

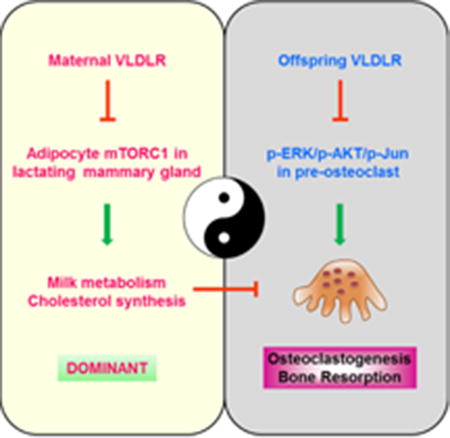
Summary
It is unknown whether and how VLDLR impacts skeletal homeostasis. Here we report that maternal and offspring VLDLR play opposite roles in osteoclastogenesis and bone resorption. VLDLR deletion in the offspring augments osteoclast differentiation by enhancing RANKL signaling, leading to osteoporosis. In contrast, VLDLR deletion in the mother alters milk metabolism, which inhibits osteoclast differentiation and causes osteopetrosis in the offspring. The maternal effects are dominant. VLDLR-null lactating mammary gland exhibits higher mTORC1 signaling and cholesterol biosynthesis. Pharmacological probing reveals that rapamycin but not statin treatment of the mother can prevent both the low bone resorption and our previously described inflammatory fur-loss in their offspring. Genetic rescue reveals that maternal mTORC1 attenuation in adipocytes but not in myeloid cells prevents offspring osteopetrosis and fur-loss. Our studies uncover functions of VLDLR and mTORC1 in lactation and osteoclastogenesis, illuminating key mechanisms and therapeutic insights for bone and metabolic diseases.
Graphical abstract
Introduction
Mother's breast milk is the ideal nourishment for infants before they are able to eat and digest solid foods. Evolution has perfected the composition of breast milk so that it confers multiple key functions essential for newborn development. It provides not only macro- and micro-nutrients for the growth and development the newborns (Gribble, 2006; Walker, 2010), but also hormones, digestive enzymes and immune-defensive factors including antibodies and cytokines (Bertotto et al., 1990; Gartner et al., 2005). Given breast milk is a pristine source of energy, nutrient and immunity for infants, defects in milk composition will exert detrimental consequences on the offspring. Therefore, understanding of the mechanisms by which mother's milk nourishes and protects the newborns is fundamentally important in order to design better infant formula and diagnostic/therapeutic strategies for infantile and childhood diseases.
Very-low-density-lipoprotein receptor (VLDLR) is a member of the LDL receptor family (Herz et al., 2009). Discovered in 1992, VLDLR is highly expressed in tissues with active triglyceride metabolism such as adipose tissue, heart, and skeletal muscle, but absent from the liver (Oka et al., 1994; Takahashi et al., 1992). It plays an important role in the delivery of free fatty acids from VLDL-triglycerides to various tissues as energy source (Tiebel et al., 1999; Webb et al., 1994). VLDLR and apolipoprotein receptor 2 (APOER2) also form a heterodimeric receptor for reelin to regulate brain development (Trommsdorff et al., 1999). However, little is known about VLDLR functions in other biological processes.
Our previous work shows that maternal VLDLR deletion causes milk defects, leading to systemic inflammation in the nursing neonates manifested as transient fur loss (Du et al., 2012). Here we investigate if these maternal and milk defects exert any long-term consequences on offspring traits such as bone. Furthermore, studies from our group and others reveal that lipid metabolism intimately connect with skeletal homeostasis through several factors, including PPARγ and ERRα (Wan, 2010; Wan et al., 2007b; Wei et al., 2016; Wei et al., 2010). In this study, we uncover VLDLR as an important dual regulator of skeletal remodeling that plays opposite roles in the offspring and in the lactating mother.
Results
Offspring VLDLR suppresses osteoclastogenesis and bone resorption
We crossed VLDLR+/− with VDLDR+/− to obtain VLDLR-/- mice and wild-type (WT) littermate control mice (Figure 1A). Body weight or bone marrow cellularity was unaltered in VLDLR-/-mice (Figure S1A-D). To investigate the intrinsic role of VLDLR in osteoclastogenesis, we employed an ex vivo bone marrow osteoclast differentiation system (Wan et al., 2007b). In this system, bone marrow osteoclast progenitors and precursors were first expanded using M-CSF, and then differentiated into osteoclasts using RANKL, with or without further stimulation with rosiglitazone, a synthetic agonist for the pro-osteoclastogenic nuclear receptor PPARγ (Wan et al., 2007b). Compared with WT control cultures, both RANKL-induced and rosiglitazone-stimulated osteoclast differentiation was enhanced in VLDLR-/- cultures, shown by TRAP staining (Figure 1B), expression of osteoclast markers (Figure 1C) and resorptive activity (Figure 1D). Osteoclast precursor proliferation or apoptosis was unaffected (Figure S1E-F). In addition, the levels of RANKL, M-CSF, or OPG in bone were unchanged (Figure S1G). These observations indicate that VLDLR deletion mainly augments RANKL signaling during osteoclast differentiation. VLDLR was expressed not only in osteoclast (Fig. S1H) but also in osteoblast (Fig. S1I). However, ex vivo bone marrow osteoblast differentiation was normal (Figure S1J-K).
Figure 1.
