

Abstract
Purpose
To establish conditionally immortal mouse corneal endothelial cell lines with genetically matched Slc4a11+/+ and Slc4a11−/− mice as a model for investigating pathology and therapies for SLC4A11 associated congenital hereditary endothelial dystrophy (CHED) and Fuchs' endothelial corneal dystrophy.
Methods
We intercrossed H-2Kb-tsA58 mice (Immortomouse) expressing an IFN-γ dependent and temperature-sensitive mutant of the SV40 large T antigen (tsTAg) with Slc4a11+/+ and Slc4a11−/− C57BL/6 mice. The growth characteristics of the cell lines was assessed by doubling time. Ion transport activities (Na+/H+ exchange, bicarbonate, lactate, and Slc4a11 ammonia transport) were analyzed by intracellular pH measurement. The metabolic status of the cell lines was assessed by analyzing TCA cycle intermediates via gas chromatography mass spectrometry (GC-MS).
Results
The immortalized Slc4a11+/+ and Slc4a11−/− mouse corneal endothelial cells (MCECs) remained proliferative through passage 49 and maintained similar active ion transport activity. As expected, proliferation was temperature sensitive and IFN-γ dependent. Slc4a11−/− MCECs exhibited decreased proliferative capacity, reduced NH3:H+ transport, altered expression of glutaminolysis enzymes similar to the Slc4a11−/− mouse, and reduced proportion of TCA cycle intermediates derived from glutamine with compensatory increases in glucose flux compared with Slc4a11+/+ MCECs.
Conclusions
This is the first report of the immortalization of MCECs. Ion transport of the immortalized endothelial cells remains active, except for NH3:H+ transporter activity in Slc4a11−/− MCECs. Furthermore, Slc4a11−/− MCECs recapitulate the glutaminolysis defects observed in Slc4a11−/− mouse corneal endothelium, providing an excellent tool to study the pathogenesis of SLC4A11 mutations associated with corneal endothelial dystrophies and to screen potential therapeutic agents.
Keywords: Slc4a11 knockout, corneal endothelium, glutamine, ammonia
SLC4A11 mutations are associated with congenital hereditary endothelial corneal dystrophy (CHED), Fuchs' endothelial corneal dystrophy, Harboyan syndrome (CHED plus perceptive deafness), and Peters anomaly.1–4 Up to 80 distinct mutations in 17 of the 19 exons of SLC4A11 have been identified in individuals with CHED,3,5–17 which is characterized at or soon after birth by bilateral diffuse corneal edema without other significant developmental abnormalities of the anterior segment.18 Histologically, the diffusely edematous corneal stroma accounts for the marked increase in corneal thickness observed clinically.19 In addition, CHED is associated with a uniform thickening of Descemet's membrane and vacuolization of corneal endothelium.20 The Slc4a11−/− C57BL/6 mouse recapitulates the human CHED-related defects, exhibiting a similar ground-glass diffuse corneal edema, increased corneal thickness, vacuolated corneal endothelial cells, and uniformly thickened Descemet's membrane.21,22 Furthermore, similar to human CHED,23 endothelial cell density in Slc4a11−/− C57BL/6 mice is not significantly affected at early age.22 Based on these observations, both Han et al.22 and the authors of this study concluded that this Slc4a11−/− C57BL/6 mouse can serve as a good animal model for human CHED.
Recent functional studies of SLC4A11 indicate that it is permeable to H+24–26 and can act as an NH3:H+ cotransporter.24,25,27 We recently demonstrated that this ammonia permeability is essential for human corneal endothelial cells, as these cells produce ammonia as a by-product of glutamine catabolism, which is required to maintain a high level of ATP production.21 Furthermore, we found that Slc4a11−/− mouse corneal endothelium has disrupted expression of enzymes involved in glutamine metabolism.21 However, only a very limited amount of corneal endothelial material can be obtained from mice, constraining further cellular, molecular, and metabolic studies.
Of neural crest origin, differentiated corneal endothelial cells are arrested in the G1 phase of the cell cycle, and studies in both mice and humans have revealed that these cells have very low proliferative potential in vivo.28 When cultured in vitro, corneal endothelial cells manifest limited passaging ability with rapid senescence and epithelial-to-mesenchymal (EMT) transition.29 This limited proliferative potential makes obtaining sufficient corneal endothelial sample a significant challenge for studying the molecular mechanisms that underlie corneal physiology and the pathophysiology in corneal endothelial diseases.
To overcome these limitations, we intercrossed the Slc4a11+/+ and Slc4a11−/− C57BL/6 mice22 with H-2Kb-tsA58 Immortomouse, which carries a temperature-sensitive mutant form of the simian virus-40 large T antigen.30 We used the resulting progeny to generate two conditionally immortal, genetically matched mouse corneal endothelial cell lines: Slc4a11+/+ mouse corneal endothelial cells (Slc4a11+/+ MCEC) and Slc4a11−/− mouse corneal endothelial cells (Slc4a11−/− MCEC). Here we report the successful establishment of these two genetically matched wild-type and Slc4a11 knockout MCECs. We examined proliferative properties, transport activity, and Slc4a11-related glutaminolysis activity. The resultant cells retain key endothelial transport function and the Slc4a11-deficient cells show altered glutamine metabolism consistent with what was observed in Slc4a11−/− mouse corneal endothelium.21 Overall, the generation of these cell lines establishes a valuable tool for studying aspects of corneal endothelial diseases that require a large number of cells.
Methods
Animal Genotyping
All mice were housed and maintained in specific pathogen-free conditions and used in the experiments in accordance with institutional guidelines and the current regulations of the National Institutes of Health, the US Department of Health and Human Services, the US Department of Agriculture, and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
The H-2Kb-tsA58 transgenic Immortomouse (Charles River Laboratories, Wilmington, MA, USA) was intercrossed with Slc4a11+/+ and Slc4a11−/− C57BL/6 mice, respectively, to generate offspring bearing the Immortomouse transgene together with Slc4a11+/+ or Slc4a11−/−. Slc4a11−/− C57BL/6 mice have a targeted deletion of exons 9 to 13 of the murine Slc4a11 gene.31 The offspring were genotyped by PCR of genomic DNA from ear-punch sections following protocols using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Oligomer sequences (5′–3′) used for PCR were as follows: primer 1 (Immorto-F), AGT CCT CAC AGT CTG TTC ATG ATC; primer 2 (null-F), GAT CTG CCT GAG GTG TTA CTT G; primer 3 (null-R and Immorto-R, common reverse primer), GGA TGG CAT CAC TAG TCA TGA C; primer 4 (Slc4a11 wt-F), TCT GGA CTT CAA CGC CTT CT; primer 5 (Slc4a11 ko-F), GCC AAG GTA TGG AGA ACA CC; primer 6 (Slc4a11 wt-R and ko-R, common reverse primer), GCA CAA ACG TGA TGG AAA TG. Primers 1 and 3 were used to identify the SV40 transgene, Immorto (tsTAg). Primers 2 and 3 were used to identify the null allele. Primers 4 and 6 were used to identify the Slc4a11 wild-type allele, and primers 5 and 6 were used to identify Slc4a11 knockout allele. The first generations were bred, and the second-generation pups were genotyped by PCR to identify Slc4a11+/+ and Slc4a11−/− animals that carried at least one copy of the SV40 tsTAg transgene to be used for tissue isolation.
