

Abstract
Primary vitreoretinal lymphoma (PVRL) was previously termed primary intraocular lymphoma. PVRL is a potentially fatal intraocular malignancy, and 65–90% of PVRL cases eventually involve the central nervous system (CNS). The incidence of PVRL has been rising in both immunocompromised and immuno-competent populations worldwide. PVRL frequently masquerades as chronic uveitis. Advanced auxiliary examinations, such as optical coherence tomography and fundus autofluorescence have been applied in the diagnosis of PVRL. Histology and immunohistochemistry in combination with molecular tests and interleukin-10 analysis have been demonstrated as reliable in diagnosing PVRL. Despite early initiation of treatment, mortality is high with PVRL associated with CNS involvement and relapses are common. The use of systemic chemotherapy has not been proven to prevent CNS involvement; however, local therapies including intravitreal injections of methotrexate and/or rituximab and low-dose radiotherapy to the eye, has shown to be extremely effective in controlling intraocular lymphoma with encouraging results.
Keywords: lymphoma, primary, retina, vitreous
1. Introduction
Primary vitreoretinal lymphoma (PVRL) is a rare but potentially fatal intraocular malignancy,1,2,3 which is a subset of primary central nervous system lymphoma (PCNSL).4,5,6,7,8,9,10
PVRL has been previously called primary intraocular lymphoma (PIOL).10,11 PIOL involving choroid was firstly mentioned in 1920 by Triebenstein12,13; however, in 1968 PIOL involving the retina described as “reticulum cell sarcoma” was reported by Vogel.14 In the early 1980s, associations between this disease and PCNSL were recognized11,15 and the origin of the tumor cells was demonstrated to be lymphoid cells (malignant B and T lymphocytes), not histio-cytes.5,16,17 Thus the term “PIOL” was established.8 Recently, based on the World Health Organization classification of tumors, intraocular lymphoma represents a heterogeneous group of malignancies located in different tissues within the eye, each of which has different morphological, immunophenotypical, and genetic features, and completely different clinical courses.8,18 Since lym-phomas originating from the retina/vitreous and the choroid are distinctive, PIOL can be classified as “PVRL” and “primary uveal lymphoma.” PVRL is proposed and defined as “a subset of PCNSL and a heterogeneous group of malignant lymphocytic neoplasms affecting the retina with or without involvement of the vitreous or the optic nerve, and without evidence of brain or cerebrospinal fluid (CSF) involvement.”1,17,19 Most intraocular lymphomas are PVRL, while ocular manifestation of systemic lymphomas occurs commonly in the uvea; however, primary uveal lymphoma is rarely reported.12,20 Approximately 15–25% of patients with PCNSL ultimately develop an ocular manifestation of their lymphoma.6,7 With a 30-fold increase in the incidence of PCNSL in recent times, similar changes of PVRL incidence has been noted worldwide.7,21
PVRL frequently masquerades as chronic uveitis and the complexity of sample management poses a challenge not only to the ophthalmologists, but also to the pathologists. PVRL is characterized by an aggressive clinical course, despite early initiation of aggressive treatment with chemotherapyand/or radiotherapy, high relapse rate, and CNS involvement, has a poor prognosis with a 1-year overall survival of 25–40%.3,22 Due to the diverse patient populations and rarity of this disease, there are no established standards for diagnosis and therapy of PVRL.1,2 Recent advances of clinical diagnosis and management for PVRL, such as optical coherence tomographic (OCT) and intravitreal injection of medications, have been achieved with encouraging results. These new methods may greatly improve our understanding and practice in controlling PVRL.2,3,20,23,24,25,26
2. Clinical presentations and image findings
PVRL is the most common intraocular lymphoma, usually of the B-cell type as diffuse large B-cell lymphoma originally invades intraocular tissues: the retina, vitreous, and/or optic nerve.1,16,18,27
2.1. Ocular clinical manifestations
Clinically, ocular involvement occurs bilaterally in 64–83% of cases.1 PVRL is usually a disease of adulthood with few cases in infants and teenagers.6,21,28 Vitreous floaters may be noticed by the patient long before PVRL is suspected, which is the main reason for patients without CNS involvement to visit the ophthalmolo-gist.2,11,20,29,30 PVRL frequently masquerades as chronic uveitis.31 Mild anterior segment inflammation is usually observed using slit lamp examination.17,21,32,33,34 Pseudohypopyon is a rare presentation (Figure 1).12 Sheets and clusters of lymphoma cells confined to the vitreous cavity are the most common ocular findings.1,2,12,20,21,30 Lymphoma cells are homogeneous and larger than inflammatory cells and do not cluster with reactive cells resulting in an “aurora borealis” appearance from cells lining along the vitreous fibrils.1 Vitreous haze is often striking, from mild to severe.35 However, visual acuity of PVRL patients can be at least better than expected.36 The typical creamy lesions with yellow-white infiltrate deep to the retina or retinal pigment epithelium (RPE) with various sizes (Figure 2A), since lymphoma cells migrate to the sub-RPE/pre-Bruch's space.1,2,17 Islands of pigments floating on these deposits make the characteristic “leopard-spot” pigmentation.1,37 The distinctive solid detachments of the RPE with irregular yellow–white deposits are considered pathognomonic of PVRL.20 Exudative retinal detachment is present in severe PVRL cases. Spontaneous resolution is often seen with RPE atrophy and sub-retinal fibrosis. Optic nerve and orbital involvement are sporadically reported.12
Figure 1.
