

Abstract
Osteoarthritis (OA) is the most common form of arthritis and a significant cause of pain and disability in older adults. Among the risk factors for OA, age is the most prominent. This review will discuss the relationship between aging and the development of OA, with a particular focus on mechanisms relevant to cartilage degeneration and the role of excessive levels of reactive oxygen species. Rather than just causing random oxidative damage, an increase in reactive oxygen species that leads to oxidative stress disrupts specific cell signaling pathways. This disruption in cell signaling affects the ability to maintain the cartilage extracellular matrix and eventually causes cell death. By understanding the specific cell signaling pathways that lead to OA through altered redox signaling, novel targets will be discovered that will be an advance over the current non-targeted anti-oxidant approach that has not been successful in treating chronic diseases of aging such as OA.
INTRODUCTION
Osteoarthritis (OA) is one of the most common causes of pain and disability in adults (1). There are a host of risk factors for the development of OA that include joint injury, obesity, genetic predisposition, and abnormal joint shape and alignment. However, the factor that has the greatest influence on the incidence and prevalence of OA is age. More than 30 million adults in the United States have been diagnosed with OA (2). The aging of our population, along with the increase in obesity seen at all ages, is expected to more than double the numbers of individuals with OA by 2030 to 67 million with greater than 50% of OA cases appearing in the 65-years and older age group (3). The loss of productivity due to the pain and disability of OA, along with the costs of caring for individuals with OA, has been estimated to cost $27 billion annually in the United States (4).
A major limitation in the management of patients with OA is the lack of any therapy that can slow the progression of the disease. Non-pharmacologic approaches such as exercise and weight loss can improve symptoms but to date have not been found to impact disease progression (5,6). The same is true for non-steroidal anti-inflammatory drugs, including cyclooxygenase-2 inhibitors, intra-articular injections of hyaluronic acid or corticosteroids, nutritional supplements including glucosamine and chondroitin sulfate, and arthroscopic surgery including anterior cruciate ligament or meniscus repair as well as joint debridement and/or irrigation. Patients will also try numerous home remedies for OA, such as gin-soaked raisins or wearing copper bracelets. The typical OA patient will often try many of these interventions and can sometimes experience temporary and intermittent improvement in symptoms, but over time will note that “nothing seems to help anymore,” resulting in a need for joint replacement surgery. Thus, the lack of any intervention that targets the disease process has resulted in a substantial increase in joint replacement surgery (7). Clearly, a safe, effective and less-expensive treatment that can alter the course of the disease will have a major impact on both quality of life and future health-care expenditures.
The obvious billion dollar question is how do we slow or stop the progression of joint damage and improve symptoms in individuals with OA. Failed attempts have been reported with not only the interventions noted above, but with other therapies ranging from vitamin D supplementation to bisphosphonates, calcitonin, and inhibitors of nitric oxide synthase, to name just a few (6,8–10). There are a number of reasons that might explain why attempts at disease modification have not met with success. First, although the understanding of the pathogenesis of OA has come a long way over the past two decades, there is still much to be learned to choose better targets for therapy. Second, pre-clinical animal models of OA with better translation to the human disease are needed as are more precise imaging and blood biomarkers that can be used to predict and measure disease progression. Third, OA is a multifactorial condition and the pathway to OA from one particular risk factor, such as joint injury, is likely quite different from that of a different risk factor, such as obesity. This suggests that interventions will need to target a specific OA phenotype rather than the current approach of treating all OA patients in a similar manner.
The most common and perhaps most obvious OA phenotypes to consider for targeted therapies would include post-traumatic OA that occurs after a joint injury, OA associated with obesity that appears to be related not only to increased joint loading from greater body weight but also systemic metabolic disturbances due to increased fat mass, and OA that is related to aging changes occurring within the joints but likely systemically as well. Because aging may contribute to the development of OA no matter the inciting cause, there has been a growing interest in determining the basic mechanisms by which aging and OA are related to target the aging aspects of the condition (11).