Generation of MCEC Cultures of Slc4a11+/+ and Slc4a11−/− Genotypes
MCECs were prepared from 12-week-old mice. Briefly, the globes were aseptically enucleated followed by cornea dissection and corneal endothelium peeling. Then corneal endothelial sheets with attached Descemet's membrane were placed in OptiMEM-I medium (#51985; Thermo Fisher Scientific, Canoga Park, CA, USA) supplemented with 8% heat-inactivated fetal bovine serum (FBS) (#10082139; Thermo Fisher Scientific), EGF 5 ng/mL (#01-107, Millipore, Darmstadt, Germany), pituitary extract 100 μg/mL (Hyclone Laboratories, Logan, UT, USA), calcium chloride 200 mg/L, 0.08% chondroitin sulfate (#G6737; Sigma-Aldrich Corp., St. Louis, MO, USA), gentamicin 50 μg/mL (#15710072; Thermo Fisher Scientific), antibiotic/antimycotic solution diluted 1:100 (#15240062; Thermo Fisher Scientific) and 44 units/mL IFN-γ (#485-MI; R&D Systems, Minneapolis, MN, USA). IFN-γ was used to stimulate the MHC promoter for tsTAg expression. Cells were further selected based on morphology via single-cell cloning for colonies with corneal endothelial hexagonal shape and contact inhibition. Cells were incubated at 33°C with 5% carbon dioxide.
The two lines, Slc4a11+/+ MCEC and Slc4a11−/− MCEC, were genotyped by PCR following protocols using QIAamp DNA Mini Kit (Qiagen). Primer sequences used were the same as the primers used for mouse genotyping listed above.
Cell Expansion and Continuous Propagation
MCECs were cultured at 33°C (5% CO2) or 37°C (5% CO2) in OptiMEM-I medium (51985; Thermo Fisher Scientific) supplemented with 8% heat-inactivated FBS (10082139; Thermo Fisher Scientific), EGF 5 ng/mL (01-107; Millipore), pituitary extract 100 μg/mL (Hyclone Laboratories), calcium chloride 200 mg/L, 0.08% chondroitin sulfate (G6737, Sigma-Aldrich Corp.), gentamicin 50 μg/mL (15710072; Thermo Fisher Scientific), antibiotic/antimycotic solution diluted 1:100 (15240062; Thermo Fisher Scientific) and with or without 44 units/mL IFN-γ (485-MI; R&D Systems). Culture at 33°C with IFN-γ is defined as permissive growth condition, whereas culture at 37°C without IFN-γ is defined as nonpermissive growth condition.
Light Microscopy
Cell morphology images were acquired with an Infinity I camera (Lumenera Corp., Ottawa, ON, Canada) attached to an inverted phase-contrast microscope.
Growth Curve and Doubling Time
MCEC cells were seeded at 5 × 103 or 1 × 104/mL (total 2500 cells/well or 5000 cells/well) in 24-well plates. Cells in four wells of 24-well plates were trypsinized to suspension and counted with a Cellometer Auto T4 (Nexcelom Bioscience, Lawrence, MA, USA). Doubling time was calculated using GraphPad Prism 6.1c (GraphPad Software, Inc., La Jolla, CA, USA).
Western Blot of SV40 Large T Antigen
MCECs were seeded at 5 × 104/mL (total 1 × 105 cells/well) in six 35-mm dishes and cultured with IFN-γ at 33°C to subconfluence. While one dish was subject to protein extraction as the sample for the permissive condition, the remaining five dishes were switched to 37°C without IFN-γ for the subsequent nonpermissive culture. In the following 5 days, one dish was subjected to protein extraction on each day (24, 48, 72, 96, and 120 hours) in the nonpermissive condition. Cells in each dish were washed two times with ice-cold PBS and suspended in 100 μL RIPA buffer (50 mM Tris base, 150 mM NaCl, 0.5% deoxycholic acid-sodium salt, 2% SDS, and 1% NP40, pH 7.5) with Complete Protease Inhibitor Cocktail (#4693159001; Roche Diagnostics, Indianapolis, IN, USA). Then the sample was sonicated and centrifuged for 20 minutes at 12000g at 4°C. Cell lysate samples (15 μL) were mixed with Protein Loading Buffer Blue (2X) (EC-886, National Diagnostics, Atlanta, GA, USA) and a total volume of 30 μL was resolved on 1.5-mm-thick 10% SDS-polyacrylamide gels and wet-transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 5% nonfat milk in TBST (25 mM Tris base, 137 mM NaCl, 0.1% Tween20) and probed with primary antibodies in the same buffer overnight at 4°C. The following primary antibodies were used: rabbit anti-SV40 T Ag 1:5000 (sc-20800; Santa Cruz Biotechnology, Dallas, TX, USA); and anti–glyceraldehyde 3-phosphate dehydrogenase antibody 1:1000 (sc-32233; Santa Cruz Biotechnology). Next, membranes were probed with secondary antibody (goat anti-rabbit IgG peroxidase-conjugated antibody, #A0545, or goat anti-mouse IgG peroxidase-conjugated antibody, #A8924; Sigma-Aldrich Corp.) for an hour at room temperature. Bound secondary antibodies were detected using an enhanced chemiluminescence assay (Supersignal West Pico, #34080; Thermo Fisher Scientific). Band densities with background subtraction were quantified using ImageJ (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA).