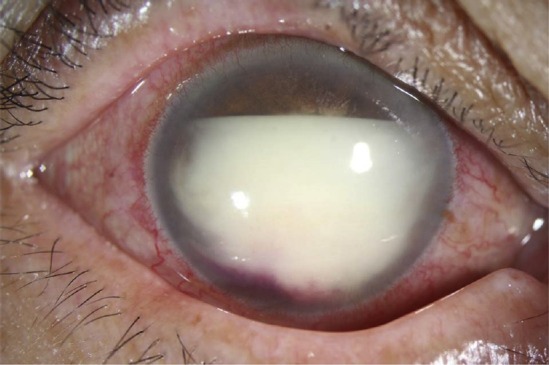
Pseudohypopyon in the right eye of a 62-year-old female patient diagnosed with primary vitreoretinal lymphoma through aqueous aspiration.
Figure 2.
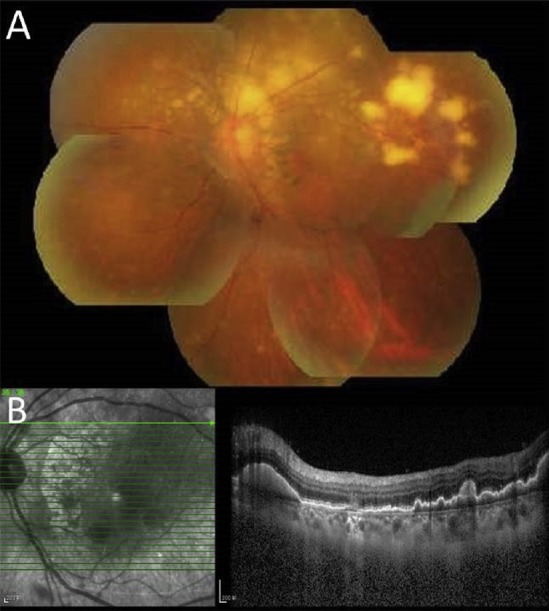
(A) Fundus photograph of the left eye of the patient presents typical yellow–white creamy lesions infiltrated deep into the retina or retinal pigment epithelium with various sizes. (B) Correlation with the optical coherence tomographic section through the yellow–white lesions shows multiple pre-Bruchs/subretinal pigment epithelium deposits that protrude anteriorly to the outer retina.
2.2. Ocular imaging
In addition to a thorough clinical examination, more and more advanced auxiliary examinations such as spectral-domain OCT(SD-OCT) and fundus autofluorescence (FAF) have been increasingly used to aid in identifying PVRL cases.2,23
2.2.1. OCT
Recently, OCT has been reported to be helpful in the diagnosis of retinal abnormalities in PVRL.23 Lymphoma cells directly infiltrate the retina and proliferate focally in the pre-Bruch’s/sub-RPE space presenting a semiopaque grey spot in a fundus photograph that appear as nodular hyper-reflective infiltrations at the level of RPE on SD-OCT (Figure 2B).23,25 Nodular or band hyper-reflective spots were noted in 43% of PVRL eyes.38 RPE damage, disruption of the photoreceptor inner segment/outer segment junction, multiple hyper-reflective infiltrations in the inner retina, and exudative retinal detachment with subretinal hyporeflective fluid may be revealed by SD-OCT in severe PVRL. Cystoid macular edema is rarely reported.39 However, these hyper-reflective signals between the Bruch's membrane and RPE need to be differentiated from those seen in age-related macular degeneration (AMD) or polypoidal choroidal vasculopathy.12,21 Dramatic resolution of the lymphoma deposits along Bruch's membrane was seen on the OCT after intravitreal chemotherapy.20 This noninvasive method may be convenient for further surveillance of relapse or resolution of PVRL.
2.2.2. FAF
FAF assesses the function of the RPE by detecting the lipofuscin distribution in various retinal disorders such as AMD.38 Recently, FAF has been noted to detect the active status of PVRL. On FAF images, hypofluorescent areas suggest RPE atrophy or lymphoma cells above the RPE, and hyperfluorescent spots indicate the overlapping of PVRL and RPE cells (Figure 3).21,23,38 Blockage by mass lesion was also seen in 11% of eyes with PVRL.38 Hyperautofluorescent spots on FAF may correlate with the hypofluorescent spots on fluorescein angiography (FA; 36%) and the nodular hyper-reflective spots on OCT (43%).38,40 This concordance was observed in 62% of patients.38 Granular patterns on FAF were identified in various locations in the retina; however, this was not limited to visible tumor location. Remarkably, the corresponding changes from hyperautofluorescent spots to hypoautofluorescent spots in retinal lesions are seen after intravitreal methotrexate treatment.23,38
Figure 3.

(A) Fundus photograph showing clusters of granular or round whitish lesions; (B) fundus autofluorescence image revealing corresponding areas of hyperautofluorescence and hypofluorescent lesions; and (C) spectral-domain optical coherence tomography of a 50-year-old primary vitreoretinal lymphoma patient after receiving systemic chemotherapy with nodular hyper-reflective spots on spectral-domain optical coherence tomography. The diagnosis of primary vitreoretinal lymphoma was confirmed by cytology and magnetic resonance imaging with concurrent central nervous system involvement.
