireland in 1992 and has been the subject of a variety of laboratory and clinical studies. Four SMRT cells on a RSII machine by Pacific Biosystems were used to produce a complete, closed genome assembly. Sequence data were obtained for a total of 30,180,391 bases from 2621 reads and assembled using the HGAP algorithm. The assembly was corrected using short reads obtained from an Illumina HiSeq 2000instrument. This resulted in a 2,146,723 bp assembly with approximately 460 fold mean coverage depth and a GC ratio of 52.3%.
Keywords: SMRT cell sequencing, Neisseria, Short read sequencing, Bacteria, Genome assembly, Nasopharyngeal microflora, Commensal
Introduction
Neisseria lactamica (henceforth, Nla) is a Gram negative, diplococcoid, commensal organism that colonises the human nasopharynx. In common with other commensal Neisseria spp., including Neisseria mucosa , Neisseria. sicca and Neisseria cinerea , carriage of Nla very rarely leads to invasive disease, and then only in severely immunocompromised individuals [1]. Examples of more commonly pathogenic Neisseria lineages include the causative agents of gonorrhoea and invasive meningococcal disease (IMD), N. gonorrhoeae (Ngo) and N. meningitidis (Nme), respectively [2]. Nla is biochemically differentiated from other members of the genus Neisseria by its ability to produce β-D-galactosidase and therefore ferment lactose.
Neisseria lactamica Y92–1009 was originally isolated during a school carriage study in Northern Ireland in 1992 and has been assigned sequence type 3493 and belongs to the ST-613 clonal complex. The strain has been used for various purposes over the past 15 years. For example, it has been used to manufacture an OMV based vaccine intended to protect against against Nme [3] and used in experimental human challenge studies [4, 5] where it was shown to inhibit co-colonisation carriage rates of the potentially pathogenic meningococcus. The general properties of the genus, species and strain are presented in Table 1.
Table 1.
Classification and general features of Neisseria lactamica strain Y92–1009 according to MIGS specification [36]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [37] | |
| Phylum Proteobacteria | TAS [37] | ||
| Class Betaproteobacteria | TAS [37] | ||
| Order Neisseriales | TAS [37] | ||
| Family - Neisseriaceae | TAS [37] | ||
| Genus Neisseria | TAS [37] | ||
| Species lactamica | TAS [37] | ||
| Strain: Y92–1009 (Accession) | |||
| Gram stain | Negative | IDA | |
| Cell shape | Diplococcus | IDA | |
| Motility | Non-motile but piliated | TAS [38] | |
| Sporulation | Not reported | NAS | |
| Temperature range | 32–39 °C | IDA | |
| Optimum temperature | 37.0 °C | IDA | |
| pH range; Optimum | 3.5–6.5 °C; 5 °C | TAS | |
| Carbon source | Glucose, Maltose, Lactose | TAS [39] | |
| MIGS-6 | Habitat | Human Nasopharynx | TAS [37, 39] |
| MIGS-6.3 | Salinity | 0.9% | TAS [39] |
| MIGS-22 | Oxygen requirement | Aerobe | TAS [39] |
| MIGS-15 | Biotic relationship | commensal | TAS [39] |
| MIGS-14 | Pathogenicity | Non-pathogen | TAS [39] |
| MIGS-4 | Geographic location | Londonderry, Northern Ireland | TAS [40]b |
| MIGS-5 | Sample collection | 1992 | TAS [40]b |
| MIGS-4.1 | Latitude | 54.9966 N | NAS |
| MIGS-4.2 | Longitude | 7.3086 W | NAS |
| MIGS-4.4 | Altitude | 128 m | NAS |
aEvidence codes - IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [2]
bData for isolate geographic location and sample collection was acquired by searching for N. latamica Y92–1009 (ID number: 4945) on pubMLST Neisseria
A study of asymptomatic carriage of Nme and Nla in 2969 healthy infants and children demonstrated that carriage rates of Nla were highest in 18 month old infants and declined to a much lower rate in teenage children [6]. Conversely, a low level of meningococcal carriage was detected in infants during the first 4 years of life with increased carriage in those aged between 14 and 17. These findings have been confirmed by further studies including the Stonehouse survey [7], which reported that Nla carriage was six times higher in children up to the age of 5 years old, with relatively lower carriage rates for Nme. Similar results were also observed in a more recent study [8]. Taken together, these studies suggest a protective role for Nla against meningococcoal disease by preventing meningococcal colonization in younger children. Harnessing this natural carriage dynamic has been proposed as a potential strategy to reduce Nme carriage, a pre-requisite for invasive meningococcal disease [9].