Intracellular pH (pHi) Measurement
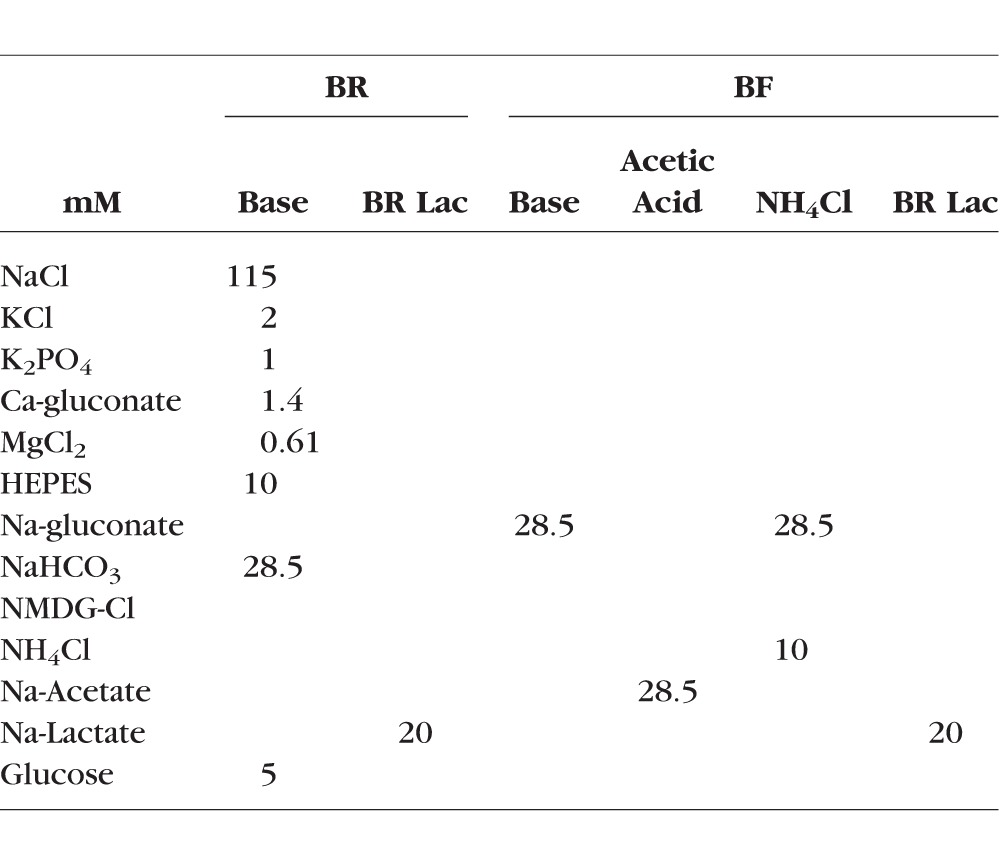
pHi measurements were performed as described previously.32 Briefly, MCECs were cultured on poly-L-lysine and fibronectin precoated 25-mm diameter glass coverslips (GG-25-pdl; Neuvitro Corporation, Vancouver, WA, USA) for 2 to 3 days in semipermissive condition. Before each experiment, cells were incubated with 10 μM of pH-sensitive fluorescent dye BCECF-AM (2′7′-bis(carboxyethyl)-5(6)-carboxyfluorescein-acetoxymethyl ester, B1170; Thermo Fisher Scientific) in Ringer's solution for 30 minutes at room temperature, and washed in dye-free Ringer's solution for another 30 minutes. The Ringer's solution constitution is listed in Table 1. All of the experimental solutions were equilibrated with air (or 5% CO2 for bicarbonate-rich [BR] solutions) and adjusted to pH 7.5 with 1N NaOH at 37°C. Osmolarity of all solutions was adjusted to 295 to 300 mOsm with mannitol. Coverslips with subconfluent cells were mounted into a perfusion chamber, and the chamber was then placed on a stage warmer (37°C) of an inverted microscope (Eclipse TE200; Nikon, Tokyo, Japan). Solutions were kept at 37°C in a warming box, and the flow of the perfusate (∼0.5 mL/min) was achieved by gravity. Cells were imaged with an oil-immersion objective (×40; Nikon). BCECF fluorescence was excited alternately at 495 ± 10 nm and 440 ± 10 nm, and the emitted light was collected through a bandpass filter (520–550 nm). Fluorescence ratios (495/440) were obtained at 1 Hz, and converted into pHi using the high K+/nigericin calibration approach.33
Table 1.
Composition of Extracellular Solutions, mM
RNA Extraction, RT-PCR, and Real-Time Quantitative PCR
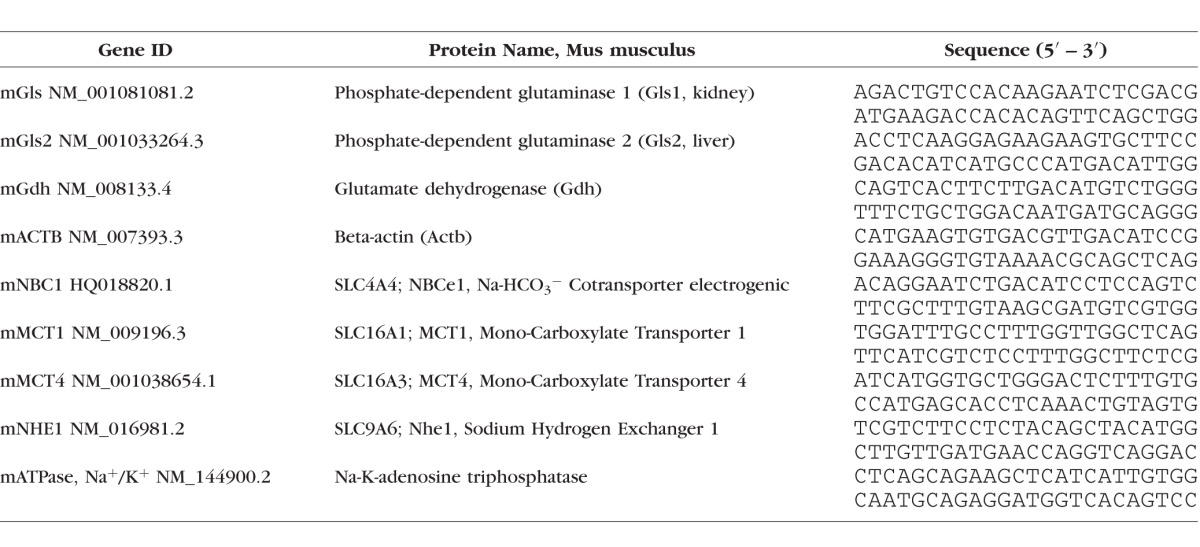
Total RNA from the MCEC cell lines (cultured in 33°C, IFN-γ [+]) was extracted and purified using the RNeasy mini kit (#74104; Qiagen) with DNase digestion (#79254; Qiagen). Complementary DNA was generated with a High Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA, USA) at 10 ng RNA/μL reverse transcription reaction concentration. Real-time quantitative PCR reactions were set up in triplicate using PowerUp SYBR Green Master Mix (A25741; Thermo Fisher Scientific). Reactions were performed with murine gene primers listed in Table 2. A 2−ΔΔCt experimental design was used for relative quantification and normalized to mouse ACTB (β-actin) for differential expression levels of target genes.
Table 2.
Murine Gene Primers
Metabolites Extraction and Gas Chromatography Mass Spectrometry (GC-MS) Measurements
Slc4a11+/+ and Slc4a11−/− MCEC in 8% FBS OptiMEM-I in T-75 flasks seeded at 5 × 104/mL (total 2.5 × 105 cells) were grown to confluence for 4 days at 33°C with IFN-γ then switched to serum-free OptiMEM-I supplemented with 200 mg/L CaCl2 and 0.08% chondroitin sulfate for 12 hours. Then the cells were incubated with serum-free Dulbecco's modified Eagle's medium (DMEM) conditional medium with 2.5 g/L U-13C6-D-glucose (CLM-1396-0; Cambridge Isotope Laboratories, Tewksbury, MA, USA) + 4 mM L-glutamine and incubated for 12 hours. Serum-free DMEM conditional medium was made using DMEM (glucose-free, glutamine-free, pyruvate-free, GIBCO #A1443001; Thermo Fisher Scientific) supplemented with CaCl2 200 mg/L, 0.08% chondroitin sulfate (Sigma-Aldrich Corp.), and the isotope-labeled glucose and nonlabeled glutamine (Sigma-Aldrich Corp.). After 12-hour incubation, isotope-labeled cells were washed three times with ice-cold 0.9% NaCl, quenched with 2 mL ice-cold 100% methanol, scraped off the plate and removed together with 2 mL quenching methanol into centrifuge tubes, vortexed thoroughly, and centrifuged at 8000g for 2 minutes. Supernatants were collected and the cell pellet was resuspended with 900 μL 90% methanol twice following vortex and centrifugation. Total supernatant was kept stationary at −20°C for 1 hour, then centrifuged at 15,000g for 5 minutes at 4°C. The supernatant was collected to new tubes and dried in a centrifugal evaporator at room temperature overnight. The level of each metabolite isotopologue was measured using GC-MS as reported previously.34 The fraction of each isotopologue and the contribution of 13C to the total carbon pool of each metabolite was calculated as previously reported.35 Briefly, the isotopologue distributions were corrected based on the natural abundance of elements, and the fraction is the contribution of each isotopologue to the total abundance of all the isotopologues. The contribution of 13C to the total carbon pool of each metabolite is the weighted average of all the labeled isotopologues according to the fraction distribution.