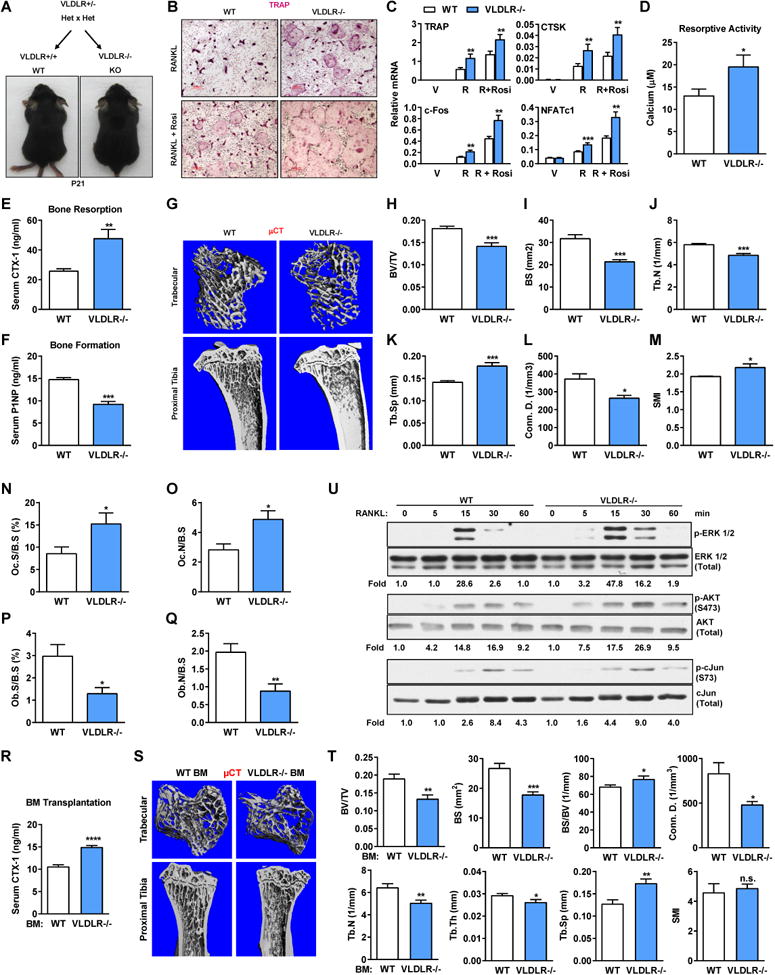
Offspring VLDLR suppresses osteoclastogenesis and bone resorption. (A) Schematic of breeding, and images of the pups on postnatal day 21 (P21). (B) TRAP staining of bone marrow osteoclast differentiation cultures (2 month old offspring). (C) Osteoclast marker expression (n=5). (D) Osteoclast resorptive activity (n=16). (E) Serum CTX-1 (n=7). (F) Serum P1NP (n=7).(G) μCT images of the trabecular bone of the tibial metaphysis (top) and the entire proximal tibia (bottom). (H-M) μCT quantification of trabecular bone parameters (n=4). (H) BV/TV, bone volume/tissue volume ratio; (I) BS, bone surface; (J) Tb.N, trabecular number; (K) Tb.Sp, trabecular separation; (L) Conn. D., connectivity density; (M) SMI, structure model index. (N-Q) Bone histomorphometry of distal femurs (n=6). Oc.S, osteoclast surface; B.S, bone surface; Oc.N, osteoclast number; Ob.S, osteoblast surface; Ob.N, osteoblast number. (R-T) 6 weeks post bone marrow transplantation (n=10). (R) Serum CTX-1; (S) μCT images; (T) μCT quantification. (U) Phosphorylation of ERK1/2, AKT and c-Jun upon RANKL treatment. Ratios of p/total are shown as fold compared with 0 min time point in the same genotype.
We next compared the in vivo bone phenotype. Consistent with our ex vivo osteoclastogenesis assay, ELISA analyses showed that serum bone resorption marker CTX-1 (carboxy-terminal telopeptides of type I collagen) was higher in VLDLR-/- mice (Figure 1E). Interestingly, despite the unaltered intrinsic ability of VLDLR-/- bone marrow mesenchymal stem cells to differentiate into osteoblasts (Figure S1J-K), serum bone formation marker P1NP (amino-terminal propeptide of type I procollagen) (Figure 1F), as well as bone formation rate and mineral apposition rate measured by double calcein labeling dynamic histomorphometry (Figure S1L-N), were decreased in VLDLR-/- mice, possibly due to changes in non-cell-autonomous signals.
Micro-computed tomography (μCT) of the trabecular bones in proximal tibiae showed that VLDLR-/- mice exhibited a low-bone-mass/osteoporosis phenotype (Figure 1G-M), with decreased bone volume/tissue volume ratio (BV/TV) (Figure 1H), bone surface (BS) (Figure 1I), trabecular number (Tb.N) (Figure 1J) and connectivity (Conn.) (Figure 1L), as well as increased trabecular separation (Tb.Sp) (Figure 1K) and structure model index (SMI), which quantifies the relative amount of plates (SMI=0, strong) and rods (SMI=3, fragile) (Figure 1M).
In accordance with the serum bone marker ELISA results, static bone histomorphometry showed that femurs of VLDLR-/- mice exhibited higher osteoclast number and surface (Figure 1N-O), but lower osteoblast number and surface (Figure 1P-Q). To determine whether the increased bone resorption originated from hematopoietic cells or stromal cells, we performed bone marrow transplantation in which total bone marrow from WT or VLDLR-/- donors was transferred into lethally irradiated WT recipients. The results showed that VLDLR-/- bone marrow was sufficient to confer increased bone resorption and decreased bone mass (Figure 1R-T), indicating an osteoclast-intrinsic role of VLDLR. Dissection of RANKL downstream signaling events showed that VLDLR-/- osteoclast differentiation cultures displayed a faster and stronger activation of ERK1/2, AKT and c-Jun (Figure 1U). These results indicate that VLDLR in osteoclast suppresses osteoclastogenesis and bone resorption.