Organism information
Classification and features
Nla is a Gram negative, non-sporulating, diplococcoid bacterium. Bacterial cells are approximately 1 μM in diameter. An electron micrograph, generated by staining with a 1% potassium phosphotungstate (PTA) for 5–10 s and captured using a Philips EM300 with an accelerating voltage of 60 kV, is shown in Fig. 1. On solid media the organism forms unpigmented to yellow, smooth and transparent colonies. The cells excrete outer membrane vesicles (OMVs) approximately 100 nm in diameter. In liquid culture, this species is likely to aggregate. Like other members of its genus, Nla is oxidase positive, catalase positive and successfully reduces nitrite (NO2 −) ions. Nla can be differentiated from the other members of the genus by its ability to ferment lactose and produce β-D-galactosidase. Nla, like Nme, dwells within the human nasopharynx but is more commonly found in infants and young children. It has been isolated from the urogenital tract on one occasion [10] but is almost never associated with disease [1].
Fig. 1.
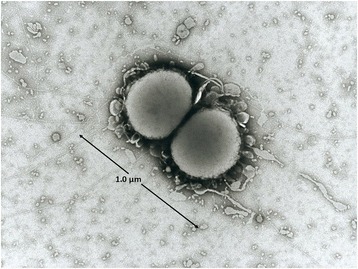
Photomicrograph of N. lactamica Y92–1009. This image was obtained with transmission electron micrography and displays the diplococcoid nature of the N. lactamica Y92–1009 cell. The size of the cell is indicated in micrometres (μm)
Nla unlike Nme is known to autoagglutinate and as such cannot be classified into serogroups. A multi locus sequence typing approach (MLST) uses the allelic profiles of seven, indexed housekeeping genes (abcZ, adk, aroE, fumC, gdh, pdhC & pgm) to classify a Neisseria isolate into a sequence types (ST) as reported in the database Pubmlst.org/neisseria [11].
At the time of writing a total of 21 unique ST’s of Nla have been described, which have been sub-classified into 6 clonal complexes (ccs). The cc640 includes ST’s 10,984, 10,326, 11,143 & the Nla 020–06 reference genome and the cc613 contains ST 11653 and Nla Y92–1009, the isolate described here. The cc595 contains ST 595, and cc624 contains ST’s 6206, 624, & 11,383. Finally, cc1494 contains ST’s 642, 1494 & 1495. To differentiate Neisseria strains further typing schemes have been developed using variable regions of hypervariable loci such as porA and fetA [12].
A core, gene by gene (n = 1629) MAAFT alignment of a collection of 32 whole genome Neisseria assemblies was done using the genome comparator tool available on Pubmlst.org/neisseria (Fig. 2). This included an example of the most complete representative assembly for every identified ST of Nla (n = 26) as well as the closed, reference genome Nla 020–06. These Nla isolates were compared to a cohort of pathogenic Neisseria reference genomes including Nme isolate MC58 (Serogroup B), Nme isolate FAM-18 (Serogroup C) and Ngo isolate FA1090. In addition to this the phylogenetic tree was rooted with another commensal Neisseria of interest, Nci 346 T. The comparison resolved the pathogenic Neisseria by their respective species and further sub-defined isolates belonging to the six clonal complexes of Nla. The comparison was calculated based on a core genome of 1629 genes. The maximum likelihood tree was generated using FastTree v.2.0 [13] (gtr nt model) and edited using Figtree v.1.4.3 [14].
Fig. 2.