Statistical Analysis
Statistical analysis was carried out using GraphPad Prime 6.0c (GraphPad Software, Inc., La Jolla, CA, USA). Student's t-test or paired t-test were used for two-group comparison. Two-way ANOVA was used for metabolite data.
Results
Conditional Immortal Slc4a11+/+ MCEC and Slc4a11−/− MCEC Growth Characteristics
The genotypes of both the mice used for cell expansion (Fig. 1A, left panel) and of the derived MCECs (Fig. 1A, right panel), Slc4a11+/+ MCECs, and Slc4a11−/− MCECs, were determined by PCR analysis of mouse or cell line genomic DNA. For the mice used, Slc4a11 wild-type alleles are present in Slc4a11+/+ mice (left two lanes) and in Slc4a11+/− mice (right two lanes), whereas Slc4a11 knockout alleles are present in Slc4a11−/− mice (middle two lanes) and in Slc4a11+/− mice. For Immorto (tsTAg) gene, all of the mice we used carry only one allele of the gene, and the other allele is detected as a null allele. We selected Slc4a11+/+ Immorto+/− mice and Slc4a11−/− Immorto+/− mice for cell expansion. Genotypes of derived cell lines were confirmed by PCR of the cell line genomic DNA for Slc4a11 and Immorto (tsTAg) genes (Fig. 1A, right panel). Verification of Slc4a11 mRNA expression in the cell lines was carried out by RT-PCR showing only Slc4a11+/+ but not Slc4a11−/− MCEC expresses Slc4a11 mRNA (Fig. 1B).
Figure 1.

Characterization of Slc4a11+/+ and Slc4a11−/− MCECs. (A) Genotypes of mice used for corneal endothelial cell collection, and genotypes of cultured MCECs. Slc4a11 wild-type (wt) and knockout (ko) alleles were identified with specific primers yielding products of 353 bp and 386 bp, respectively. Immorto (tsTAg) and null alleles yielded PCR products of 500 bp and 300 bp, respectively. Slc4a11+/+ and Slc4a11−/− mice with one allele of Immorto gene were used for cell derivation. Slc4a11+/+ and Slc4a11−/− MCECs show the expected genotype of Slc4a11 and tsTAg gene. (B) RT-PCR of Slc4a11+/+ and Slc4a11−/− MCECs verified that no Slc4a11 mRNA is expressed in ko. (C) Morphology of Slc4a11+/+ and Slc4a11−/− MCECs cultured at 33°C with and without IFN-γ. (D) Growth curve of Slc4a11+/+ and Slc4a11−/− MCECs cultured at 33°C with and without IFN-γ.
Next, we examined the cell morphology and growth doubling time of Slc4a11+/+ MCECs and Slc4a11−/− MCECs. The two cell lines remained proliferative to passage 49 and are still propagating. We noticed a fibroblastic-like cell morphology in Slc4a11+/+ MCECs cultured under permissive conditions (IFN-γ [+], 33°C), in which the cells are elongated and lacking contact inhibition at high density. However, Slc4a11−/− MCECs had a more hexagonal shape closely resembling corneal endothelial primary culture and form a monolayer when confluent in the same IFN-γ (+) medium (Fig. 1C). We asked if IFN-γ stimulated SV40 large T antigen (tsTAg) expression changes the MCEC morphology, so IFN-γ was removed, but the cells were kept at 33°C to see if the morphology was affected in Slc4a11+/+ MCECs. We define this 33°C IFN-γ (−) culture condition as “semipermissive.” Indeed, in the semipermissive condition, Slc4a11+/+ MCECs presented with a more hexagonal shape and cell monolayer was formed once confluent (Fig. 1C). In contrast, Slc4a11−/− MCEC morphology in semipermissive culture was similar to that in permissive culture (Fig. 1C). In terms of growth, although there is a small difference (P = 0.013) in the doubling time between Slc4a11+/+ MCECs (0.78 day) and Slc4a11−/− MCECs (0.79 day) in the permissive condition, Slc4a11−/− MCECs (doubling time: 0.93 day) grow significantly slower than Slc4a11+/+ MCECs (0.76 day) in the semipermissive condition (P < 0.0001) (Fig. 1D).
Removal of IFN-γ reduces stimulation of the HMC promoter constructed upstream of the Immorto (tsTAg) gene. Given the dynamic SV40 large T antigen expression changes expected from IFN-γ withdrawal and change in culture temperature, we next analyzed the growth behavior of the MCECs under permissive (33°C, IFN-γ [+]) and subsequent nonpermissive conditions (37°C, IFN-γ [−]). In nonpermissive culture, in addition to the lack of IFN-γ stimulation on the HMC promoter, a shift to the 37°C incubation temperature induces degradation of the tsTAg mutant protein. As a result, MCEC proliferation gradually stopped once the culture was removed from the permissive condition into the nonpermissive condition (Fig. 2A). In addition, we also performed Western blot to analyze the SV40 large T antigen level in this series in comparison to SV40 large T level in semipermissive culture (33°C, IFN-γ [−]) (Fig. 2B). Both Slc4a11+/+ and Slc4a11−/− MCECs showed very little SV40 large T antigen at 4 weeks of semipermissive culture and temperature maneuver minimally affected the SV40 large T level (Fig. 2B, first three lanes). The SV40 large T antigen level was highest in the permissive culture (33°C, IFN-γ [+]). When immediately moved to the nonpermissive culture (37°C, without IFN-γ), there was a time-dependent decline in large T antigen level, in parallel with the decline of cell proliferation (Fig. 2A, 2B). To summarize, semipermissive culture of Slc4a11+/+ MCECs and Slc4a11−/− MCECs maintains cell proliferation and corneal endothelial morphology with declining level of SV40 large T antigen expression. Due to the rapid decline of cell proliferation and accompanied large cell loss within the first 2 days of moving from 33°C permissive to 37°C nonpermissive culture, which could introduce large variances in experimental results, together with good endothelial morphology and declining large T antigen levels in the semipermissive condition (33°C, IFN-γ [−]), we chose to test the ion transporter properties of these cells in the semipermissive state.
Figure 2.