2.2.3. Fundus FA and indocyanine green angiography
The most common pattern on FA was hypofluorescent round spots with a “leopard-spot” appearance (43%), which is better appreciated on photographs enhanced with venous FA (Figure 4).21,25,36,37,41,42 In a study of 44 PVRL patients, punctate hyperfluorescent window defects were revealed in 55%, round hypofluorescent lesions in 34%, and vasculitis in 14%, although only 2% cystoid macular edema was found in this study.21,43 In another study of 53 PVRL patients, clusters of round stable hypofluorescent lesions on FA that corresponded to punctate whitish lesions in the fundus and round-clustered hypofluorescent lesions revealed scarce by indocyanine green angiography, warrants further biopsies for differential diagnosis of PVRL with a positive predictive value of 88.9% and negative predictive value of 85%.41
Figure 4.
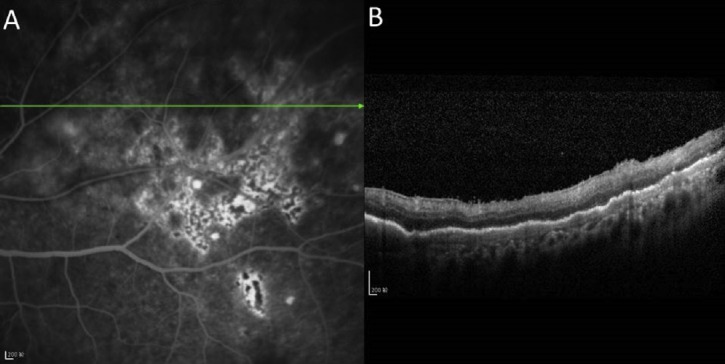
(A) Fluorescein angiography showing lesion stains with fluorescein and retinal pigment epithelial changes; and (B) optical coherence tomographic image of a 59-year-old female PVRL patient showing nodular and band hyper-reflective spots at the level of retinal pigment epithelium which seem to correspond with the lesions on fluorescein angiography.
2.3. CNS features and neuro-imaging
At the time of diagnosis of PVRL, detectable CNS involvement is present in 16–34% of patients.1,19,43,44 CNS involvement may occur at any time of the disease course, especially in the late stage when widespread dissemination of PCNSL occurs in 7–8% of patients.44 Within a mean interval of 8–29 months, 42–92% of PVRL cases presented with intracranial lymphoma.45,46 Patients with CNS involvement can present with neurological symptoms depending on the tumor location in the brain.44 The common presenting symptom is personality change and new-onset seizures as an indicator of CNS involvement.6,31
Sixty-two percent of PVRL patients have CNS lesions, especially in relapsed patients.47 Therefore, use of contrast-enhanced magnetic resonance imaging every 3 months during the disease course after the definitive diagnosis to detect CNS involvement is imper-ative.1,32 Lymphoma lesions with discrete or diffuse borders can be identified, which are hypodense on T1-weighted and hyperdense on T2-weighted images.44 (Positron emission tomography/computed tomography has been used to identify not only CNS lesions, but also ocular activity.2,20
3. Diagnosis
3.1. Biopsy
Biopsy for the cytological assessment of suspected intraocular lymphoma is invaluable not only in confirming the diagnosis, but also in providing information of prognostic importance.34,48,49 However, lymphoma cells are fragile that easily undergo necrosis, and systemic corticosteroid use for treating a presumed “uveitis” also may cause false negative results.46 The corticosteroid treatment must be discontinued as least 2 weeks before biopsy.21 Surgical intervention to obtain sufficient material includes pars planavitrectomy, chorioretinal biopsy, aqueous aspiration, and rarely diagnostic enucleation.49,50 Before biopsy, it is crucial for surgeons to contact the pathologist to ensure use of the correct containers and preservative solutions.1,50 If prolonged delays of transportation are anticipated, the samples should be placed in culture medium or a mild cytofixative, such as herpes-glutamic acid buffer mediated organic solvent protection effect fixation or Cytolyt, which provides superior preservation of cytomorphology and immunoreactivity.51,52
3.1.1. Vitreous and retina
Clinically, pars planavitrectomy is performed most for PVRL biopsy because its diagnostic yield is superior to that of vitreous aspirate.49 A 25-gauge transconjunctival sutureless vitrectomy is a safe and effective technique for the diagnosis of PVRL.50 The effect of different vitrectomy cut-rates on lymphoma cell viability and diagnostic yield was investigated and a cut rate of 600 cpm is recommended for PVRL diagnosis.39 Routinely, it is useful to obtain undiluted vitreous (1 –2 mL) and diluted vitreous samples as much as possible after turning on the infusion.35,50 Retinal biopsy is seldom used clinicallyand retinal samples should be taken from the deeper part of the lesion, near the choriocapillaris where viable lymphoma cells are most likely to be found.49 Aqueous aspiration may be adequately used in special conditions, such as pseudohy-popyon. However, more than one biopsy is often needed to make a confident diagnosis if hampered by paucicellular specimens mixed with necrotic lymphoma cells.17 A study of 12 patients found that 30% of PVRL cases had a previous false-negative biopsy.1,2
3.1.2. CSF biopsy
Despite a low cellular yield for diagnosis, CSF biopsy is highly recommended to identify atypical lymphoid cells, which spares the patient from further invasive diagnostic procedures.16,17
3.2. Cytology and histopathology