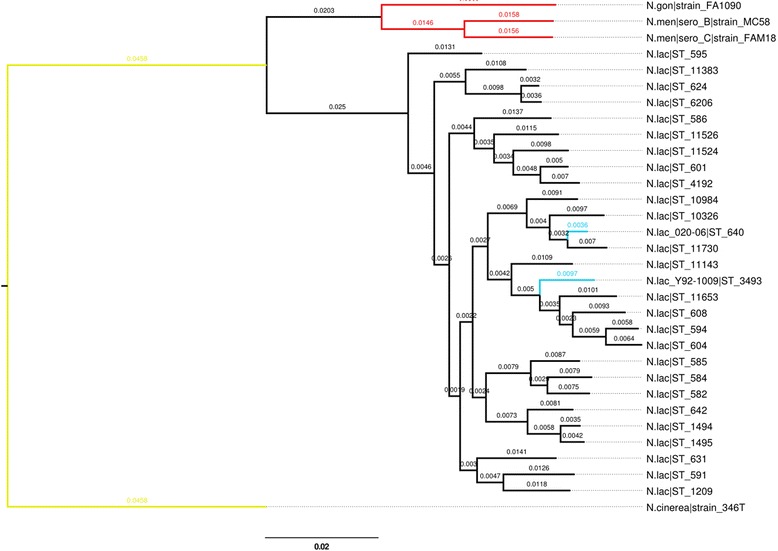
Phylogenetic tree indicating the position of N. lactamica Y92–1009 amongst other pathogenic and commensal Neisseria. This tree was constructed based on a core genome comparison of a collection of 32 Neisseria assemblies generated using the genome comparator tool available on pubmlst.org/neisseria. The reconstructed evolutionary relationships among N. meningitidis (Red, n = 2), N. gonnorhoeae (Red, n = 1), N. cinerea (Yellow, n = 1) and N. lactamica (Black, n = 27) are shown. The genome sequenced here, N. lactamica Y92–1009 is outlined in cyan (Nla_PacBio|ST_3493). This analysis included the sequenced genome and the best representative assembly for every identified sequence type (ST) of this species. The tree was generated using FastTree v.2 [13] and edited using Figtree v.1.4.3 [14]. The tree is drawn to scale, with branch length units being expressed as an overall proportion of divergence based on the comparison of 1629 genes
Genome sequencing information
Genome project history
Nla Y92–1009 GMP stocks were generated in 2011 [4]. Chromosomal DNA was extracted at the University of Southampton and sent to TGAC, Norwich for SMRT cell sequencing (PacBio RS II) and the Wellcome Trust Genomics Centre, Oxford for short read sequencing using HiSeq 2000. The genome was assembled using the internal pipeline at TGAC. The assembly was analysed, error-corrected, annotated and utilised for downstream applications by the authors. The assembly process and genome statistics are summarized in Tables 2 and 3.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Complete |
| MIGS-28 | Libraries used | SMRTbell Template prep kit |
| MIGS 29 | Sequencing platforms | Pacific Biosciences RSI |
| MIGS 31.2 | Fold coverage | 470× |
| MIGS 30 | Assemblers | HGAP |
| MIGS 32 | Gene calling method | Prokka, Blast2GO |
| Locus Tag | ||
| Genbank ID | CP019894 | |
| GenBank Date of Release | 17/02/2017 | |
| GOLD ID | - | |
| BIOPROJECT | PRJNA331097 | |
| MIGS 13 | Source Material Identifier | - |
| Project relevance | Medical, Biotechnological |
Table 3.
Genome statistics
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 2,146,723 | 100 |
| DNA coding (bp) | 1,831,541 | 85.3 |
| DNA G + C (bp) | 1,123,594 | 52.3 |
| DNA scaffolds | 1 | 100 |
| Total genes | 2053 | 100 |
| Protein coding genes | 1980 | 96.4 |
| RNA genes | 72 | 3.5 |
| Pseudo genes | 16 | 0.8 |
| Genes in internal clusters | 16 | 0.8 |
| Genes with function prediction | 1918 | 93.4 |
| Genes assigned to COGs | 1527 | 74.3 |
| Genes with Pfam domains | 5 | 0.2 |
| Genes with signal peptides | 0 | 0 |
| Genes with transmembrane helices | 0 | 0 |
| CRISPR repeats | 3 | 0.1 |
The accession numbers associated with this genome are Bioproject (PRJNA331097) and Biosample (SAMN05437355) and Genbank (SUB1713102: pending assignation of genbank ID).
Growth conditions and genomic DNA preparation
Frozen Nla Y92–1009 stock was plated onto Columbia agar supplemented with horse blood and grown overnight at 37 °C, 5% CO2. β-galactosidase activity, and therefore identity as Nla, was confirmed by exposure of colonies to 5-bromo-4chloro-3-indolyl-β-D-galactopyranoside (X-Gal) in phosphate buffered saline. Blue colonies were sub-cultured into Trypticase soy broth +0.2% yeast extract. The cultures were grown overnight at 37 °C, 5% CO2 at 320 rpm. The Gentra Puregene yeast/bacteria kit and protocol (Qiagen, UK) was used per manufacturer’s instructions to extract high molecular weight (>40 kb) DNA, assessed via gel electrophoresis and improving sample quality for long read sequencing.