SV40 expression and growth characteristics in permissive, semipermissive, and nonpermissive conditions. (A) Growth profile of Slc4a11+/+ MCECs and Slc4a11−/− MCECs when transferred from permissive culture (33°, IFN-γ [+]) to nonpermissive culture (37°C, IFN-γ [−]). (B) Western blot analysis of SV40 large T antigen expression in Slc4a11+/+ MCECs and Slc4a11−/− MCECs under semipermissive culture (33°, IFN-γ [−], 4 weeks), and from permissive culture (33°, IFN-γ [+]) to nonpermissive culture (37°, IFN-γ [−]), showing a decrease of SV40 in nonpermissive culture over time.
Slc4a11+/+ and Slc4a11−/− MCEC Have Similar Active Ion Transport Activity
To verify that these MCECs still carry significant ion transport activities, we next examined the mRNA expression and transport activity of selected ion transporters. RT-PCR analysis of Slc4a11+/+ and Slc4a11−/− MCECs revealed that these cells express mRNA encoding the essential transporters Na+-K+-ATPase, Na+/H+ exchanger (SLC9A6; NHE1, Sodium Hydrogen Exchanger 1), Na+-HCO3− cotransporter (SLC4A4; NBCe1, Sodium Bicarbonate Cotransporter electrogenic), H+-Lactate− cotransporters (SLC16A1; MCT1, Mono-Carboxylate Transporter 1; and SLC16A3; MCT4, Mono-Carboxylate Transporter 4) (Fig. 3A). Similarly, BCECF-based continuous pHi monitoring of apparent proton fluxes indicate the activity of bicarbonate transporters (Fig. 3B), Na+/H+ exchangers (Fig. 3C), and lactate transporters (Fig. 3D) are present and comparable between the Slc4a11+/+ and Slc4a11−/− MCECs (see below).
Figure 3.

Na+/H+ exchanger, Na+-HCO3− cotransporter, and H+-lactate cotransporter activity in Slc4a11+/+ and Slc4a11−/− MCECs. (A) RT-PCR analysis of selected transporter mRNA expression in Slc4a11+/+ MCECs and Slc4a11−/− MCECs, including Na+-K+-ATPase, Na+/H+ exchanger (NHE1), Na+-HCO3− cotransporter (NBC1), and H+-lactate cotransporter (MCT1 and MCT4). (B) Analysis of apparent bicarbonate transporter activity. (C) Analysis of apparent Na+/H+ exchanger activity. (D) Analysis of apparent lactate transporter activity. Bar graphs show summary statistics.
Sodium Bicarbonate Cotransporter: In Figure 3B left panel, cells were initially perfused with bicarbonate-free (BF) Ringer's and then changed to BR ([HCO3−] 28.5 mM) Ringer's. Due to simple diffusion of dissolved CO2 inward across the plasma membrane, the cell rapidly acidifies as CO2 reacts with intracellular H2O to form H2CO3 and further release one H+ as a weak acid. Then pHi slowly rises, which indicates the Na-HCO3− cotransporters are moving weak base HCO3− inward using Na+ inward transmembrane electrochemical gradient. We determined the initial slope of this pHi rise as an indirect measure of the apparent Na+-HCO3− cotransport activity. This assay revealed no significant difference between Slc4a11+/+ and Slc4a11−/− MCECs in apparent bicarbonate transporter activity (Fig. 3B, Student's t-test, P = 0.48, n = 3 in each group). The overshoot after CO2/HCO3− removal is an additional indication that there was HCO3− accumulation inside the cell during BR perfusion.
Na+/H+ exchanger: In Figure 3C left panel, cells were initially perfused with BF Ringer's and then switched to BF Ringer's containing 28.5 mM Na-Acetate. Once dissolved in solution, a small fraction of Na-Acetate becomes noncharged acetic acid that readily diffuses across the membrane and rapidly acidifies the cell cytosol. Then pHi recovers due to the activity of Na+/H+ exchangers and Slc4a11 proton permeability.25 Here, we measured the initial slope of this pHi recovery as an indirect measure of the apparent Na+/H+ exchanger activity, revealing that there is a borderline significant difference between Slc4a11+/+ and Slc4a11−/− MCECs (Fig. 3C, paired t-test, 2-tailed P = 0.10, 1-tailed P = 0.05, n = 3 in each group) that can be attributed to Slc4a11 proton permeability.25
H+-lactate− cotransporter: Because we recently demonstrated that lactate transport is a significant component of the corneal endothelial pump,36 we next examined the lactate-dependent H+ fluxes in MCECs. Cells were initially perfused with BR Ringer's, and where indicated (horizontal bars), 20 mM lactate (dissolved in BR Ringer's, pH 7.4) was applied twice for 60 seconds (Fig. 3D, upper panel). The cells were then switched to perfusion with BF Ringer's and again 20 mM lactate (dissolved in BF Ringer's, pH 7.4) was applied twice for 60 seconds. Lactate exposure (pH 7.4) induces intracellular acidification due to the activity of H+-lactate cotransporters (Mono-Carboxylate Transporters, MCTs),37 in which the lactate transmembrane inward gradient favors the inward movement of lactate and H+ together causing intracellular H+ accumulation. The acidification is less prominent in BR than in BF Ringer's because HCO3−/CO32− offers extra H+ buffering power. We measured the initial slope of the induced acidification on lactate perfusion as an indirect measure of the apparent H+-lactate cotransporter activity. Figure 3D lower panel shows there was no significant difference between Slc4a11+/+ and Slc4a11−/− MCECs in both BF and BR Ringer's (t-test, in BR P = 0.25, in BF P = 0.35, n = 3 in each group).