Cytopathological evaluation was performed by an experienced cytopathologist on the vitreous or aqueous specimen to identify morphological evidence of PVRL characterized by large (2–5 times the diameter of a small lymphocyte), atypical lymphoid cells with large, irregular nuclei, prominent nucleoli, scanty basophilic cytoplasm, and rare mitoses.35 Using either Giemsa or Diff-Quick staining is recommended to identify lymphoma cells.1,16 Smaller, round lymphocytes, macrophages, and polymorphonuclear lymphocytes presenting as a reactive inflammatory infiltrate may outnumber the malignant cells, which is often misread as evidence of inflammation.19
Histologically, according to World Health Organization classification, the majority of PVRL is a high-grade, malignant non-Hodgkin lymphoma, and can be subtyped in most cases as diffuse large B-cell lymphoma.5,19
3.3. Immunocytochemistry and flow cytometry
Monoclonality, either a B-cell population stain positively for B-cell markers (CD19, CD20, or CD22) with restricted expression of either κ or λ, or a T-cell population stain positively for T-cell mar- kers(CD3, CD4), can support B or T cell PVRL.20 Concomitant expression of BCL6 and MUM1 has also been reported by Wallace et al48 in five PVRL patients. Flow cytometry can analyze larger numbers of different cell surface markers simultaneously to differentiate uveitis from lymphoma. A ratio of κ:λ of >3 or <0.6 is useful as a marker for clonality.20
3.4. Molecular analysis of gene rearrangements
Molecular technique with microdissection and polymerase chain reaction to detect a clonally expanded population of lymphocytes is an important adjunct to diagnose PVRL.53 The immu-noglobulin heavy chain (IgH; FR2, FR3, and/or CDR3 primers) and T-cell receptor (TCR) gene rearrangements (TCR-γ) provide a molecular diagnosis of B- and T-cell lymphoma, respectively.54 Without microdissection to select a relatively pure lymphoma cell population, molecular testing may be less sensitive and specific.53 Positive IgH gene rearrangements reached 80.6% in a Japanese study including 67 PIOL patients.33 One-hundred percent positive of either IgH or TCR gene rearrangements in a study of 114 PIOL patients was reported; however, sampling of too few cells from microdissection may result in a false-positive result.20,52
3.5. Other laboratory tests of PVRL
A high level of interleukin (IL)-10 produced by B-cell lym-phomas, is a Th2 cytokine and linked to rapid disease progression. A high level of IL-6 is secreted by normal inflammatory cells.1,2 In 1995, Chan et al55 first reported that high levels of vitreous IL-10 were detected in three B-cell PVRL patients. Recently, higher levels of IL-10 (mean values: 543.4 pg/mL) in PIOL patients than in uveitis (21.9 pg/mL) in aqueous humor was revealed in a study of 51 PIOL.43 IL-10-1082 A allele is a risk factor for higher IL-10 levels in PVRL and higher IL-10 levels have been correlated with more aggressive disease.56 Currently, a ratio of IL-10:IL-6 greater than 1 in ocular fluid have become adjunctive and supportive biomarkers for the diagnosis and prognosis of PVRL, particularly the B-cell PVRL.1,20
3.6. Differential diagnosis of PVRL
Clinical manifestations of PVRL frequently masquerade as both infectious and noninfectious uveitis, such as tuberculosis, acute retinal necrosis syndrome, retinochoroidal toxoplasmosis, syphilitic retinitis, endophthalmitis, or idiopathic uveitis.21,31,51,57 Ocular degeneration diseases, such as AMD and polypoidal choroidal vasculopathy, need to be differential diagnosed due to similar OCT imaging. Metastasis and amelanotic melanoma must be identified from a PVRL cohort.21
4. Treatment
At present, the overall survival rate of PVRL is still quite low and mortality rates range between 9% and 81% in follow-up periods ranging from 12 months to 35 months, even though PVRLs are highly radiosensitive and chemosensitive.8,45,51,58 Because no standard therapy for PVRL has been established yet, a symposium on PVRL was organized by the International Primary CNS Lym-phoma Collaborative Group in 2011, and the following therapeutic guidelines was recommended.1 A professional team of experts in ophthalmology, oncology (particularly neuro-oncology or hemato-oncologist), and pathology are essential for optimizing patient management.2
Without CNS or systemic involvement: local treatment (intra-vitreal methotrexate, intravitreal rituximab, or low-dose (30–35 Gy) stereotactic external beam radiotherapy to the eye with close follow up and ongoing collaboration between neu-rooncologists and ophthalmologists. If both eyes are involved, systemic chemotherapy has been suggested in addition to intravitreal medications for bilateral PVRL.1,59
Involves the CNS: a high-dose methotrexate based systemic therapy (possibly with systemic rituximab) was suggested in conjunction with local ocular therapy. If the patient has failed systemic therapy and does not meet the criteria for a more aggressive therapy such as autologous stem cell transplantation, whole brain radiotherapy in conjunction with ocular radiotherapy should be considered.