DNA purity was initially assessed using a nanodrop 1000 spectrophotometer and then quantified using the Qubit 2.0 fluorometer and BR dsDNA kit (Invitrogen, UK). The resulting DNA samples were stored at four assessed by examining both the trace and absorbance levels of the 260/280 and 260/230 absorbance ratios in a nanodrop 1000 spectrophotometer. The resulting DNA samples were placed in −20 °C cryostorage.
Purified DNA samples were collected until a threshold of 30 μg DNA was reached. The sample (260/280: 1.83, 260/230: 1.86) was collated and sent to TGAC, Norwich for de novo long read sequencing.
Genome sequencing and assembly
Following sample collection, TGAC reassessed the sample purity and performed the long read sequencing using the PacBio RSII instrument. Four SMRT cells, each sequencing 50,000 × 8500 bp length reads, were used. The HGAP assembler [15] was used to generate a closed genome sequence of 2191181 bases.
Illumina paired-end, short read (151 bp) HiSeq 2000 sequencing was carried out using the same stock as that sent for Pacific Biosciences RS II long read sequencing. Sequencing libraries for paired end sequencing were constructed using the EBNext DNA sample Prep Master Mix Set 1 Kit (New England Biolabs).
Following generation of sequencing reads, nextera adapter sequence was trimmed using trimmomatic V.0.36 [16]. These reads were mapped to the unedited HGAP assembly and used to detect and correct errors present in repetitive regions using Pilon v.1.17 [17]. The reads were also used to trim low coverage areas present at the beginning and end of the circular genome sequence using the Breseq pipeline v.0.26a [18]. This reduced the assembly size down to 2,146,723 bp. Assembly metrics were evaluated using Quast [19].
Genome annotation
The Prokka pipeline [20] was used to putatively assign genetic function and identify RNA and pseudogenes. As part of this annotation pipeline, prodigal [21] was initially used to identify all co-ordinates of CDSs from the assembly but did not assign a putative gene product. Once all CDSs were detected, gene prediction is normally inferred by comparing an unknown protein to a database containing known protein sequences. To ensure maximum possible accuracy, this sequence-database homology comparison is staggered hierarchically in the following way by Prokka.
All putative CDSs are matched with a trusted list of proteins. From the only manually curated Nlatamica reference genome [22]. All unannotated proteins are then compared to the uniprot bacterial database, a Neisseria specific RefSeq database (enabled with the –genus and –usegenus flags) Interproscan searches were conducted using BLAST2GO software and searched against the MMMPFAM [23] SignalPHMM [24] and THHMM [25] databases to identify genes with PFAM domains, signal peptides and transmembrane helices respectively.
Any genes in internal clusters were identified using CD-HIT [26] using file of in silico translated proteins from identified ORFS. Any CRISPRs were found using CRISPRfinder [27].
Genome properties
The Nla Y92–1009 genome assembly contains 2,146,723 bp with approximately 460 fold mean coverage depth and 52.3% GC ratio. The assembly was predicted to contain 2053 putative ORFs, 1980 of which code for proteins. There were 72 genes predicted to encode RNA genes and three CRISPR repeats were detected.
Furthermore, 74.3% of total putative ORFs matched with the COG database; these results are presented in Table 4 and displayed in a circular genome diagram in Fig. 3.
Table 4.
Number of predicted genes associated with general COG functional categories
| Code | Value | %age | Description |
|---|---|---|---|
| J | 148 | 7.21 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.05 | RNA processing and modification |
| K | 56 | 2.73 | Transcription |
| L | 137 | 6.67 | Replication, recombination and repair |
| B | 1 | 0.05 | Chromatin structure and dynamics |
| D | 24 | 1.17 | Cell cycle control, Cell division, chromosome partitioning |
| V | 23 | 1.12 | Defense mechanisms |
| T | 25 | 1.22 | Signal transduction mechanisms |
| M | 130 | 6.33 | Cell wall/membrane biogenesis |
| N | 20 | 0.97 | Cell motility |
| U | 42 | 2.05 | Intracellular trafficking and secretion |
| O | 75 | 3.65 | Posttranslational modification, protein turnover, chaperones |
| C | 109 | 5.31 | Energy production and conversion |
| G | 48 | 2.34 | Carbohydrate transport and metabolism |
| E | 129 | 6.28 | Amino acid transport and metabolism |
| F | 45 | 2.19 | Nucleotide transport and metabolism |
| H | 76 | 3.70 | Coenzyme transport and metabolism |
| I | 51 | 2.48 | Lipid transport and metabolism |
| P | 77 | 3.75 | Inorganic ion transport and metabolism |
| Q | 10 | 0.49 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 137 | 6.67 | General function prediction only |
| S | 163 | 7.94 | Function unknown |
| - | 526 | 25.62 | Not in COGs |
The total is based on the total number of protein coding genes (1980) putatively discovered in the genome
Fig. 3.