Slc4a11 NH3:H+ Flux Is Intact in Slc4a11+/+ MCECs But Impaired in Slc4a11−/− MCECs
We recently reported that human SLC4A11 mediates electrogenic transmembrane NH3:H+ fluxes,27 and this result has been verified by two other independent groups.25,26 Furthermore, a recent report from a third group shows murine Slc4a11 mediates similar electrogenic transmembrane NH3:H+ fluxes.24 So we next set out to determine if the Slc4a11-mediated NH3:H+ flux is impaired in Slc4a11−/− MCECs (Fig. 4A). Cells were initially incubated with BF Ringer's, pulsed with 10 mM NH4Cl in BF Ringer's, and then switched back to BF Ringer's. Once dissolved in solution, a small fraction of NH4Cl forms NH3, a small noncharged molecule that is readily membrane diffusible. On entry, NH3 instantaneously reacts with intracellular H2O to form NH4+ and releases OH−. The latter rapidly alkalinizes the cell, causing the initial rapid alkalinizing phase (100–120 seconds) on NH4Cl application. Eventually the cell will reach an equilibrium where intracellular [NH3] equals extracellular [NH3]. Then a slow acidification occurs in the midphase (120–400 seconds) of the NH4Cl pulse, indicating there is an additional weak acid NH4+ (or NH3:H+ equivalently) flux entering the cell.38 In this case, Slc4a11 activity brings more NH3:H+ into the cell. Figure 4B shows that the rate of this slow acidification is significantly faster in Slc4a11+/+ MCECs compared with Slc4a11−/− MCECs, consistent with the additional NH3:H+ influx provided by Slc4a11. Then, when the NH4Cl was washed away by BF Ringer's, there is a pronounced and rapid acidification on NH4Cl removal (400–440 seconds) (Fig. 4B). This is due to the rapid exit of NH3 gas and the conversion of accumulated NH4+ to NH3 + H+. The rapid acid loading immediately after NH4Cl removal is a reflection of the amount of weak acid (NH4+ or NH3:H+) that has entered the cell during the NH4Cl pulse.38 And even though NH4Cl was removed in this phase on the outside, there is NH4+ temporarily trapped intracellularly.38 The amount of NH4+ trapped is directly correlated with the extent of the acidification according to the Henderson-Hasselbalch equation.38 Figure 4A shows that the depth of this acid load is much greater in Slc4a11+/+ MCECs. The pHi recovery (440–520 seconds) from this acid load is a phenomenon of the collective effect from Slc4a11-mediated NH3:H+ efflux and Na+/H+ exchanger-mediated H+ extrusion. We measured the initial rate of pHi recovery as a measure of apparent NH3:H+ efflux primarily attributed to Slc4a11 activity. We observed a rapid recovery in Slc4a11+/+ MCECs, but significantly slower recovery in Slc4a11−/− MCECs (Fig. 4B). To more accurately represent the Slc4a11-mediated NH3:H+ efflux, we performed further analysis by subtracting the Na+/H+ exchanger-mediated apparent pHi recovery (Fig. 3C), to obtain the adjusted NH3:H+ efflux. The average of apparent Na+/H+ exchanger activity (0.0014/s) between Slc4a11+/+ and Slc4a11−/− MCECs was used given there was no statistical significance between the two cell lines. Figure 4C shows that the adjusted NH3:H+ efflux is 0.0044 ± 0.0015/s (n = 5) in Slc4a11+/+ MCECs, and significantly smaller 0.0005 ± 0.0003/s (n = 5, P = 0.0347) in Slc4a11−/− MCECs.
Figure 4.

NH3:H+ Flux in Slc4a11+/+ and Slc4a11−/− MCECs. (A) Analysis of NH3:H+ cotransporter activity in Slc4a11+/+ MCECs and Slc4a11−/− MCECs. Cells loaded with the pH-sensitive dye BCECF were perfused in BF Ringer's and 10 mM NH4Cl was applied for 5 minutes. (B) Apparent NH3:H+ influx (P = 0.009, gradual acidification phase during NH4Cl pulse) and NH3:H+ efflux (P = 0.035, intracellular pH recovery phase after NH4Cl pulse) were both impaired in Slc4a11−/− MCECs compared with Slc4a11+/+ MCECs. (C) Adjusted NH3:H+ efflux was calculated from apparent NH3:H+ efflux subtracted by apparent Na+/H+ exchanger-mediated H+ efflux. Adjusted NH3:H+ efflux is close to zero in Slc4a11−/− MCECs. *P < 0.05.
Impaired Glutaminolysis Is Present in Slc4a11−/− MCECs as Seen in Slc4a11−/− CHED Mouse Model
Many of the enzymes involved in glutamine metabolism exhibit an aberrant expression pattern in Slc4a11−/− mouse corneal endothelium,21 although the kidney-type glutaminase 1 (Gls1) is upregulated 3-fold in Slc4a11−/− mouse corneal endothelium, the liver-type glutaminase 2 (Gls2) can no longer be detected in these cells. Therefore, we set out to determine if the Slc4a11−/− MCEC recapitulates this expression difference and manifests as functional differences in glutamine metabolism. Consistent with the observation in mouse corneal endothelial tissue in vivo, we observed an upregulation of Gls1, and a downregulation of Gls2 mRNA expression in Slc4a11−/− MCECs (Fig. 5A). These changes in gene expression were also evident at the level of metabolic flux. Our previous studies demonstrated that glutamine supplies approximately 50% of TCA cycle carbon chains in human corneal endothelium.21 Consistent with this earlier observation, we find that 50% of citrate and approximately 35% of α-ketoglutarate (α-KG), fumarate, and malate carbon chains were labeled with U-13C-glucose-sourced carbon in Slc4a11+/+ MCECs in the presence of unlabeled glutamine (Fig. 5C). Figure 5B shows the schematic of TCA cycle intermediates analyzed, where 13C isotope-labeled green in the setting where cells were cultured with U-13C-glucose and unlabeled glutamine. In contrast to wild type, Slc4a11−/− MCECs exhibit a significant increase in the percentage of TCA cycle intermediates that contain carbons derived from U-13C-glucose, with approximately 75% of citrate and approximately 50% of α-KG, fumarate, and malate carbon chains being labeled with U-13C-glucose-sourced carbon. These results indicate that the contribution of glutamine to the TCA cycle is decreased in Slc4a11−/− MCECs relative to Slc4a11+/+ MCECs and support our previous observation that SLC4a11 is essential for facilitating glutaminolysis.
Figure 5.

Analysis of glutaminolysis in Slc4a11+/+ and Slc4a11−/− MCECs. (A) Real-time quantitative PCR of glutaminolysis enzymes in Slc4a11+/+ and Slc4a11−/− MCECs. There is a 3-fold increase of Gls1 (P = 0.013) and a 20% decrease of Gls2 (P = 0.031) expression in Slc4a11−/− MCECs. Gdh, glutamate dehydrogenase. (B) Schematic of the TCA cycle. Green dots indicate 13C, whereas gray dots are 12C. Dark green–colored 13C enters the TCA cycle as acetyl-CoA through pyruvate dehydrogenase, while light green–colored 13C potentially enters the TCA cycle as oxaloacetate through pyruvate carboxylase, and gray-colored 12C enters the TCA cycle through glutamine. (C) Fraction of 13C-labeled TCA cycle intermediates (green shading) from cells fed with U-13C-glucose in the presence of unlabeled glutamine is significantly higher in Slc4a11−/− MCECs than in Slc4a11+/+ MCECs (33°C, IFN-γ [+]), indicating the glutamine-originated carbon source (gray) is significantly reduced. *P < 0.05; ****P < 0.0001.
Discussion
Here we report the generation of the first immortalized MCECs. The use of H-2Kb-tsA58 transgenic Immortomouse not only circumvents the limitations and uncertainties associated with in vitro transfection-based immortalization (e.g., initial requirement of large number of cells, different sites of gene integration, multiple copy numbers), but also allows for the production of genetically matched cell lines directly from a transgenic mouse model: Slc4a11+/+ MCECs and Slc4a11−/− MCECs. The conditional immortalization approach provided another advantage that the expression of SV40 large T antigen can be eliminated by simple temperature maneuver and/or removal of IFN-γ. We found that the SV40 large T antigen declined and could be virtually eliminated (Fig. 2B) in the semipermissive condition (IFN-γ [+], 33°C). This condition was preferred because it allowed repeated expansion of MCECs, avoided large cell losses, yet retained endothelial morphology and transport function.