CARTILAGE AGING AND OA
OA is a slowly progressive disease of synovial joints characterized pathologically by focal destruction of the articular cartilage, a hypertrophic response in neighboring bone that results in osteophyte formation and subchondral sclerosis, variable degrees of synovial inflammation, a thickening of the joint capsule, and damage to soft tissue structures including ligaments and, in the knee, the meniscus (12). The destruction and loss of the articular cartilage is central to the development of OA and most of the research to date on aging mechanisms relevant to OA has focused on changes in the cartilage (11). It is important to note that joint aging and OA are not one and the same but rather aging changes can make the development of OA more likely to occur. With normal aging the cartilage appears slightly brown due to an accumulation of advanced glycation end-products (13) and is thinner than in young adults but is otherwise smooth and intact. The accumulation of advanced glycation end-products has been found to alter the biomechanical properties of cartilage making it more “brittle” and susceptible to degeneration (14). In contrast, in joints affected by OA there is marked destruction and loss of the cartilage accompanied by osteophytes and subchondral bone thickening.
The destruction and loss of the articular cartilage in OA is driven by an imbalance in the production and activity of pro-inflammatory and catabolic mediators, including a host of cytokines and chemokines, relative to the activity of anabolic factors, including the growth factors insulin-like growth factor 1 (IGF-1) and osteogenic protein 1 (OP-1), also known as bone morphogenetic protein -7 (15). The imbalance in catabolic and anabolic signaling results in overproduction of matrix degrading enzymes including the matrix metalloproteinases (MMPs) and aggrecanases. MMP-13 is important because of its ability to degrade type II collagen, the major structural protein in cartilage which provides the tissue’s tensile strength, whereas the aggrecanases are notable for their ability to degrade aggrecan, the large proteoglycan that is responsible for the resiliency of cartilage.
Aging processes that promote an imbalance in chondrocyte signaling resulting in increased production of MMPs and aggrecanases would be central to the development and progression of OA. A focus of our research efforts, and others in the field, has been on gaining a better understanding of the basic molecular mechanisms driving this imbalance in signaling. This could be due to cellular senescence and the development of what has been termed the senescence-associated secretory phenotype (11). The senescence-associated secretory phenotype is characterized by increased production of many of the same cytokines, chemokines, and MMPs found in OA cartilage, suggesting that OA chondrocytes assume a senescent phenotype. More studies are needed to define the underlying mechanisms of chondrocyte senescence and to determine if removal of senescent cells using compounds that have been called “senolytics”(16) would slow the progression of OA and be disease and/or symptom modifying.
AGING, OXIDATIVE STRESS, AND OA
Hallmarks of aging have been proposed, such as cellular senescence and telomere attrition, that are believed to represent key mechanisms by which aging contributes to the development of age-related conditions (17). One of the hallmarks is mitochondrial dysfunction which can promote age-related disorders in part through increased levels of reactive oxygen species (ROS). Age-related mitochondrial dysfunction has been suggested as a contributing factor in the development of OA (18–20). To obtain in vivo support for the hypothesis that mitochondrial dysfunction promotes the development of OA through increased levels of ROS, we evaluated the severity of naturally occurring OA in transgenic mice engineered to express human catalase targeted to the mitochondria (MCAT mice). These mice, developed by Peter Rabinovitch, have been shown to have reduced markers of oxidative stress with aging, accompanied by a reduction in age-related pathology and increased lifespan (21,22). Compared to age-matched wild-type controls, we found that the 18- to 33-month-old male MCAT mice had a modest but significant reduction in the severity of OA changes in knee articular cartilage (23).
Increased levels of ROS can result in oxidative damage which is one mechanism by which ROS can promote age-related disease. Immunohistochemical studies of human, non-human primates, and mouse articular cartilage, have shown the presence of the oxidative damage marker nitrotyrosine in aged and OA cartilage (20,24). However, ROS also have a normal physiologic function as regulators of cell signaling (25). It is thought that elevated levels of ROS, found in oxidative stress conditions, could promote age-related conditions through the disruption of physiologic signaling (26,27).