Maternal VLDLR enhances offspring osteoclastogenesis and bone resorption via milk
Surprisingly, we found that VLDLR-/- mice from VLDLR-/- x VLDLR-/- breeding - KO (KOxKO) (Figure 2A) instead showed decreased ex vivo bone marrow osteoclast differentiation (Figure 2B-C) and in vivo osteoclast number/surface (Figure 2D), compared with WT mice from WT x WT breeding – WT (WTxWT). As we previously reported, pups nursed by VLDLR-/- mothers develop transient furloss (Figure 2A) and growth retardation (Figure S2A-B) due to a systemic inflammatory response to defective milk (Du et al., 2012). This suggests that the blunted osteoclastogenesis in the offspring may result from maternal defects and milk disorders. As often observed in osteopetrotic mice with low bone resorption (Wan et al., 2007b), KO (KOxKO) mice also displayed splenomegaly possibly due to compromised hematopoiesis in the marrow (Figure S2C-D). Indeed, KO (KOxKO) mice had a lower total bone marrow cell number (Figure S2E) without overtly affected hematopoietic lineage allocation (Figure S2F), suggesting a reduction of hematopoiesis in the marrow but not any lineage shift. Osteoclast precursor proliferation or apoptosis was unaltered in KO (KOxKO) bone marrow cultures (Figure S2G-H), indicating that the defects mainly reside in osteoclast differentiation.
Figure 2.
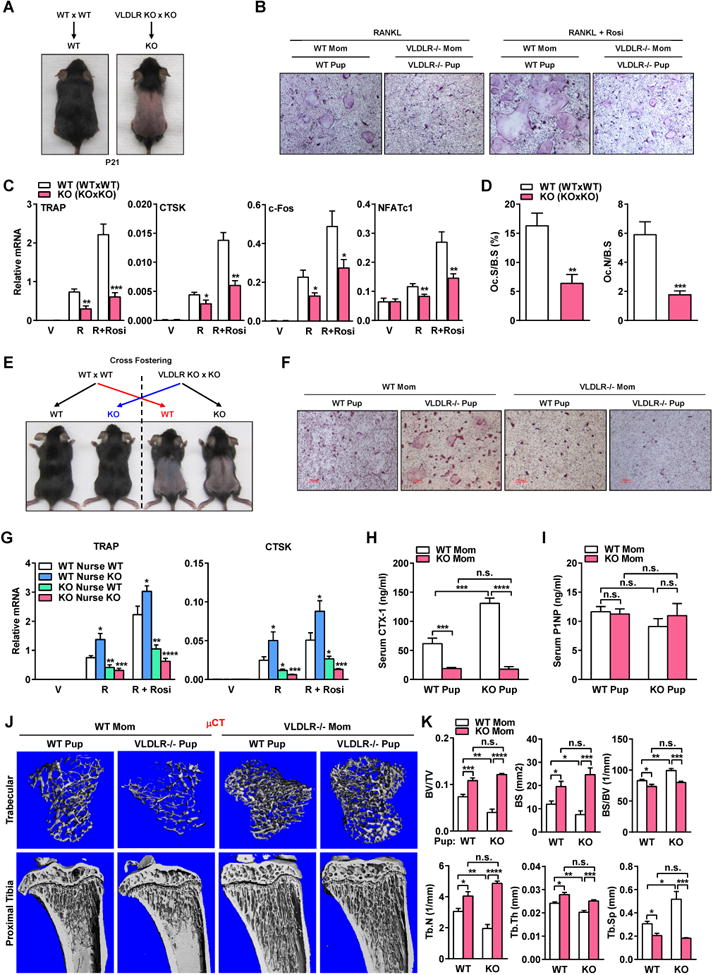
Maternal VLDLR enhances offspring osteoclastogenesis and bone resorption via milk. (A) Schematic of breeding, and images of the pups. (B) TRAP staining of bone marrow osteoclast differentiation cultures (2 month old offspring). (C) Osteoclast marker expression (n=5). (D) Bone histomorphometry of distal femurs (n=6). (E) Schematic of cross-fostering, and images of the pups. (F) TRAP staining of osteoclast differentiation cultures (2 month old offspring). (G) Osteoclast marker expression (n=4). (H) Serum CTX-1 (n=4). (I) Serum P1NP (n=4). (J) μCT images. (K) μCT quantification.
To determine whether the bone phenotype in KO (KOxKO) mice originate from maternal or offspring genotype and prenatally or postnatally, we performed cross-fostering experiments (Figure 2E). WTxWT and KOxKO breeding were set up at the same time, and mothers that gave birth around the same day were used as foster pairs. On postnatal day 0-1, 50% of WT (WTxWT) pups were transferred to and nursed by KO lactating females; at the same time, 50% of KO (KOxKO) pups were transferred to and nursed by WT counterparts. Compared with WT pups nursed by WT moms, both ex vivo osteoclast differentiation (Figure 2F-G) and in vivo bone resorption (Figure 2H) were enhanced for KO pups nursed by WT moms, but blunted in WT pups nursed by KO moms, indicating that the reduced osteoclastogenesis was due to a postnatal maternal defects in the KO moms likely through milk disorders. Moreover, KO pups nursed by KO moms showed a similar phenotype as WT pups nursed by KO moms rather than KO pups nursed by WT moms, indicating that the maternal effects were dominant (Figure 2E-H). In contrast, bone formation marker was unaffected by the maternal defects (Figure 2I). μCT showed that compared with WT pups nursed by WT moms, KO pups nursed by WT moms exhibited low bone mass causing osteopenia/osteoporosis, whereas both WT and KO pups nursed by KO moms exhibited high bone mass causing osteopetrosis (Figure 2J-K). Our data indicate that maternal VLDLR plays an opposite and dominant role over offspring VLDLR to enhance offspring osteoclastogenesis and bone resorption by maintaining a normal milk composition.
Maternal VLDLR deletion accumulates cholesterol precursors in milk
VLDLR is highly expressed in adipocyte-containing tissues (Yagyu et al., 2002) including the mammary glands (Macias and Hinck, 2012). To pinpoint the lactation defects, we collected lactating mammary glands on lactation day 2 (early lactation), day 10 (peak lactation), and day 18 (late lactation/early involution). Initial examination of the expression of genes associated with milk production revealed no significant changes between KO and WT glands (Figure S3A). Because VLDLR is closely related to LDLR – an important modulator of cholesterol metabolism, we tested whether cholesterol biosynthetic pathway was affected. We found that both mRNA and protein of the rate-limiting enzyme 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) was higher in the KO glands during peak lactation on day 10 (Figure 3A-B). The elevated HMGCR expression was absent during early lactation on day 2 but continued to late lactation on day 18 (Figure S3B-D). In addition, the expression of several other intermediate enzymes in the cholesterol biosynthetic pathway was also increased on day 10 (Figure 3A). Interestingly, the expression of lathosterol 5-desaturase (SC5D) was decreased in the KO glands throughout lactation (Figure 3A, S3B-C). SC5D catalyzes the second to the last step of cholesterol biosynthesis to convert lathosterol to 7-dehydrocholesterol in the Kandutsch-Russel pathway, or convert cholesta-7,24-dien-3β-ol to 7-dehydrodesmosterol in the Bloch pathway (Porter and Herman, 2011).