Circular map of N. lactamica Y92–1009 genome and features generated with Cgview Comparison Tool. The arrows in the outermost ring indicate putative genes (with the arrow indicating the 5′ to 3′ direction on the positive strand.) identified and assigned to Clusters of Orthologous Groups (COGs). The 20 COG categories are indicated by different colours from Red to Grey according to the colour key. The second and third rings indicate regions containing coding sequence (Blue), tRNA (Orange), and other RNAs (Grey), with the second ring running 5′ to 3′ (positive strand) and the third ring running 3′to 5′ (negative strand). The fourth ring indicates open reading frames assigned as COGs and encoded on the negative strand. The fifth, black, graph ring displays GC content while the last ring (purple and green) shows positive (Green) and negative (Purple) GC skew
Insights from the genome sequence
This genome sequence was typical for a Neisseria genome, being small (2.2 Mb) with a high amount of coding content (85.3%).
Nla remains relatively poorly defined at the genomic level, with the COG analysis (Table 4) demonstrating that approximately a third (33.5%) of all open reading frames (ORFs) in this genome have either unknown, predicted function or did not possess sufficient homology to be sorted into a COG category. Additionally, there were only five matches when comparing Nla Y92–1009 translated ORFs against the PFAM database as well as a total lack of matches with SignalP and THHMM databases. Despite this, five ORFs presenting were annotated as putative signal peptides by Prokka.
Repetitive sequences play important roles in Neisseria genome modification and gene expression. The ten base pair DNA uptake sequence (DUS) has been shown to be pre-requisite for transformation in Neisseria species [28]. DUS-containing sequences have permeated the Neisseria genus core genome, indicating these sequences can survive genome diversification via recombination [29]. Another repeat type is named dRS3. This is an abundantly recurring 20 bp repeat sequence known to flank larger repeat sequences and act as a site for phage integration [30]. Finally, the transposon-like Correia repeat enclosed elements (CREEs) may combine with native sequence to form gene promotors as well as affect post transcriptional gene expression [31]. Therefore these elements have often been observed as hot spots of DNA rearrangement and recombination [32]. Repeat sequence content also reflects on the evolutionary history of an organism. Nme possesses many more CREEs than any other member of the genus. This is thought to have arisen after the species diversified away from a common ancestor [33].
Motifs for these repetitive sequences were taken from a genus wide study of Neisseria that reported DUS, CRE and dRS3 motifs among ten species [34] and a study on the overepresentation of DUS motifs (dialects)among the Neisseriaceae [28]. These motifs were searched in the genome using fuzznuc as part of the EMBOSS [35] package. The frequency of these repetitive sequence motifs observed in Nla Y92–1009 is described in Table 5. In comparison to isolates examined in the first study cited [34], Nla isolate Y92–1009 has a higher number (n = 454) of dSR3 elements than all other neisserial species bar Nme isolate MC58 (n = 689). This value also exceeded the number those detected in another Nla isolate ATCC 23970 (n = 197) and N. gonnorhoeae isolate FA 1090 (n = 208). Nla isolate Y92–1009 exhibited a lower numbers of CREE repeats (n = 86) than all other known Neisseria except for Nme isolate MC58 (n = 524) (Table 5).
Table 5.