In a series of studies, we have shown that oxidative stress occurs with aging in articular cartilage and this promotes an imbalance in catabolic and anabolic signaling that could play a key role in the development of OA (Figure 1). IGF-1 and OP-1 are key cartilage growth factors and we showed a reduced response of human articular chondrocytes to IGF-1 or the combination of IGF-1 and OP-1, resulting in reduced matrix gene expression and matrix protein synthesis (28,29). The reduced response to these growth factors appears to be due to altered cell signaling mediated by oxidative stress. Although low physiologic levels of ROS promote IGF-1 signaling, we have found that the levels of ROS that occur under oxidative stress conditions inhibit IGF-1–mediated Akt activation (necessary for chondrocyte matrix synthesis and survival) and increase activation of catabolic mitogen-activated protein kinase pathways (29–31). Chondrocyte ROS-mediated signaling that regulates MMP expression has been noted in response to cytokines including interleukin 1β and tumor necrosis factor α (32–35) and to stimulation of chondrocytes by fibronectin fragments that have been found to accumulate in OA cartilage (36). Unlike anabolic signaling that declines with age, we found that chondrocytes from older adults become more responsive to stimulation by interleukin-1 and fibronectin fragments (37).
Fig. 1.
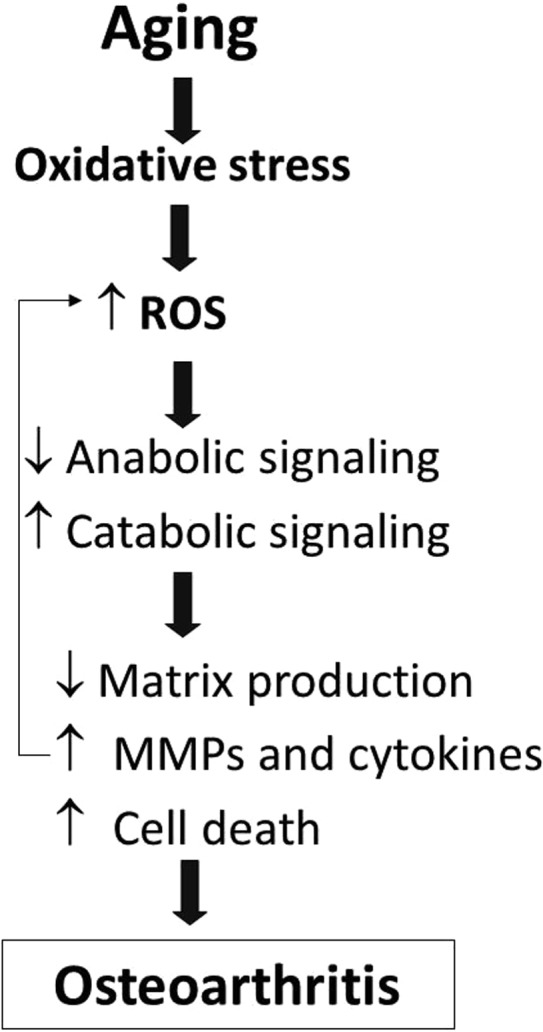
Oxidative stress due to aging contributes to the development of osteoarthritis. Age-related oxidative stress results in increased levels of reactive oxygen species (ROS) in articular chondrocytes. Excessive ROS disrupts cell signaling to inhibit anabolic and promote catabolic signaling that results in reduced matrix production and increased production of matrix metalloproteinases (MMPs) and pro-inflammatory cytokines as well as increased chondrocyte death. These changes in turn lead to cartilage destruction and osteoarthritis.
Normally, the cellular levels of ROS are controlled by the balance of ROS production and the activity of various anti-oxidants. Glutathione is a major intracellular anti-oxidant and we noted an age-related increase in the amount of oxidized relative to reduced glutathione consistent with an age-related increase in ROS (38). The peroxiredoxin (Prx) family of thiol peroxidases is responsible for the control of intracellular levels of the H2O2. Because H2O2 is the ROS that is most important in the regulation of cell signaling, the control of H2O2 by the Prxs is thought to be central to redox signaling (39). When levels of ROS become excessive, Prxs are inactivated through hyperoxidation. Using an antibody that recognizes hyperoxidized Prxs (Prx-SO2/3), we noted the presence of increased levels of hyperoxidized Prxs in human cartilage from older adults with a further increase noted in OA cartilage (23). In that study, we showed that chondrocytes from older adults were more susceptible to Prx hyperoxidation when exposed to ROS and this was associated with inhibition of IGF-1–mediated phosphoinositide 3 (PI-3) kinase–Akt signaling and promotion of p38 activation that resulted in chondrocyte cell death (Figure 2). We went on to show that expression of catalase targeted to the mitochondria could inhibit these effects, resulting in restoration of normal IGF-1 signaling and promotion of cell survival.