Figure 3.
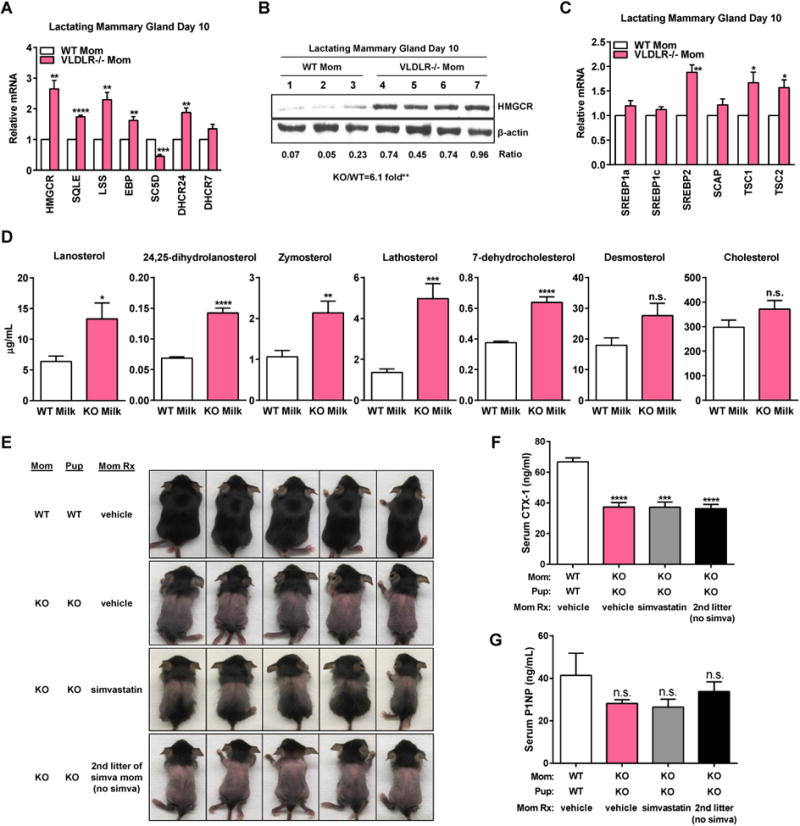
Maternal VLDLR deletion accumulates cholesterol precursors in the milk. (A-C) mRNA (A, C) and protein (B) expression of enzymes (A, B) and transcription factors (C) regulating cholesterol biosynthesis in day 10 lactating mammary glands (WT n=3; KO n=4). (D) Quantification of sterol precursors and cholesterol in milk by GC-MS (n = 6). (E) Lactating moms were treated with simvastatin (simva) or vehicle every other day for 21 days since parturition. Images of P21 pups, representative of 3 independent experiments. (F) Serum CTX-1 (n=5). (G) Serum P1NP (n=5).
Because HMGCR is critically regulated by SREBP family of transcription factors (Horton et al., 2002), we next examined the expression of SREBP1-a, -c, and -2 in lactating mammary glands. SREBP-2 was up-regulated in the KO glands, whereas SREBP1-a and SREBP1-c were unaltered (Figure 3C, S3E-F). This suggests that VLDLR inhibits SREBP-2, but not SREBP-1, to suppress HMGCR and cholesterol biosynthesis.
The combination of higher HMGCR and lower SC5D in the KO glands suggests an accumulation of cholesterol precursors but not necessarily cholesterol itself. Intriguingly, cholesterol precursor accumulation has been reported to cause hair growth defects (Evers et al., 2010), suggesting that this may contribute to the milk disorders in question. To test this hypothesis, we collected milk from WT and KO moms on lactation day 11, and performed gas chromatography-mass spectrometry (GC-MS) to quantify cholesterol and precursors (Figure 3D). Lanosterol, 24,25-dihydrolanosterol, zymosterol, lathosterol and 7-dehydrocholesterol were increased in the KO milk compared with WT control milk, whereas desmosterol and cholesterol were not different (Figure 3D). Consistent with the comparable milk cholesterol between WT and KO, serum cholesterol were also unaltered in KO lactating moms (Figure S3G).
To evaluate if cholesterol precursor accumulation in the KO milk mediates the phenotype in the pups, we treated lactating KO moms with 100 mg/kg simvastatin every other day for 21 days since parturition via intra-peritoneal injection. Maternal treatment with simvastatin partially rescued pup fur-loss (Figure 3E), but the rescuing effect was short term as the next litter from the same mom without further simvastatin treatment reverted back to full fur-loss (Figure 3E). However, maternal simvastatin treatment could not rescue the low bone resorption in the pups (Figure 3F, G). These results suggest that cholesterol precursor accumulation in milk may contribute to fur-loss but does not play a major role in the osteopetrosis in the offspring.
Maternal mTORC1 inhibition by rapamycin rescues offspring osteopetrosis and alopecia
Gene expression analysis also revealed a marked reduction of lipin 1 (LPIN1) in the KO glands on day 10 and 18 (Figure S4A). Lipin 1, a phosphatidic acid phosphatase, is regulated by mTOR complex 1 (mTORC1) to control the SREBP pathway (Peterson et al., 2011). Moreover, an induction of tuberous sclerosis 1 & 2 (TSC1/2) in the KO glands on day 10 (Figure 3C) suggests a feedback inhibition of mTORC1. Akt is an upstream activator of mTORC1, while S6K1 and S6 are downstream targets of mTORC1 (Laplante and Sabatini, 2012). Indeed, higher levels of phosphorylated Akt, S6K1 and S6 were observed in the KO glands on day 10 and 18 (Figure 4A, S4B), indicating higher mTORC1 activity. This activated mTORC1 signaling was not due to changes in ERK1/2 MAP kinases (Figure S4C-D).