Frequency of repeat sequences in N. lactamica Y92–1009 genome
| Repeat type | Repeat sequence | Value |
|---|---|---|
| AT-DUS | ‘ATGCCGTCTGAA’ | 1718 |
| AG-DUS | ‘AGGCCGTCTGAA’ | 262 |
| AG-mucDUS | ‘AGGTCGTCTGAA’ | 45 |
| DSR3 | ‘ATTCCCNNNNNNNNGGGAAT’ | 454 |
| Correia | ‘ATAG[CT]GGATTAACAAAAATCAGGAC’ | 50 |
| ‘TATAG[CT]GGATTAAATTTAAACCGGTAC’ | 1 | |
| ‘TATAG[CT]GGATTAACAAAAACCGGTAC’ | 17 | |
| ‘TATAG[CT]GGATTAAATTTAAATCAGGAC’ | 17 |
In comparison to isolates examined in the Neisseria -wide DNA uptake sequence study, Nla isolate Y92–1009 possessed an overrepresentation of AT-DUS sequence (n = 1718) which was typical of other Nla isolates and the pathogenic Neisseria . It also possessed low levels of AG-DUS and AG-mucDUS dialects which are found more prominently in species such as N. polysaccherea, N. cinerea (Nci) and N. mucosa . Due to the interspecific barrier to transformation that exists between Neisseria spp. with uncomplimentary DUS dialects, it is more likely that Nla would engage in transformation with Neisseria spp. possessing the same DUS dialect.
A number of phage related genes and atypical GC content were seen in a region starting 1, 350, 054 bp and running until 1,399,045 bp. The presence of a prophage was investigated by PHAST (Phage Search Tool; http://phast.wishartlab.com/index.html). PHAST detected an intact prophage sequence 49.8Kb in length. This contained 53.98% GC content and possessed 81 proteins, 46 of which were phage associated. These proteins include two attachment sites, two coat proteins, two tail-fiber proteins, an integrase, two plate, a portal, a tail shaft and terminase subunits (Fig. 4 ). The prophage sequence scored a completeness score of 130, where 150 is the maximum and a minimum score of 90 indicates intactness of prophage. The proteins from the putative, Nla Y92–1009 prophage sequence were compared against the PHAST-prophage database. This revealed that at least one predicted protein from this prophage sequence was detected in over 23 different species of bacteriophage. Three of the four bacteriophages with the greatest number of shared proteins with the Nla Y92–1009 prophage were found in Acinetobacter phages. While these prophages only shared 18, 17 and 16 proteins respectively out of a potential 81, this stretch of sequence was found to be highly conserved.
Fig. 4.
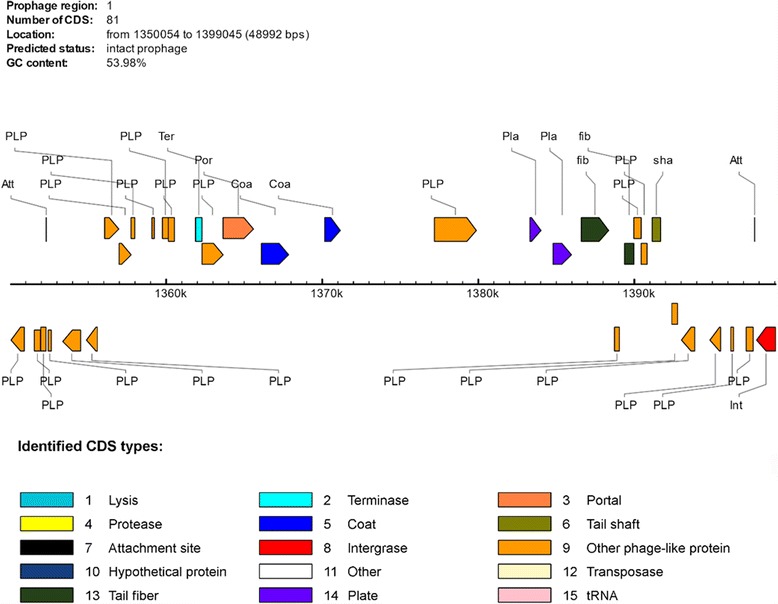
Graphic showing phage related proteins identified in the intact prophage by PHAST. The diagram above was annotated by and imported from the PHAST-prophage database. Twenty-six hypothetical proteins were removed from the schematic to increase the clarity of the phage related proteins. The proteins above the black line indicating genome position are encoded 5′ to 3′ while the proteins under it are encoded 3′ to 5′. The abbreviations in the diagrams are as follows Att (phage attachment site), Coa (Phage coat protein), fib (Phage Tail Fiber), Int (Phage integrase) Pla (Phage plate protein), PLP (Phage like protein), Por (Portal protein) sha (Phage tail shaft protein) Ter (Terminase)
Conclusions
This closed whole genome assembly is the first for this specific strain of Nla isolate Y92–1009 and the second for the species as a whole and contains 2,146,723 bp, that encode 1980 predicted proteins, 72 RNA genes and three CRISPR repeats. The profile of repeat sequence patterns discovered in this genome compared to other Neisseria spp. indicates that it possesses DUS, CREE motifs and numbers typical to other Nla isolates but contains an unexpectedly high amount of dRS3 repeats for a commensal Neisseria , similar to the number seen in Nme isolate MC58. This genome will form the reference for studies on the microevolution of commensal Neisseria among individuals experimentally challenged with this strain.