Fig. 2.

Mechanism by which increased levels reactive oxygen species (ROS) disrupt signaling through hyperoxidation of peroxiredoxins. Low physiologic levels of ROS are controlled by the peroxiredoxins (Prxs) and promote phosphoinositide 3 (PI-3) kinase-Akt signaling. As levels of ROS rise, Prxs are hyperoxidized to the PrxSO2/3 state which causing their inactivation and loss of redox control. This results in the inhibition of PI-3 kinase-Akt signaling and activation of specific mitogen-activated protein (MAP) kinase signaling pathways that increase production of matrix degrading enzymes and eventually cause cell death resulting in cartilage destruction and osteoarthritis.
CONCLUSIONS
There are likely multiple factors related to aging that promote the development of age-related conditions such as OA. A central feature of OA is the imbalance in catabolic and anabolic signaling in cartilage that results in progressive matrix destruction. Our studies are providing evidence that age-related oxidative stress plays a key role in this catabolic-anabolic imbalance. Studies on the molecular mechanism are revealing excessive oxidation of key anti-oxidant systems in chondrocytes including glutathione and the peroxiredoxins. Oxidative inactivation of anti-oxidant systems allows for rising levels of intracellular ROS that cause disruption of physiologic signaling. The failure of simple anti-oxidants to impact aging and age-related disease may be related to their inability to specifically target this disrupted signaling. Future interventions that can restore proper redox signaling in aging chondrocytes hold promise for the treatment of OA.
Footnotes
Potential Conflicts of Interest: This work was supported by grants from the National Institute on Aging (RO1 AG044034) and the National Institute of Arthritis, Musculoskeletal, and Skin Disease (R37 AR049003). Dr. Loeser serves as a consultant for Unity Biotechnology.
DISCUSSION
Billings, Baton Rouge: Last year, President Anne Moore commented on what was called a Pointe Vedra syndrome (a favorite location of ACCA members). That is familiar to everyone in the audience. And that was reported in 1981 by Daniel Deykin, and it showed that the bleeding time was prolonged not only by aspirin. It was not prolonged by 125 cc’s of vodka. But when they were added together there was potentiation and bleeding time was even more prolonged by the two together. So I am asking if vodka has anything to do with the gin-raisin issue that you discussed earlier. And is a joint affected directly by the alcohol, or is the cognition of what is going on in the joint affected.
Loeser, Chapel Hill: Well that’s a very interesting question. You’re supposed to let the raisins dry out so that the alcohol content drops off. I know there is an interest in this topic, because if you heard of the people’s pharmacy, they had recent[ly] a series of questions given to them and it was about whether golden raisins were as effective as the dark raisins for this particular therapy. So I think there is a lot of work that we still need to be doing.
Williams, Wilmington: As a geriatrician I advise my patients to walk at least 30 continuous minutes a day with the rationale that you’re stimulating the growth and repair side of the cascade and therefore inhibiting the decline in decay in inflammatory pathways. And it has to be 30 continuous minutes as opposed to two, 15-minute segments. Is there any rationale to that?
Loeser, Chapel Hill: That’s a good question. The other side of the research that I do on the clinical side is actually looking at the effects of exercise and weight loss for people with osteoarthritis in the knee. We have done several large randomized controlled trials to show that exercise and weight loss are a benefit. But when we’ve looked at biomarkers for cartilage degradation, and MRI changes, we haven’t been able to actually show any effect of exercise and weight loss directly on the tissue. So it’s probably more in terms of improving muscle function, reducing some of the mediators that come from fat by the weight loss side. But whether or not that’s directly impacting the tissue it’s been hard to show. But the point is important in terms of activity, because our joint tissues need a certain level of activity to stay healthy. The nutrition that you get to your cartilage comes in from the diffusion when you load your joint. So, for example, when they have done MRIs on people that have been paralyzed, you start to see a thinning and loss of joint tissue. So the activity probably plays an important role but demonstrating how that might benefit the arthritic joint has been hard to do so far.