Figure 4.
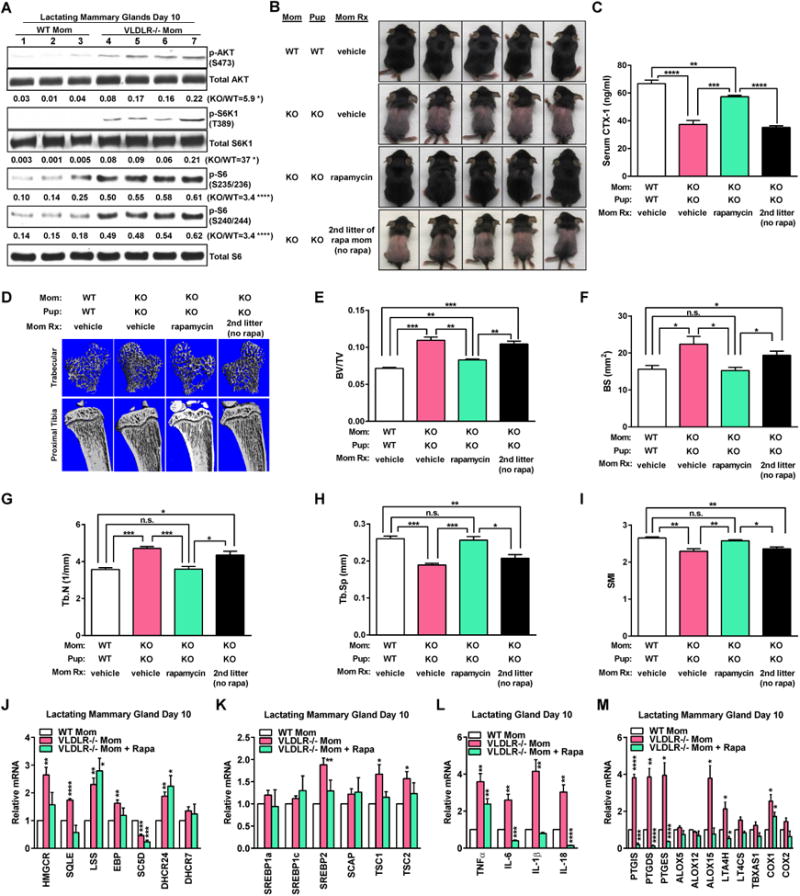
Maternal mTORC1 inhibition by rapamycin rescues osteopetrosis and alopecia in the offspring. (A) Western blot for mTORC1 signaling in day 10 lactating mammary glands. (B) Lactating moms were treated with rapamycin (rapa) or vehicle every other day for 21 days since parturition. Images of P21 pups, representative of 3 independent experiments. (C) Serum CTX-1 (n=5). (D) μCT images. (E-I) μCT quantification (n=4). (J-M) Gene expression for enzymes (J) and transcription factors (K) regulating cholesterol synthesis, cytokines (L) and enzymes (M) regulating inflammation in day 10 lactating mammary glands (WT n=3; KO n=4; KO+Rapa n=3).
To test if pharmacologic inhibition of mTORC1 could rescue pup phenotype, we treated lactating KO moms with 25 mg/kg rapamycin every other day for 21 days since parturition via intra-peritoneal injection. Maternal rapamycin treatment effectively prevented fur-loss (Figure 4B), and remarkably, also the low bone resorption and high bone mass in their pups (Figure 4C-I). The rescue was short term without lasting until the next litter (Figure 4C-E).
Rapamycin reversed the elevated expression of HMGCR, SREBP2 and TSC1/2 in the KO glands (Figure 4J-K, S4E-F), as the result of mTORC1 signaling suppression (Figure S4I-K). Moreover, rapamycin also reversed the elevated expression of pro-inflammatory adipokines TNFα, IL-6, IL-1β and IL-18 in the KO glands (Figure 4L, S4G). In addition, we examined the expression of several enzymes of the arachidonic acid pathway, including prostaglandin synthases, lipoxygenases and cyclooxygenases, as a recent study showed that prostaglandins promote alopecia (Garza et al., 2012). PTGIS, PTGDS and PTGES were elevated in the KO glands on day 10, but reduced back to control levels on day 18. COX1 and ALOX15 were also higher in the KO glands on both day 10 and 18. Rapamycin treatment also effectively abolished the up-regulation of these genes in the KO glands (Figure 4M, S4H). Therefore, mTORC1 inhibition by rapamycin exerted multiple actions to more effectively prevent milk disorders and offspring defects compared with HMGCR inhibition by simvastatin alone.
Genetic mTORC1 attenuation in adipocyte rescues offspring osteopetrosis and alopecia
To confirm our pharmacological findings that excessive mTORC1 mediates the milk disorder, and to dissect the key cell type in which mTORC1 acts, we next performed genetic rescue experiments. Since adipocyte and macrophage are two major cell types in the lactating mammary gland, we disrupt mTORC1 functions in these two cell types by crossing Raptor flox mice with lysozyme (Lyz)-Cre or adiponectin (Adipo)-Cre to generate macrophage- and adipocyte-specific Raptor KO mice, respectively (Figure S5). These mice were then crossed with VLDLR-/- mice to generate compound mutants in order to assess rescuing effects. Both fur-loss and osteopetrosis in the offspring were effectively rescued by adipocyte Raptor deletion, but not by macrophage Raptor deletion, in the VLDLR-KO moms (Figure 5A-I). Although acute maternal rapamycin treatment did not significantly improve the body weight in the pups (Figure S4L), maternal genetic Raptor deletion in adipocyte, but not in macrophages, did significantly prevent the growth retardation (Figure 5J). These results not only confirmed mTORC1 as a key VLDLR downstream signaling to control milk metabolism and offspring traits, but also pinpoint adipocyte as the crucial site of regulation.
Figure 5.