Analysis of ion transporter activity in the two lines of MCECs show that apparent bicarbonate transport, Na+/H+ exchanger, and lactate transport activities were not significantly changed by Slc4a11 knockout (Fig. 3). As expected, Slc4a11−/− MCECs showed significantly less NH3:H+ flux relative to Slc4a11+/+ MCECs (Fig. 4), consistent with the known NH3:H+ permeability provided by SLC4A11.27
The impaired ability to facilitate NH3:H+ transport in Slc4a11−/− MCECs is likely the cause of observed changes in glutamine metabolism. Ammonia was reported to inhibit both the N-ethylmaleimide-sensitive and -insensitive fraction of glutaminase.39,40 In Slc4a11−/− MCECs, expression changes of glutaminolysis enzymes is consistent with our observation in Slc4a11−/− CHED mouse corneal endothelium tissue.21 Further analysis of TCA cycle intermediates in Slc4a11+/+ MCECs indicated that 50% of citrate and approximately 35% of other TCA cycle intermediates were derived from glucose, which is similar to what was found in an immortalized human corneal endothelial cell line.21 However, in Slc4a11−/− MCECs, we found a significantly increased fraction from glucose-sourced carbon, indicating reduced fraction from glutamine (Fig. 5C), providing evidence that there are functional metabolic changes associated with the enzyme expression changes. Given that ammonia inhibits glutaminase activity,39 this observed reduced flux of glutaminolysis suggests that the increased expression of Gls1 found in the Slc4a11−/− MCECs (Fig. 5A) may be a compensatory response to glutaminase inhibition.
Interestingly, both human SLC4A11 and murine Slc4a11 genes were reported to be a target of p53,41 a main cell metabolism regulator.42 Conversely, knock-down of Slc4a11 in bovine nucleus pulposus cells inhibits p53 activity by abolishing p53 phosphorylation.43 This suggests that the SLC4A11 transporter could indirectly regulate glutaminolysis enzyme expression via p53 phosphorylation, which is consistent with the finding that glutaminase GLS2 is a target of p53.44
Additionally, we observed fibroblast-like morphology changes only in Slc4a11+/+ MCECs when tsTAg was induced by IFN-γ (Fig. 1C). Given the direct inhibition from tsTAg on Rb and p53 protein,45 it will be of interest to study if the SLC4A11 transporter plays a role in corneal endothelial cell proliferation, polarity formation, and EMT transformation.
In summary, we successfully established two genetically matched conditionally immortal MCECs: Slc4a11+/+ MCECs and Slc4a11−/− MCECs. These cells remain proliferative with reasonable endothelial morphology after prolonged culture, and present key active ion transport activities as expected from corneal endothelial cells. Slc4a11−/− MCECs recapitulate glutaminolysis enzyme expression changes as seen in Slc4a11−/− mouse corneal endothelial tissue. These cell lines allowed us to carry out further investigations using cellular-based approaches requiring a significantly larger sample volume than mouse tissue can achieve. Further analysis of TCA cycle intermediates suggests functionally impaired glutaminolysis in Slc4a11−/− MCECs. These Slc4a11+/+ MCEC and Slc4a11−/− MCEC cell lines provide an excellent tool for future cell-based experiments to study the pathophysiological changes resulting from loss of Slc4a11 function and for potential therapeutic pharmaceutical reagent screening.
Acknowledgments
The authors thank Shimin Li for the technical support of experimental procedures. We thank Eranga N. Vithana from the Singapore Eye Research Institute for Slc4a11−/− C57BL/6 mouse as a gift. We thank Keith Johnson from the University of Nebraska Medical Center for introducing us to H-2Kb-tsA58 Immortomouse.
Supported by National Institutes of Health Grants 5R01EY008834 (JAB), 5P30EY019008 (Vision Science Core Grant), 5R00GM101341-05, and 1R35GM119557-01 (JMT), and an American Optometric Foundation William C. Ezell Fellowship (WZ).
Disclosure: W. Zhang, None; D.G. Ogando, None; E.T. Kim, None; M.-J. Choi, None; H. Li, None; J.M. Tenessen, None; J.A. Bonanno, None
References
- 1. Siddiqui S,, Zenteno JC,, Rice A,, et al. Congenital hereditary endothelial dystrophy caused by SLC4A11 mutations progresses to Harboyan syndrome. Cornea. 2014; 33: 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Riazuddin SA,, Vithana EN,, Seet LF,, et al. Missense mutations in the sodium borate cotransporter SLC4A11 cause late-onset Fuchs corneal dystrophy. Hum Mutat. 2010; 31: 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hemadevi B,, Veitia RA,, Srinivasan M,, et al. Identification of mutations in the SLC4A11 gene in patients with recessive congenital hereditary endothelial dystrophy. Arch Ophthalmol. 2008; 126: 700–708. [DOI] [PubMed] [Google Scholar]
- 4. Weh E,, Reis LM,, Happ HC,, et al. Whole exome sequence analysis of Peters anomaly. Hum Genet. 2014; 133: 1497–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiao X,, Sultana A,, Garg P,, et al. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet. 2007; 44: 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vithana EN,, Morgan P,, Sundaresan P,, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet. 2006; 38: 755–757. [DOI] [PubMed] [Google Scholar]
- 7. Sultana A,, Garg P,, Ramamurthy B,, Vemuganti GK,, Kannabiran C. Mutational spectrum of the SLC4A11 gene in autosomal recessive congenital hereditary endothelial dystrophy. Mol Vis. 2007; 13: 1327–1332. [PubMed] [Google Scholar]
- 8. Kumar A,, Bhattacharjee S,, Prakash DR,, Sadanand CS. Genetic analysis of two Indian families affected with congenital hereditary endothelial dystrophy: two novel mutations in SLC4A11. Mol Vis. 2007; 13: 39–46. [PMC free article] [PubMed] [Google Scholar]
- 9. Ramprasad VL,, Ebenezer ND,, Aung T,, et al. Novel SLC4A11 mutations in patients with recessive congenital hereditary endothelial dystrophy (CHED2). Mutation in brief #958. Online. Hum Mutat. 2007; 28: 522–523. [DOI] [PubMed] [Google Scholar]
- 10. Aldave AJ,, Yellore VS,, Bourla N,, et al. Autosomal recessive CHED associated with novel compound heterozygous mutations in SLC4A11. Cornea. 2007; 26: 896–900. [DOI] [PubMed] [Google Scholar]
- 11. Desir J,, Moya G,, Reish O,, et al. Borate transporter SLC4A11 mutations cause both Harboyan syndrome and non-syndromic corneal endothelial dystrophy. J Med Genet. 2007; 44: 322–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shah SS,, Al-Rajhi A,, Brandt JD,, et al. Mutation in the SLC4A11 gene associated with autosomal recessive congenital hereditary endothelial dystrophy in a large Saudi family. Ophthalmic Genet. 2008; 29: 41–45. [DOI] [PubMed] [Google Scholar]
- 13. Aldahmesh MA,, Khan AO,, Meyer BF,, Alkuraya FS. Mutational spectrum of SLC4A11 in autosomal recessive CHED in Saudi Arabia. Invest Ophthalmol Vis Sci. 2009; 50: 4142–4145. [DOI] [PubMed] [Google Scholar]