Barondess, New York City: Thank you very much for clarifying the origin of the term gin-joint, which has perplexed me for a long time. My question has to do with the fact that most of the chronic diseases that dominate the causes of death in this and other developed countries appear particularly at older age. But the fact that they appear in older age does not mean that old age is causative. That association, in fact, turns out to be false for most of them, which have been present by a large for decades before they become clinically apparent. My question is do you have any information about how long before the clinical appearance of evidence of osteoarthritis, some of these metabolic and cellular changes appear?
Loeser, Chapel Hill: Yes, that’s a very good question because when patients come to see us with joint pain and loss of function they’re most commonly at a more advanced stage. There are imaging studies that are being done, particularly with MRI, to try to pick up earlier stages of diseases that are pre-symptomatic. And you can definitely find that 5 to 10 years before someone is symptomatic in longitudinal studies, that they have had some early changes within their joint tissues. The things that seem to be most predictive are for the knee joint and the meniscus. So, early degeneration or tears in the meniscus and early changes in the synovium, some inflammation in the synovium, seem to be predictive of disease occurring later. So, I do agree that it probably is starting at an earlier time in life. The other important issue is the risk factors for OA. If you have a joint injury — you tear your ACL — OA clearly develops earlier in life than if you haven’t had that type of joint injury. But comparing young and old adults with an ACL injury — when an older adult tears their ACL, OA develops much more rapidly than it would occur in a young adult. So, I think aging is probably interacting with many of these other risk factors.
Weinblatt, Boston: Richard, you talked today about potential targets that could be focused on for the management of osteoarthritis. As you know, we have no disease-modifying therapy outside of the orthopedic surgeon in OA, and with the population aging and the extreme costs to society of total joint replacements, which are exponentially increasing at a higher rate actually than some of the pharmaceutical products, where are we going to go with disease-modifying therapy for OA?
Loeser, Chapel Hill: Great question. There is a lot of interest again in disease-modifying therapy from the pharmaceutical industry. The bottleneck so far has been the FDA requirement that for registration of a drug being disease-modifying, you had to show that you slowed progression by measuring joint space width, which is a very insensitive measure. You have to have a large number of people to be able to see that. And that held a lot of companies back from going into that area. The Osteoarthritis Research Society (Marc Hochberg is up there to talk next) has been involved in trying to come up with new recommendations to the FDA for other measures such as MRI that might be better for drug companies to be able to use, rather plain films. So that is sort of where the field is now. There are disease modifying drugs that are in clinical trials right now and we’ll wait and see how those turn out. But the limitation has really been the outcome measures to register a drug that way.
Hochberg, Baltimore: So, a brief comment and a question. So, the comment is on Dr. Billings’ question: I think it’s the botanicals in gin as compared to the fact that vodka is just distilled from grain. But it would be interesting to have our ACCA group maybe do a randomized trial comparing different types of gin with different botanical mixtures to see if that might make a difference in their raisins and then a multifactorial design with sultanas versus dark raisins. So, the question is that the incidence of osteoarthritis as measured by radiographs tends to drop off in the 8th and 9th decades. It appears, then, that if people are going to get the radiographic changes of osteoarthritis, that they usually get it by a certain age. But they are then protected against it if they live longer. And more individuals are living longer, particularly the members of this society (which I think has a protective effect on survival). Do you have any speculations as to why some people are protected from getting osteoarthritis based on your research?
Loeser, Chapel Hill: I think that is a fantastic question and I don’t have the answer, but people are living longer and longer who don’t seem to get OA. Other aging changes may be also less pronounced in that particular population. So, it may be that these are people that have healthy aging. We know that aging is playing an important role in osteoarthritis, so they may have a number of different reasons why they have lived that long. And it may be related to some healthy genes and other things that also protect them from getting an age-related disease such as osteoarthritis.