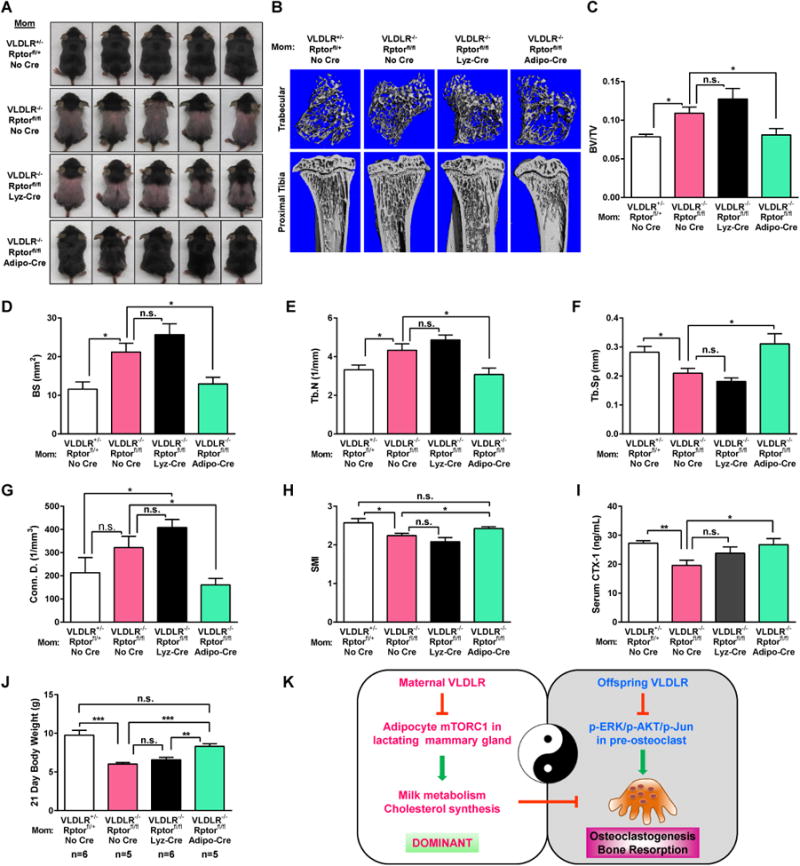
Genetic mTORC1 attenuation in adipocyte rescues osteopetrosis and alopecia in the offspring. (A) Images of P21 pups, representative of 3 independent experiments. (B) μCT images. (C-H) μCT quantification. (I) Serum CTX-1 (n=7). (J) Body weight of P21 pups. (K) A schematic diagram.
Discussion
This study reveals a yin-yang dual role of VLDLR in the regulation of osteoclastogenesis and bone resorption (Figure 5K). On one hand, VLDLR in the offspring suppresses osteoclast differentiation by inhibiting RANKL signaling such as ERK/Akt/c-Jun phosphorylation. The cell-autonomous regulation of osteoclast differentiation by offspring VLDLR is demonstrated by 1) VLDLR-/- osteoclast precursors exhibit significantly higher ability for osteoclast differentiation in the absence of stromal cells (Figure 1B, D); 2) VLDLR-/- osteoclast differentiation cultures exhibit hypersensitivity to RANKL signaling including augmented activation of NFATc1, c-Fos, ERK, AKT and c-Jun in the absence of stromal cells (Figure 1C, U); 3) RANKL, M-CSF and OPG levels in bone were unaltered, indicating that the enhanced osteoclastogenesis in vivo is due to increased cytokine sensitivity rather than cytokine abundance (Figure S1G); 4) bone marrow chimera with VLDLR-/- bone marrow cells showed that the elevated bone resorption mainly originates from hematopoietic cells rather than stromal cells (Figure 1R-T). On the other hand, VLDLR in the nursing mother dominantly promotes offspring osteoclastogenesis by attenuating mTORC1 signaling in the lactating mammary gland to prevent milk metabolic and inflammatory disorders (Figure 5K). Consequently, maternal VLDLR deletion causes decreased osteoclast differentiation and bone resorption in the offspring, leading to higher bone mass (Figure 2), owing to excessive maternal mTORC1 activities (Figure 4). The functional significance of mTORC1 is illustrated by the ability of maternal rapamycin treatment to effectively abrogate the bone defects in the offspring (Figure 4). Moreover, adipocyte is the major cell type for the excessive maternal mTORC1 activities because Raptor deletion therein rescues the offspring bone phenotype (Figure 5). Together, these findings unveil VLDLR as an important yet previously unrecognized player in skeletal homeostasis and lactation metabolism that exerts cross generation regulation.
The transient osteopetrosis and alopecia in the offspring nursed by VLDLR-KO mothers are mainly due to milk defects. Fostering experiments show that these offspring traits are strictly conferred by VLDLR-KO lactating mother but independent of the genotype of birth mother, father or pups. Moreover, the impaired osteoclastogenesis in offspring nursed by VLDLR-KO mothers persists for 2 months after weaning but becomes rescued by 3 months after weaning (Figure S2I-K). This is likely due to the dilution of the milk effects and the turnover of bone cells, as the literature shows that the lifespan of osteoclast and osteoblast is approximately 2 weeks and 3 months, respectively (Manolagas, 2000). Initially, we hypothesized that the transmission of maternal defects to the offspring may be also contributed by microbiota. However, when we cross-transferred cage bedding and feces between WT and KO moms, we did not observe the switch of pup phenotype, indicating that microbiota do not play a major role in this context. Together, our findings support that milk is responsible for the maternal VLDLR control of offspring traits.
Initially, the observation of a dramatic increase of HMGCR expression in the lactating mammary gland prompted us to investigate the significance of milk cholesterol in the offspring bone and fur loss phenotype. However, our data indicate that cholesterol pathway may be a contributing factor but not the major mediator of the phenotype in question. Based on our maternal pharmacological experiments showing a stronger rescue by rapamycin than by simvastatin, we decided to instead focus on mTORC1 signaling. Our findings suggest that multiple pathways in the mom may contribute to the milk defect and pup phenotype, but mTORC1 is the master regulator because rapamycin treatment corrected several defects in the lactating mammary gland including cholesterol pathway and inflammatory signaling (Fig. 4J-M). Our study focuses on the moms and the lactating mammary gland, thus we treated the lactating moms (rather than pups) with simvastatin or rapamycin. It is possible that the extent to which cholesterol defects contribute to these phenotypes was not fully revealed by the simvastatin treatment due to dosage limitation, which will require future studies using genetic approaches to further elucidate.