Another published recommendation from the British Neuro-Oncology Society (National Cancer Action Team Rare Tumor Guidelines, June 2011) differed from the International Primary CNS Lymphoma Collaborative Group that specify systemic chemotherapy with high-dose methotrexate followed by whole-globe irradiation for concurrent or ocular only patients and intravitreal methotrexate as an effective treatment option for isolated ocular recurrences.20
In one retrospective study to investigate the function of prophylactic therapy, 17 PVRL patients treated for ocular only disease with chemotherapy and/or irradiation lived an average of 60 months after onset of ocular symptoms until death compared with 35 months for 14 patients treated only after CNS disease developed.60 Another study also demonstrated that prophylactic systemic chemotherapy did not inhibit the onset of CNS involvement in PVRL patients, but it significantly prolonged brain involvement.61
4.1. Chemotherapy
The efficiency of high-dose methotrexate is demonstrated to produce a response rate of up to 72% when used alone and up to 94–100% in combinations.1,2,21 Combined intravitreal methotrexate and systemic high-dose methotrexate treatment has been reported effective in 19 PIOL patients that the 5-year overall survival rate reached 55.8%.62 Intensive chemotherapy with thiotepa, busulfan, and cyclophosphamide followed by hematopoietic stem cell rescue in relapsed PCNSL and PIOL improved the 5-year survival probability to 62% and obtained complete remission in 66 out of 79 pa-tients.12,21 Cytarabine has also been used with variable success rates, especially intrathecal use combined with methotrexate for recurrent cases.4 However, a recent 17-Center European collaborative study found that systemic chemotherapy in isolated PVRL patients was not proven to prevent CNSL and was associated with more severe adverse effects, such as acute renal failure.3 Macul-opathy without significantly affecting visual acuity is also reported to be associated with chemotherapy.21
4.2. Radiotherapy
At present, low-dose (30–35 Gy) stereotactic external beam radiotherapy to the eye is recommended as local treatment for ocular only PVRL patients.1 No ocular relapse was reported in any of 12 patient received radiotherapy (30–35 Gy in 15 fractions) during a mean 19 months follow up.58 If PVRL patients concurrent with CNS involvement have failed systemic chemotherapy and also unavailable to aggressive therapy, whole brain and eye radiotherapy may be added. Currently, whether to use intravitreal chemotherapy or ocular radiation as first-line therapy is still controversial. The complication of whole brain radiation often induces a delayed neurotoxicity with decline in cognitive function, ataxia, and even death, whereas ocular complications were cataract, dry eye, and radiation retinopathy.58,63
4.3. Ocular therapy
Besides radiotherapy to the eye, another two local treatments are intravitreal methotrexate and rituximab.1
4.3.1. Intravitreal methotrexate
Intravitreal methotrexate therapy is extremely effective in inducing clinical remission of vitreoretinal involvement in PVRL with acceptable morbidity (Figure 5). In a 10-year study using intravitreal injections of methotrexate as first line treatment in PVRL, none of the total 26 patients had an intraocular recurrence.26 Injections of 0.4 mg/0.1 mL-methotrexate intravitreal are performed twice weekly for 4 weeks, once weekly for 8 weeks, and once monthly for 9 months(total of 25 injections). Remission was observed after a mean of 6.4 injections. The common complications were transient intraocular pressure elevations and corneal epi-theliopathy which subsided when the interval between injections increased.2,3
Figure 5.

Fundus photographs of a 62-year-old male with primary vitreoretinal lymphoma confirmed using vitreous biopsy who received intravitreal methotrexate treatment. (A) Before treatment; (B) 2 months after treatment; (C) 6 months after treatment; (D) 1 year after treatment. Note the corresponding changes from the significant yellowewhite lesion to retinal scar after successful intravitreal methotrexate treatment.
4.3.2. Intravitreal rituximab
Intravitreal injections of rituximab, an anti-CD20 monoclonal antibody, have been used recently to treat CD20-positive PVRL.61 Twenty eyes with CD20-positive PVRL discontinued intravitreal methotrexate because of severe corneal epitheliopathy, and received weekly intravitreal injections of 1 mg/0.1 mL-rituximab for 4 weeks as a one-course protocol with encouraging results.64 However, half recurrence was present in this study. Rituximab may be a less toxic alternative to methotrexate.
5. Conclusions
Currently, diagnostic and therapeutic dilemmas of PVRL still frequently occur in clinical practice. High relapse rates and CNS involvement are common, which result in poor prognosis of PVRL despite aggressive treatment with chemotherapy and/or radio-therapy.3 Recent advances of diagnostic techniques, such as OCT, FAF, molecular test for clonality, and cytokine analysis, are proven to improve the diagnostic yield.20,21 Multiple clinical therapies are proven to be efficacious in controlling this malignancy. Intravitreal chemotherapy with methotrexate and/or rituximab is promising to control the ocular involvement. In PVRL, despite multiple encouraging diagnostic and therapeutic advances have been achieved, the optimal standard clinical diagnosis and treatment strategies still merit further research.
Acknowledgments
Publication of this article was supported in part by research grants from the National Natural Science Foundation of China (81570854) and the National Key Basic Research Program of China (2013CB967503).
Footnotes
Conflicts of interest: The authors have no financial/conflicting interests to disclose.