Acknowledgments
Funding
This project was supported by funding from the MRC: MR/N013204/1 and MR/026993/1, The Sir Christopher Benson Studentship (University of Southampton), the Wessex Institute of Virology and Infectious Disease and the Southampton NIHR Biomedical Research Centre.
Abbreviations
- Bp
Base pairs
- Cc
Clonal complex
- CDS(s)
CoDing sequence
- COG
Clusters of Orthologous Groups
- CREEs
Correia repeat enclosed elements
- CRISPR
Clusters of regularly intersperced short palindromic repeats
- DUS
DNA uptake sequence
- GC
Guanine-cytosine ratio
- GMP
Good manufacturing practice
- MLST
Multi locus sequence typing
- Nci
Neisseria cinerea
- Ngo
Neisseria gonorrhoeae
- Nla
Neisseria lactamica
- Nme
Neisseria meningitidis
- ORFs
Open reading frames
- PacBio
Pacific biosciences
- SMRT
Smart cell sequencing
- ST
Sequence type
- V.(#)
Program Version followed by number
Authors’ contributions
AKP performed sample preparation, curation of the genome and drafted the manuscript. JRL conceived the study and helped to draft the manuscript. RCR helped conceive the study and the drafting of the manuscript. DWC participated in the design of the study and helped to revise the manuscript. AG participated in the design of the study, statistical analysis, and helped to revise the manuscript. MCM participated in the design of the study, statistical analysis, and helped to revise the manuscript. XD participated in the design of the study, statistical analysis, and helped to revise the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Anish K. Pandey, Email: anishkumarpandey89@gmail.com
Robert C. Read, Email: r.c.read@soton.ac.uk
References
- 1.Wilson HD, Overman TL. Septicemia due to Neisseria lactamica. J Clin Microbiol. 1976;4:214–215. doi: 10.1128/jcm.4.3.214-215.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Criss AK, Seifert HS. A bacterial siren song: intimate interactions between Neisseria and neutrophils. Nat Rev Microbiol. 2012;10:178–190. doi: 10.1038/nrmicro2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorringe AR, Taylor S, Brookes C, Matheson M, Finney M, Kerr M, Hudson M, Findlow J, Borrow R, Andrews N, Kafatos G, Evans CM, Read RC. Phase I safety and immunogenicity study of a candidate meningococcal disease vaccine based on Neisseria lactamica outer membrane vesicles. Clin Vaccine Immunol. 2009;16:1113–1120. doi: 10.1128/CVI.00118-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans CM, Pratt CB, Matheson M, Vaughan TE, Findlow J, Borrow R, Gorringe AR, Read RC. Nasopharyngeal colonization by Neisseria lactamica and induction of protective immunity against Neisseria meningitidis. Clin Infect Dis. 2011;52:70–77. doi: 10.1093/cid/ciq065. [DOI] [PubMed] [Google Scholar]
- 5.Deasy AM, Guccione E, Dale AP, Andrews N, Evans CM, Bennett JS, Bratcher HB, Maiden MCJ, Gorringe AR, Read RC. Nasal inoculation of the commensal Neisseria lactamica inhibits carriage of Neisseria meningitidis by young adults: a controlled human infection study. Clin Infect Dis. 2015;60:1512–20. [DOI] [PubMed]
- 6.Gold R, Goldschneider I, Lepow ML, Draper TF, Randolph M. Carriage of Neisseria meningitidis and Neisseria lactamica in infants and children. J Infect Dis. 1978;137:112–121. doi: 10.1093/infdis/137.2.112. [DOI] [PubMed] [Google Scholar]
- 7.Cartwright KA, Stuart JM, Jones DM, Noah ND. The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica. Epidemiol Infect. 1987;99:591–601. doi: 10.1017/S0950268800066449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakir M, Yagci A, Ulger N, Akbenlioglu C, Ilki A, Soyletir G. Asymtomatic carriage of Neisseria meningitidis and Neisseria lactamica in relation to Streptococcus pneumoniae and Haemophilus influenzae colonization in healthy children: apropos of 1400 children sampled. Eur J Epidemiol. 2001;17:1015–1018. doi: 10.1023/A:1020021109462. [DOI] [PubMed] [Google Scholar]
- 9.Read RC. Neisseria meningitidis; clones, carriage, and disease. Clin Microbiol Infect. 2014;20:391–395. doi: 10.1111/1469-0691.12647. [DOI] [PubMed] [Google Scholar]
- 10.Telfer Brunton WA, Young H, Fraser DR. Isolation of Neisseria lactamica from the female genital tract. A case report. Br J Vener Dis. 1980;56:325–6. [DOI] [PMC free article] [PubMed]