Our previous histological analysis of the skin of the pups nursed by VLDLR-/- moms revealed that the transient fur loss was due to an early ejection of the hair shaft upon inflammation rather than hair follicle formation defects (Du et al., 2012). Although the pups lost their fur and developed follicular cyst in the skin, the hair bulbs/follicles appeared normal and similar to the pups nursed by WT moms; hypodermal adipocytes in the skin were also comparable as controls (Du et al., 2012). In contrast, the skin histology from published mouse models that lose their fur due to cholesterol defects, such as epidermal-specific (Epi)-Insig1/2-DKO (Evers et al., 2010), LDLR/ApoAI double KO (Zabalawi et al., 2007), ACAT1 KO, ApoE KO, or ACAT1/LDLR double KO mice (Yagyu et al., 2000), showed that hair bulbs/follicles, as well as subcutaneous adipocytes, were largely absent. In addition, VLDLR is known to mediate VLDL uptake and free fatty acids delivery; however, it does not play as an important role as LDLR in cholesterol metabolism (Nimpf and Schneider, 2000; Oka et al., 1994), supporting the differential functions by VLDLR and LDLR. Collectively, these findings indicate that the fur loss in the pups nursed by VLDLR-/- moms may be contributed by milk cholesterol metabolic defects but may be distinct from or less severe than the fur loss in these published genetic mouse models with cholesterol defects. In Epi-Insig-DKO mice, topical treatment with simvastatin reduced sterol precursors in skin and corrected the hair and skin defects (Evers et al., 2010). Since our current study focuses on VLDLR-/- lactating moms, future study will be needed to determine in their pups whether topical simvastatin treatment can rescue the fur loss and systemic simvastatin treatment can rescue the low bone resorption.
We have investigated extensively the milk factors that may confer the pup phenotype by injecting WT lactating moms with individual inflammatory cytokines or lipids such as cholesterol precursors. The results show that none of them alone is sufficient to replicate the pup phenotype, indicating that multiple inflammatory and metabolic milk factors from VLDLR-KO mom are simultaneously required to exert these consequences in the pups. Both inflammatory pathways and lipid metabolism are known downstream targets of mTORC1 signaling, revealing mTORC1 as a master regulator of lactation that confer these pleotropic changes.
It is well known that Akt activates mTORC1 by inhibiting mTORC1 suppressors TSC1/2 (Guertin and Sabatini, 2007; Zoncu et al., 2011). It is also known that acute rapamycin inhibition of mTORC1/S6K often increases Akt due to compensatory effects by alleviating a negative feed-back loop (Chaturvedi et al., 2009; Harrington et al., 2004; Manning, 2004; Um et al., 2004; Wan et al., 2007a). Indeed, we found that short-term treatment of VLDLR-/- moms with rapamycin suppressed mTORC1 activity but increased Akt signaling (Fig. S4J).
Virgin mammary glands are essentially fat pads such as inguinal fat; during pregnancy and lactation, mammary glands/fat pads undergo epithelial expansion and the glands turn into largely mammary epithelial cells (Inman et al., 2015). As an important stromal cell type, adipocyte regulates mammary epithelial cell functions by both providing energy source and acting as an endocrine organ (Hovey and Aimo, 2010; Inman et al., 2015). Moreover, there are evidence that adipocyte and mammary epithelial cells undergo reversible trans-differentiation (Morroni et al., 2004). Therefore, Raptor deletion in adipocytes likely modulates mammary epithelial functions during lactation via both direct and indirect effects, by controlling multiple metabolic and inflammatory pathways.
Our current study reveals adipocyte as a key cell type for mTORC1 regulation during lactation. Our previous studies show that macrophage is also important for maternal PPARγ and VLDLR modulation of milk composition and offspring phenotype. These findings reveal that cellular crosstalk in the lactating mammary gland between adipocyte and macrophage may modulate milk composition and lactation outcome by balancing metabolism and immunity. For example, as we discussed in our earlier study (Wan et al., 2007c), PPARγ deficiency in macrophage may produce more endogenous PPARγ ligands such as HODES/HETES that can activate PPARγ in the mammary adipocytes to increase lipogenesis. Similarly, VLDLR deficiency in macrophage may alter adipocyte mTORC1 signaling and lipid homeostasis, leading to altered milk metabolism. Future studies are needed to further characterize this crosstalk.
Provocatively, our findings highlight that metabolic/inflammatory/bone diseases in the offspring may originate from maternal defects and milk disorders, which can be prevented by maternal interventions such as rapamycin treatment. Mechanistically, our work reveal a key signaling pathway in the lactating mammary gland, in which VLDLR inhibits mTORC1 activities in the adipocyte to suppress cholesterol biosynthesis by down-regulating the expression of SREBP2 and HMGCR, as well as to suppress the expression of inflammatory adipokines and prostaglandin synthases. These combined actions tightly control milk metabolism and immunity to ensure milk quality and optimal development of the offspring. Pharmacologic inhibition of HMGCR with simvastatin did not rescue low bone resorption and only partially rescued fur-loss in the pups nursed by VLDLR-KO moms, suggesting that the milk disorder was more than just altered cholesterol biosynthesis and accumulation of cholesterol precursors. In addition to regulating many cellular processes that control organismal growth and homeostasis, including protein synthesis, autophagy, metabolism and lipogenesis, mTORC1 has become increasingly appreciated to also play important roles in immunity and inflammation. Indeed, the exacerbated mTORC1 signaling in the VLDLR-KO lactating mammary glands leads to increased levels of inflammatory adipokines and prostaglandin synthases. The pleiotropic effects of mTORC1 on both metabolism and inflammation establish mTORC1 as a master regulator of lactation and VLDLR function, and rapamycin as an effective treatment of milk disorders and associated offspring diseases. Moreover, our genetic dissection pinpoints adipocyte as the main site of mTORC1 action in the lactating mammary gland, further highlighting the important roles of adipocyte at the crossroad of metabolism and immunity.