References
- 1.Chan CC, Rubenstein JL, Coupland SE, et al. Primary vitreoretinal lymphoma: a report from an International Primary Central Nervous System Lymphoma Collaborative Group symposium. Oncologist. 2011;16:1589–1599. doi: 10.1634/theoncologist.2011-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan CC, Sen HN. Current concepts in diagnosing and managing primary vit-reoretinal (intraocular) lymphoma. Discov Med. 2013;15:93–100. [PMC free article] [PubMed] [Google Scholar]
- 3.Riemens A, Bromberg J, Touitou V, et al. Treatment strategies in primary vit-reoretinal lymphoma: a 17-center European collaborative study. JAMA Ophthalmol. 2015;133:191–197. doi: 10.1001/jamaophthalmol.2014.4755. [DOI] [PubMed] [Google Scholar]
- 4.Mason JO, Fischer DH. Intrathecal chemotherapy for recurrent central nervous system intraocular lymphoma. Ophthalmology. 2003;110:1241–1244. doi: 10.1016/S0161-6420(03)00268-9. [DOI] [PubMed] [Google Scholar]
- 5.Coupland SE, Hummel M, Muller HH, Stein H. Molecular analysis of immuno-globulin genes in primary intraocular lymphoma. Invest Ophthalmol Vis Sci. 2005;46:3507–3514. doi: 10.1167/iovs.05-0401. [DOI] [PubMed] [Google Scholar]
- 6.Mochizuki M, Singh AD. Epidemiology and clinical features of intraocular lymphoma. Ocul Immunol Inflamm. 2009;17:69–72. doi: 10.1080/09273940902957305. [DOI] [PubMed] [Google Scholar]
- 7.Schabet M. Epidemiology of primary CNS lymphoma. J Neurooncol. 1999;43:199–201. doi: 10.1023/a:1006290032052. [DOI] [PubMed] [Google Scholar]
- 8.Char DH, Ljung BM, Miller T, Phillips T. Primary intraocular lymphoma (ocular reticulum cell sarcoma) diagnosis and management. Ophthalmology. 1988;95:625–630. doi: 10.1016/s0161-6420(88)33145-3. [DOI] [PubMed] [Google Scholar]
- 9.Larkin KL, Saboo US, Comer GM, et al. Use of intravitreal rituximab for treatment of vitreoretinal lymphoma. Br J Ophthalmol. 2014;98:99–103. doi: 10.1136/bjophthalmol-2013-304043. [DOI] [PubMed] [Google Scholar]
- 10.Hong JT, Chae JB, Lee JY, Kim JG, Yoon YH. Ocular involvement in patients with primary CNS lymphoma. J Neurooncol. 2011;102:139–145. doi: 10.1007/s11060-010-0303-9. [DOI] [PubMed] [Google Scholar]
- 11.Qualman SJ, Mendelsohn G, Mann RB, Green WR. Intraocular lymphomas. Natural history based on a clinicopathologic study of eight cases and review of the literature. Cancer. 1983;52:878–886. doi: 10.1002/1097-0142(19830901)52:5<878::aid-cncr2820520523>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 12.Mulay K, Narula R, Honavar SG. Primary vitreoretinal lymphoma. Indian J Ophthalmol. 2015;63:180–186. doi: 10.4103/0301-4738.156903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Triebenstein O. Ein beitrag zur frage der aleukämischen augenveränderungen. Klin Monatsbl Augenheilkd. 1920;64:825–836. [Google Scholar]
- 14.Vogel MH, Font RL, Zimmerman LE, Levine RA. Reticulum cell sarcoma of the retina and uvea. Report of six cases and review of the literature. Am J Oph-thalmol. 1968:66, 205–215. doi: 10.1016/0002-9394(68)92065-5. [DOI] [PubMed] [Google Scholar]
- 15.Char DH, Margolis L, Newman AB. Ocular reticulum cell sarcoma. Am J Oph-thalmol. 1981;91:480–483. doi: 10.1016/0002-9394(81)90236-1. [DOI] [PubMed] [Google Scholar]
- 16.Chan CC. Molecular pathology of primary intraocular lymphoma. Trans Am Ophthalmol Soc. 2003;101:275–292. [PMC free article] [PubMed] [Google Scholar]
- 17.Coupland SE, Chan CC, Smith J. Pathophysiology of retinal lymphoma. Ocul Immunol Inflamm. 2009;17:227–237. doi: 10.1080/09273940903168696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Char DH, Ljung BM, Deschenes J, Miller TR. Intraocular lymphoma: immuno-logical and cytological analysis. Br J Ophthalmol. 1988;72:905–911. doi: 10.1136/bjo.72.12.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coupland SE, Heimann H, Bechrakis NE. Primary intraocular lymphoma: a review of the clinical, histopathological and molecular biological features. Graefes Arch Clin Exp Ophthalmol. 2004;242:901–913. doi: 10.1007/s00417-004-0973-0. [DOI] [PubMed] [Google Scholar]
- 20.Davis JL. Intraocular lymphoma: a clinical perspective. Eye (Lond) 2013;27:153–162. doi: 10.1038/eye.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sagoo MS, Mehta H, Swampillai AJ, et al. Primary intraocular lymphoma. Surv Ophthalmol. 2014;59:503–516. doi: 10.1016/j.survophthal.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Sierra Del Rio M, Ricard D, Houillier C, et al. Prophylactic intrathecal chemotherapy in primary CNS lymphoma. JNeurooncol. 2012;106:143–146. doi: 10.1007/s11060-011-0649-7. [DOI] [PubMed] [Google Scholar]