- 14. Paliwal P,, Sharma A,, Tandon R,, et al. Congenital hereditary endothelial dystrophy—mutation analysis of SLC4A11 and genotype-phenotype correlation in a North Indian patient cohort. Mol Vis. 2010; 16: 2955–2963. [PMC free article] [PubMed] [Google Scholar]
- 15. Kodaganur SG,, Kapoor S,, Veerappa AM,, et al. Mutation analysis of the SLC4A11 gene in Indian families with congenital hereditary endothelial dystrophy 2 and a review of the literature. Mol Vis. 2013; 19: 1694–1706. [PMC free article] [PubMed] [Google Scholar]
- 16. Park SH,, Jeong HJ,, Kim M,, Kim MS. A novel nonsense mutation of the SLC4A11 gene in a Korean patient with autosomal recessive congenital hereditary endothelial dystrophy. Cornea. 2013; 32: e181–e182. [DOI] [PubMed] [Google Scholar]
- 17. Kim JH,, Ko JM,, Tchah H. Fuchs endothelial corneal dystrophy in a heterozygous carrier of congenital hereditary endothelial dystrophy type 2 with a novel mutation in SLC4A11. Ophthalmic Genet. 2015; 36: 284–286. [DOI] [PubMed] [Google Scholar]
- 18. Aldave AJ,, Han J,, Frausto RF. Genetics of the corneal endothelial dystrophies: an evidence-based review. Clin Genet. 2013; 84: 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kenyon KR,, Maumenee AE. The histological and ultrastructural pathology of congenital hereditary corneal dystrophy: a case report. Invest Ophthalmol. 1968; 7: 475–500. [PubMed] [Google Scholar]
- 20. McCartney ACE,, Kirkness CM. Comparison between posterior polymorphous dystrophy and congenital hereditary endothelial dystrophy of the cornea. Eye. 1988; 2: 63–70. [DOI] [PubMed] [Google Scholar]
- 21. Zhang W,, Li H,, Ogando DG,, et al. Glutaminolysis is essential for energy production and ion transport in human corneal endothelium. EBioMedicine. 2017; 16: 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Han SB,, Ang HP,, Poh R,, et al. Mice with a targeted disruption of Slc4a11 model the progressive corneal changes of congenital hereditary endothelial dystrophy. Invest Ophthalmol Vis Sci. 2013; 54: 6179–6189. [DOI] [PubMed] [Google Scholar]
- 23. Ehlers N,, Módis L,, Møller-Pedersen T. A morphological and functional study of congenital hereditary endothelial dystrophy. Acta Ophthalmol Scand. 1998; 76: 314–318. [DOI] [PubMed] [Google Scholar]
- 24. Myers EJ,, Marshall A,, Jennings ML,, Parker MD. Mouse Slc4a11 expressed in Xenopus oocytes is an ideally selective H+/OH- conductance pathway that is stimulated by rises in intracellular and extracellular pH. Am J Physiol Cell Physiol. 2016; 311: C945–C959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kao L,, Azimov R,, Shao XM,, et al. Multifunctional ion transport properties of human SLC4A11: comparison of the SLC4A11-B and SLC4A11-C variants. Am J Physiol Cell Physiol. 2016; 311: C820–C830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loganathan SK,, Schneider HP,, Morgan PE,, Deitmer JW,, Casey JR. Functional assessment of SLC4A11, an integral membrane protein mutated in corneal dystrophies. Am J Physiol Cell Physiol. 2016; 311: C735–C748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang W,, Ogando DG,, Bonanno JA,, Obukhov AG. Human SLC4A11 is a novel NH3/H+ co-transporter. J Biol Chem. 2015; 290: 16894–16905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joyce NC. Cell cycle status in human corneal endothelium. Exp Eye Res. 2005; 81: 629–638. [DOI] [PubMed] [Google Scholar]
- 29. Peh GSL,, Beuerman RW,, Colman A,, Tan DT,, Mehta JS. Human corneal endothelial cell expansion for corneal endothelium transplantation: an overview. Transplantation. 2011; 91: 811–819. [DOI] [PubMed] [Google Scholar]
- 30. Jat PS,, Noble MD,, Ataliotis P,, et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A. 1991; 88: 5096–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vilas GL,, Loganathan SK,, Liu J,, et al. Transmembrane water-flux through SLC4A11: a route defective in genetic corneal diseases. Hum Molec Genet. 2013; 22: 4579–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ogando DG,, Jalimarada SS,, Zhang W,, Vithana EN,, Bonanno JA. SLC4A11 is an EIPA-sensitive Na(+) permeable pHi regulator. Am J Physiol Cell Physiol. 2013; 305: C716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thomas JA,, Buchsbaum RN,, Zimniak A,, Racker E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry. 1979; 18: 2210–2218. [DOI] [PubMed] [Google Scholar]
- 34. Tennessen JM,, Barry WE,, Cox J,, Thummel CS. Methods for studying metabolism in Drosophila. Methods. 2014; 68: 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nanchen A,, Fuhrer T,, Sauer U. Determination of metabolic flux ratios from 13C-experiments and gas chromatography-mass spectrometry data. : Weckwerth W, Metabolomics: Methods and Protocols. Totowa, NJ: Humana Press; 2007: 177–197. [DOI] [PubMed] [Google Scholar]
- 36. Li S,, Kim E,, Bonanno JA. Fluid transport by the cornea endothelium is dependent on buffering lactic acid efflux. Am J Physiol Cell Physiol. 2016; 311: C116–C126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nguyen T,, Bonanno JA. Lactate-H+ transport is a significant component of the in vivo corneal endothelial pump. Invest Ophthalmol Vis Sci. 2012; 53: 2020–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boron WF,, De Weer P. Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. J Gen Physiol. 1976; 67: 91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kvamme E,, Lenda K. Regulation of glutaminase by exogenous glutamate, ammonia and 2-oxoglutarate in synaptosomal enriched preparation from rat brain. Neurochem Res. 1982; 7: 667–678. [DOI] [PubMed] [Google Scholar]
- 40. Curthoys NP,, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr. 1995; 15: 133–159. [DOI] [PubMed] [Google Scholar]
- 41. Younger ST,, Kenzelmann-Broz D,, Jung H,, Attardi LD,, Rinn JL. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res. 2015; 43: 4447–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berkers CR,, Maddocks OD,, Cheung EC,, Mor I,, Vousden KH. Metabolic regulation by p53 family members. Cell Metab. 2013; 18: 617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mavrogonatou E,, Papadimitriou K,, Urban JP,, Papadopoulos V,, Kletsas D. Deficiency in the alpha1 subunit of Na+/K+-ATPase enhances the anti-proliferative effect of high osmolality in nucleus pulposus intervertebral disc cells. J Cell Physiol. 2015; 230: 3037–3048. [DOI] [PubMed] [Google Scholar]
- 44. Suzuki S,, Tanaka T,, Poyurovsky MV,, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010; 107: 7461–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ahuja D,, Saenz-Robles MT,, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005; 24: 7729–7745. [DOI] [PubMed] [Google Scholar]