REFERENCES
- 1.Prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation–united states, 2003–2005. MMWR Morb Mortal Wkly Rep. 2006;55:1089–92. [PubMed] [Google Scholar]
- 2.Cisternas MG, Murphy L, Sacks JJ, et al. Alternative methods for defining osteoarthritis and the impact on estimating prevalence in a us population-based survey. Arthritis Care Res. 2016;68:574–80. doi: 10.1002/acr.22721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hootman JM, Helmick CG. Projections of us prevalence of arthritis and associated activity limitations. Arthritis Rheum. 2006;54:226–9. doi: 10.1002/art.21562. [DOI] [PubMed] [Google Scholar]
- 4.Losina E, Paltiel AD, Weinstein AM, et al. Lifetime medical costs of knee osteoarthritis management in the united states: impact of extending indications for total knee arthroplasty. Arthritis Care Res. 2015;67:203–15. doi: 10.1002/acr.22412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunter DJ, Beavers DP, Eckstein F, et al. The intensive diet and exercise for arthritis (idea) trial: 18-month radiographic and mri outcomes. Osteoarthritis Cartilage. 2015;23:1090–8. doi: 10.1016/j.joca.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu SP, Hunter DJ. Emerging drugs for the treatment of knee osteoarthritis. Expert Opin Emerg Drugs. 2015;20:361–78. doi: 10.1517/14728214.2015.1037275. [DOI] [PubMed] [Google Scholar]
- 7.Cram P, Lu X, Kates SL, et al. Total knee arthroplasty volume, utilization, and outcomes among medicare beneficiaries, 1991-2010. JAMA. 2012;308:1227–36. doi: 10.1001/2012.jama.11153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bingham CO, 3rd, Buckland-Wright JC, Garnero P, et al. Risedronate decreases biochemical markers of cartilage degradation but does not decrease symptoms or slow radiographic progression in patients with medial compartment osteoarthritis of the knee: results of the two-year multinational knee osteoarthritis structural arthritis study. Arthritis Rheum. 2006;54:3494–507. doi: 10.1002/art.22160. [DOI] [PubMed] [Google Scholar]
- 9.Hellio le Graverand MP, Clemmer RS, Redifer P, et al. A 2-year randomised, double-blind, placebo-controlled, multicentre study of oral selective inos inhibitor, cindunistat (sd-6010), in patients with symptomatic osteoarthritis of the knee. Ann Rheum Dis. 2013;72:187–95. doi: 10.1136/annrheumdis-2012-202239. [DOI] [PubMed] [Google Scholar]
- 10.McAlindon T, LaValley M, Schneider E, et al. Effect of vitamin d supplementation on progression of knee pain and cartilage volume loss in patients with symptomatic osteoarthritis: a randomized controlled trial. JAMA. 2013;309:155–62. doi: 10.1001/jama.2012.164487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nature Rev Rheumatol. 2016;12:412–20. doi: 10.1038/nrrheum.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loeser RF, Goldring SR, Scanzello CR, et al. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verzijl N, Bank RA, TeKoppele JM, et al. Ageing and osteoarthritis: a different perspective. Curr Opin Rheumatol. 2003;15:616–22. doi: 10.1097/00002281-200309000-00016. [DOI] [PubMed] [Google Scholar]
- 14.Verzijl N, DeGroot J, Ben ZC, et al. Crosslinking by advanced glycation end products increases the stiffness of the collagen network in human articular cartilage: a possible mechanism through which age is a risk factor for osteoarthritis. Arthritis Rheum. 2002;46:114–23. doi: 10.1002/1529-0131(200201)46:1<114::AID-ART10025>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 15.Loeser RF. Aging processes and the development of osteoarthritis. Curr Opin Rheumatol. 2013;25:108–13. doi: 10.1097/BOR.0b013e32835a9428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang J, Wang Y, Shao L, et al. Clearance of senescent cells by abt263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nature Rev Rheumatol. 2011;7:161–9. doi: 10.1038/nrrheum.2010.213. [DOI] [PubMed] [Google Scholar]