- 23.Egawa M, Mitamura Y, Hayashi Y, Naito T. Spectral-domain optical coherence tomographic and fundus autofluorescence findings in eyes with primary intraocular lymphoma. Clin Ophthalmol. 2014;8:335–341. doi: 10.2147/OPTH.S58114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mudhar HS, Sheard R. Diagnostic cellular yield is superior with full pars plana vitrectomy compared with core vitreous biopsy. Eye (Lond) 2013;27:50–55. doi: 10.1038/eye.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang HS, Sepah YJ, Sophie R, et al. Longitudinal spectral domain optical coherence tomography changes in eyes with intraocular lymphoma. J Ophthalmic Inflamm Infect. 2013;3:59. doi: 10.1186/1869-5760-3-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frenkel S, Hendler K, Siegal T, Shalom E, Pe’er J. Intravitreal methotrexate for treating vitreoretinal lymphoma: 10 years of experience. Br J Ophthalmol. 2008;92:383–388. doi: 10.1136/bjo.2007.127928. [DOI] [PubMed] [Google Scholar]
- 27.Chan CC. Primary intraocular lymphoma: clinical features, diagnosis, and treatment. Clin Lymphoma. 2003;4:30–31. doi: 10.1016/s1526-9655(11)70005-7. [DOI] [PubMed] [Google Scholar]
- 28.Wender A, Adar A, Maor E, Yassur Y. Primary B-cell lymphoma of the eyes and brain in a 3-year-old boy. Arch Ophthalmol. 1994;112:450–451. doi: 10.1001/archopht.1994.01090160024009. [DOI] [PubMed] [Google Scholar]
- 29.Turaka K, Bryan JS, De Souza S, et al. Vitreoretinal lymphoma: changing trends in diagnosis and local treatment modalities at a single institution. Clin Lym-phoma Myeloma Leuk. 2012;12:412–417. doi: 10.1016/j.clml.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Levy-Clarke GA, Chan CC, Nussenblatt RB. Diagnosis and management of primary intraocular lymphoma. Hematol Oncol Clin North Am. 2005;19:739–749. doi: 10.1016/j.hoc.2005.05.011. viii. [DOI] [PubMed] [Google Scholar]
- 31.Gill MK, Jampol LM. Variations in the presentation of primary intraocular lymphoma: case reports and a review. Surv Ophthalmol. 2001;45:463–471. doi: 10.1016/s0039-6257(01)00217-x. [DOI] [PubMed] [Google Scholar]
- 32.Sen HN, Bodaghi B, Hoang PL, Nussenblatt R. Primary intraocular lymphoma: diagnosis and differential diagnosis. Ocul Immunol Inflamm. 2009;17:133–141. doi: 10.1080/09273940903108544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura K, Usui Y, Goto H Japanese Intraocular Lymphoma Study G. Clinical features and diagnostic significance of the intraocular fluid of 217 patients with intraocular lymphoma. Jpn J Ophthalmol. 2012;56:383–389. doi: 10.1007/s10384-012-0150-7. [DOI] [PubMed] [Google Scholar]
- 34.Chan CC, Fisson S, Bodaghi B. The future of primary intraocular lymphoma (retinal lymphoma) Ocul Immunol Inflamm. 2009;17:375–379. doi: 10.3109/09273940903434804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faia LJ, Chan CC. Primary intraocular lymphoma. Arch Pathol Lab Med. 2009;133:1228–1232. doi: 10.5858/133.8.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitcup SM, de Smet MD, Rubin BI, et al. Intraocular lymphoma. Clinical and histopathologic diagnosis. Ophthalmology. 1993;100:1399–1406. doi: 10.1016/s0161-6420(93)31469-7. [DOI] [PubMed] [Google Scholar]
- 37.Davis JL. Diagnosis of intraocular lymphoma. Ocul Immunol Inflamm. 2004;12:7–16. doi: 10.1076/ocii.12.1.7.28072. [DOI] [PubMed] [Google Scholar]
- 38.Casady M, Faia L, Nazemzadeh M, Nussenblatt R, Chan CC, Sen HN. Fundus autofluorescence patterns in primary intraocular lymphoma. Retina. 2014;34:366–372. doi: 10.1097/IAE.0b013e31829977fa. [DOI] [PubMed] [Google Scholar]
- 39.Jiang T, Zhao Z, Chang Q. Evaluation of cytologic specimens obtained during experimental vitreous biopsy using B-cell lymphoma line. Eur J Ophthalmol. 2014;24:911–917. doi: 10.5301/ejo.5000488. [DOI] [PubMed] [Google Scholar]
- 40.Ishida T, Ohno-Matsui K, Kaneko Y, et al. Fundus autofluorescence patterns in eyes with primary intraocular lymphoma. Retina. 2010;30:23–32. doi: 10.1097/IAE.0b013e3181b408a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fardeau C, Lee CP, Merle-Beral H, et al. Retinal fluorescein, indocyanine green angiography, and optic coherence tomography in non-Hodgkin primary intraocular lymphoma. Am J Ophthalmol. 2009;147:886–894. doi: 10.1016/j.ajo.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 42.Velez G, Chan CC, Csaky KG. Fluorescein angiographic findings in primary intraocular lymphoma. Retina. 2002;22:37–43. doi: 10.1097/00006982-200202000-00007. [DOI] [PubMed] [Google Scholar]
- 43.Cassoux N, Giron A, Bodaghi B, et al. IL-10 measurement in aqueous humor for screening patients with suspicion of primary intraocular lymphoma. Invest Ophthalmol Vis Sci. 2007;48:3253–3259. doi: 10.1167/iovs.06-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herrlinger U. Primary CNS lymphoma: findings outside the brain. J Neurooncol. 1999;43:227–230. doi: 10.1023/a:1006250317940. [DOI] [PubMed] [Google Scholar]