- 11.Jolley KA, Maiden MCJ. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maiden MCJ, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, ND MC. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol. 2013;11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price MN, Dehal PS, Arkin AP. FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavlopoulos GA, Soldatos TG, Barbosa-Silva A, Schneider R. A reference guide for tree analysis and visualization. BioData Min. 2010;3:1. doi: 10.1186/1756-0381-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 16.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deatherage DE, Barrick JE. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 2014;1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 21.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett JS, Bentley SD, Vernikos GS, Quail MA, Cherevach I, White B, Parkhill J, Maiden MCJ. Independent evolution of the core and accessory gene sets in the genus Neisseria: insights gained from the genome of Neisseria lactamica isolate 020-06. BMC Genomics. 2010;11:652. [DOI] [PMC free article] [PubMed]
- 23.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. Pfam: The protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–30. [DOI] [PMC free article] [PubMed]
- 24.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 25.Krogh a, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 26.Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspace short palindromic repeats. Nucleic Acids Res. 2007;35(Web Server issue):52–57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frye SA, Nilsen M, Tønjum T, Ambur OH. Dialects of the DNA uptake sequence in Neisseriaceae. PLoS Genet. 2013;9:e1003458. doi: 10.1371/journal.pgen.1003458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Treangen TJ, Ambur OH, Tonjum T, Rocha EPC. The impact of the neisserial DNA uptake sequences on genome evolution and stability. Genome Biol. 2008;9:R60. doi: 10.1186/gb-2008-9-3-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rotman E, Seifert HS. The genetics of Neisseria species. Annu Rev Genet. 2014;48(September):405–431. doi: 10.1146/annurev-genet-120213-092007. [DOI] [PubMed] [Google Scholar]
- 31.Lin YH, Ryan CS, Davies JK. Neisserial correia repeat-enclosed elements do not influence the transcription of pil genes in Neisseria gonorrhoeae and Neisseria meningitidis. J Bacteriol. 2011;193:5728–5736. doi: 10.1128/JB.05526-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siddique A, Buisine N, Chalmers R. The transposon-like correia elements encode numerous strong promoters and provide a potential new mechanism for phase variation in the meningococcus. PLoS Genet. 2011;7:e1001277. doi: 10.1371/journal.pgen.1001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rotman E, Seifert HS. The genetics of Neisseria species. Annu Rev Genet. 2014;48:405–431. doi: 10.1146/annurev-genet-120213-092007. [DOI] [PubMed] [Google Scholar]
- 34.Marri PR, Paniscus M, Weyand NJ, Rendón MA, Calton CM, Hernández DR, Higashi DL, Sodergren E, Weinstock GM, Rounsley SD, So M. Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PLoS One. 2010;5:e11835. doi: 10.1371/journal.pone.0011835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 36.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, Ashburner M, Axelrod N, Baldauf S, Ballard S, Boore J, Cochrane G, Cole J, Dawyndt P, De Vos P, DePamphilis C, Edwards R, Faruque N, Feldman R, Gilbert J, Gilna P, Glöckner FO, Goldstein P, Guralnick R, Haft D, Hancock D, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB. Bergey’s volume 4 Bacteroidetes, Acidobacteria, Spirochaetes etc. 2010. [Google Scholar]
- 38.Aho EL, Botten JW, Hall RJ, Larson MK, Ness JK. Characterization of a class II pilin expression locus from Neisseria meningitidis: evidence for increased diversity among pilin genes in pathogenic Neisseria species. Infect Immun. 1997;65(7):2613–20. doi: 10.1128/iai.65.7.2613-2620.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lennette EH. Manual of clinical microbiology. 4. Washington DC: American Society for Microbiology; 1985. [Google Scholar]
- 40.Neisseria MLST website[http://pubmlst.org/neisseria/].