- 19.Loeser RF. Aging and osteoarthritis. Curr Opin Rheumatol. 2011;23:492–6. doi: 10.1097/BOR.0b013e3283494005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hui W, Young DA, Rowan AD, et al. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann Rheum Dis. 2016;75:449–58. doi: 10.1136/annrheumdis-2014-206295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schriner SE, Linford NJ, Martin GM, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–11. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 22.Treuting PM, Linford NJ, Knoblaugh SE, et al. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J Gerontol A Biol Sci Med Sci. 2008;63:813–22. doi: 10.1093/gerona/63.8.813. [DOI] [PubMed] [Google Scholar]
- 23.Collins JA, Wood ST, Nelson KJ, et al. Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro-survival signalling in aging chondrocytes. J Biol Chem. 2016;291:6641–54. doi: 10.1074/jbc.M115.693523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loeser RF, Carlson CS, Carlo MD, et al. Detection of nitrotyrosine in aging and osteoarthritic cartilage: correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002;46:2349–57. doi: 10.1002/art.10496. [DOI] [PubMed] [Google Scholar]
- 25.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 27.Jones DP. Redox theory of aging. Redox Biology. 2015;5:71–9. doi: 10.1016/j.redox.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin W, Park JI, Loeser RF. Oxidative stress inhibits insulin-like growth factor-i induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-kinase-akt and mek-erk mapk signaling pathways. J Biol Chem. 2009;284:31972–81. doi: 10.1074/jbc.M109.056838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loeser RF, Gandhi U, Long DL, et al. Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol. 2014;66:2201–9. doi: 10.1002/art.38641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loeser RF, Shanker G, Carlson CS, et al. Reduction in the chondrocyte response to insulin-like growth factor 1 in aging and osteoarthritis: studies in a non-human primate model of naturally occurring disease. Arthritis Rheum. 2000;43:2110–20. doi: 10.1002/1529-0131(200009)43:9<2110::AID-ANR23>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 31.Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage. 2009;17:971–9. doi: 10.1016/j.joca.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo YY, Cruz TF. Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J Biol Chem. 1995;270:11727–30. doi: 10.1074/jbc.270.20.11727. [DOI] [PubMed] [Google Scholar]
- 33.Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c-jun nh2- terminal kinases. J Biol Chem. 1996;271:15703–7. doi: 10.1074/jbc.271.26.15703. [DOI] [PubMed] [Google Scholar]
- 34.Mendes AF, Caramona MM, Carvalho AP, et al. Differential roles of hydrogen peroxide and superoxide in mediating il-1-induced nf-kappab activation and inos expression in bovine articular chondrocytes. J Cell Biochem. 2003;88:783–93. doi: 10.1002/jcb.10428. [DOI] [PubMed] [Google Scholar]
- 35.Ahmad R, Sylvester J, Ahmad M, et al. Involvement of h-ras and reactive oxygen species in proinflammatory cytokine-induced matrix metalloproteinase-13 expression in human articular chondrocytes. Arch Biochem Biophys. 2011;507:350–5. doi: 10.1016/j.abb.2010.12.032. [DOI] [PubMed] [Google Scholar]
- 36.Del Carlo M, Schwartz D, Erickson EA, et al. Endogenous production of reactive oxygen species is required for stimulation of human articular chondrocyte matrix metalloproteinase production by fibronectin fragments. Free Radic Biol Med. 2007;42:1350–8. doi: 10.1016/j.freeradbiomed.2007.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forsyth CB, Cole A, Murphy G, et al. Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J Gerontol A Biol Sci Med Sci. 2005;60:1118–24. doi: 10.1093/gerona/60.9.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Del Carlo M, Jr., Loeser RF. Increased oxidative stress with aging reduces chondrocyte survival: correlation with intracellular glutathione levels. Arthritis Rheum. 2003;48:3419–30. doi: 10.1002/art.11338. [DOI] [PubMed] [Google Scholar]
- 39.Rhee SG, Woo HA, Kil IS, et al. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem. 2012;287:4403–10. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]