- 45.Akpek EK, Ahmed I, Hochberg FH, et al. Intraocular-central nervous system lymphoma: clinical features, diagnosis, and outcomes. Ophthalmology. 1999;106:1805–1810. doi: 10.1016/S0161-6420(99)90341-X. [DOI] [PubMed] [Google Scholar]
- 46.Peterson K, Gordon KB, Heinemann MH, DeAngelis LM. The clinical spectrum of ocular lymphoma. Cancer. 1993;72:843–849. doi: 10.1002/1097-0142(19930801)72:3<843::aid-cncr2820720333>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 47.Grimm SA, Pulido JS, Jahnke K, et al. Primary intraocular lymphoma: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Ann Oncol. 2007;18:1851–1855. doi: 10.1093/annonc/mdm340. [DOI] [PubMed] [Google Scholar]
- 48.Wallace DJ, Shen D, Reed GF, et al. Detection of the bcl-2 t(14;18) translocation and proto-oncogene expression in primary intraocular lymphoma. Invest Ophthalmol Vis Sci. 2006;47:2750–2756. doi: 10.1167/iovs.05-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonzales JA, Chan CC. Biopsy techniques and yields in diagnosing primary intraocular lymphoma. Int Ophthalmol. 2007;27:241–250. doi: 10.1007/s10792-007-9065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yeh S, Weichel ED, Faia LJ, et al. 25-Gauge transconjunctival sutureless vit-rectomy for the diagnosis of intraocular lymphoma. Br J Ophthalmol. 2010;94:633–638. doi: 10.1136/bjo.2009.167940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coupland SE, Damato B. Understanding intraocular lymphomas. Clin Experiment Ophthalmol. 2008;36:564–578. doi: 10.1111/j.1442-9071.2008.01843.x. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Shen D, Wang VM, Sen HN, Chan CC. Molecular biomarkers for the diagnosis of primary vitreoretinal lymphoma. Int J Mol Sci. 2011;12:5684–5697. doi: 10.3390/ijms12095684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White VA, Gascoyne RD, Paton KE. Use of the polymerase chain reaction to detect B- and T-cell gene rearrangements in vitreous specimens from patients with intraocular lymphoma. Arch Ophthalmol. 1999;117:761–765. doi: 10.1001/archopht.117.6.761. [DOI] [PubMed] [Google Scholar]
- 54.Shen DF, Zhuang Z, LeHoang P, et al. Utility of microdissection and polymerase chain reaction for the detection of immunoglobulin gene rearrangement and translocation in primary intraocular lymphoma. Ophthalmology. 1998;105:1664–1669. doi: 10.1016/S0161-6420(98)99036-4. [DOI] [PubMed] [Google Scholar]
- 55.Chan CC, Whitcup SM, Solomon D, Nussenblatt RB. Interleukin-10 in the vitreous of patients with primary intraocular lymphoma. Am J Ophthalmol. 1995;120:671–673. doi: 10.1016/s0002-9394(14)72217-2. [DOI] [PubMed] [Google Scholar]
- 56.Ramkumar HL, Shen de F, Tuo J, et al. IL-10-1082 SNP and IL-10 in primary CNS and vitreoretinal lymphomas. Graefes Arch Clin Exp Ophthalmol. 2012;250:1541–1548. doi: 10.1007/s00417-012-2037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pahk PJ, Todd DJ, Blaha GR, et al. Intravascular lymphoma masquerading as Vogt-Koyanagi-Harada syndrome. Ocul Immunol Inflamm. 2008;16:123–126. doi: 10.1080/09273940802023810. [DOI] [PubMed] [Google Scholar]
- 58.Berenbom A, Davila RM, Lin HS, Harbour JW. Treatment outcomes for primary intraocular lymphoma: implications for external beam radiotherapy. Eye (Lond) 2007;21:1198–1201. doi: 10.1038/sj.eye.6702437. [DOI] [PubMed] [Google Scholar]
- 59.Pe’er J, Hochberg FH, Foster CS. Clinical review: treatment of vitreoretinal lymphoma. Ocul Immunol Inflamm. 2009;17:299–306. doi: 10.3109/09273940903370755. [DOI] [PubMed] [Google Scholar]
- 60.Hormigo A, Abrey L, Heinemann MH, DeAngelis LM. Ocular presentation of primary central nervous system lymphoma: diagnosis and treatment. Br J Haematol. 2004;126:202–208. doi: 10.1111/j.1365-2141.2004.05028.x. [DOI] [PubMed] [Google Scholar]
- 61.Hashida N, Ohguro N, Nishida K. Efficacy and complications of intravitreal rituximab injection for treating primary vitreoretinal lymphoma. Transl Vis Scie Technol. 2012;1:1. doi: 10.1167/tvst.1.3.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma WL, Hou HA, Hsu YJ, et al. Clinical outcomes of primary intraocular lym-phoma patients treated with front-line systemic high-dose methotrexate and intravitreal methotrexate injection. Ann Hematol. 2016;95:593–601. doi: 10.1007/s00277-015-2582-x. [DOI] [PubMed] [Google Scholar]
- 63.Rosenfeld MR, Pruitt AA. Management of malignant gliomas and primary CNS lymphoma: standard ofcareand future directions. Continuum. 2012;18:406–415. doi: 10.1212/01.CON.0000413666.88539.0b. [DOI] [PubMed] [Google Scholar]
- 64.Hashida N, Nakai K, Saitoh N, Nishida K. Association between ocular findings and preventive therapy with onset of central nervous system involvement in patients with primary vitreoretinal lymphoma. Graefes Arch Clin Exp Oph-thalmol. 2014;252:687–693. doi: 10.1007/s00417-014-2584-8. [DOI] [PubMed] [Google Scholar]