Experimental Procedures
VLDLR-/- mice (Frykman et al., 1995), Raptor flox mice (Sengupta et al., 2010), Lysozyme-Cre (Clausen et al., 1999) and Adiponectin-Cre transgenic mice (Eguchi et al., 2011) were from Jackson Laboratory and maintained on C57BL/6 background. Mice were fed standard rodent chow ad libitum (Harlan Laboratories). 9-10 week old females were used for breeding. Litter sizes were normalized to 6 pups. Milk was collected as described (Wan et al., 2007c). Sterol precursors and cholesterol in milk were analyzed by GC-MS as described (Evers et al., 2010). To obtain VLDLR-/-;raptorflox/flox;Adiponectin-Cre (or Lysozyme-Cre) triple genotype female mice, female VLDLR-/-;raptorflox/flox mice were bred with male VLDLR-/-;raptorflox/+;Adiponectin-Cre (or Lysozyme-Cre). To evaluate the genetic rescue, female VLDLR-/-;raptorflox/flox;Adiponectin-Cre (or Lysozyme-Cre) were bred with WT male mice. For treatment, lactating female mice received intraperitoneally 25 mg/kg of rapamycin, 100 mg/kg of simvastatin, or vehicle control every other day for 21 days since parturition. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center. Bone marrow osteoclast/osteoblast differentiation, bone analyses and gene expression were performed as described (Wei et al., 2011). All statistical analyses were performed with Student's t-test and represented as mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001; and n.s., nonsignificant (p > 0.05).
Supplementary Material
Highlights.
Maternal VLDLR regulates milk to enhance offspring osteoclastogenesis
Offspring VLDLR suppresses osteoclastogenesis and bone resorption
mTORC1 in adipocyte mediates maternal VLDLR regulation of milk
Rapamycin rescues milk disorder caused by maternal VLDLR loss
Acknowledgments
We thank Sean Morrison (UT Southwestern) and Jerry Feng (Baylor College of Dentistry) for assistance with μCT and histomorphometry; Fang Xu (UT Southwestern) for assistance with GC-MS. Y.W. is Lawrence Raisz Professor in Bone Cell Metabolism and a Virginia Murchison Linthicum Scholar in Medical Research. This work was in part supported by March of Dimes (#6-FY13-137, Y.W.), The Welch Foundation (I-1751, Y.W.), NIH (R01DK089113, Y.W.), CPRIT (RP130145, Y.W.), DOD (W81XWH-13-1-0318, Y.W.) and UTSW Endowed Scholar Startup Fund (Y.W.). The authors declare that they have no financial conflict of interest.
Footnotes
Author contributions: H.H. and Y.W. conceived the project, designed the experiments, and wrote the manuscript. H.H. conducted most of the experiments and data analyses. W.W. assisted with bone analyses.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bertotto A, Castellucci G, Fabietti G, Scalise F, Vaccaro R. Lymphocytes bearing the T cell receptor gamma delta in human breast milk. Arch Dis Child. 1990;65:1274–1275. doi: 10.1136/adc.65.11.1274-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi D, Gao X, Cohen MS, Taunton J, Patel TB. Rapamycin induces transactivation of the EGFR and increases cell survival. Oncogene. 2009;28:1187–1196. doi: 10.1038/onc.2008.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Du Y, Yang M, Wei W, Huynh HD, Herz J, Saghatelian A, Wan Y. Macrophage VLDL receptor promotes PAFAH secretion in mother's milk and suppresses systemic inflammation in nursing neonates. Nat Commun. 2012;3:1008. doi: 10.1038/ncomms2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E, Rosen ED. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13:249–259. doi: 10.1016/j.cmet.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers BM, Farooqi MS, Shelton JM, Richardson JA, Goldstein JL, Brown MS, Liang G. Hair growth defects in Insig-deficient mice caused by cholesterol precursor accumulation and reversed by simvastatin. J Invest Dermatol. 2010;130:1237–1248. doi: 10.1038/jid.2009.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frykman PK, Brown MS, Yamamoto T, Goldstein JL, Herz J. Normal plasma lipoproteins and fertility in gene-targeted mice homozygous for a disruption in the gene encoding very low density lipoprotein receptor. Proc Natl Acad Sci U S A. 1995;92:8453–8457. doi: 10.1073/pnas.92.18.8453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner LM, Morton J, Lawrence RA, Naylor AJ, O'Hare D, Schanler RJ, Eidelman AI. Breastfeeding and the use of human milk. Pediatrics. 2005;115:496–506. doi: 10.1542/peds.2004-2491. [DOI] [PubMed] [Google Scholar]
- Garza LA, Liu Y, Yang Z, Alagesan B, Lawson JA, Norberg SM, Loy DE, Zhao T, Blatt HB, Stanton DC, et al. Prostaglandin D2 inhibits hair growth and is elevated in bald scalp of men with androgenetic alopecia. Sci Transl Med. 2012;4:126ra134. doi: 10.1126/scitranslmed.3003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble KD. Mental health, attachment and breastfeeding: implications for adopted children and their mothers. Int Breastfeed J. 2006;1:5. doi: 10.1186/1746-4358-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Chen Y, Masiulis I, Zhou L. Expanding functions of lipoprotein receptors. Journal of Lipid Research. 2009;50:S287–S292. doi: 10.1194/jlr.R800077-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovey RC, Aimo L. Diverse and active roles for adipocytes during mammary gland growth and function. J Mammary Gland Biol Neoplasia. 2010;15:279–290. doi: 10.1007/s10911-010-9187-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman JL, Robertson C, Mott JD, Bissell MJ. Mammary gland development: cell fate specification, stem cells and the microenvironment. Development. 2015;142:1028–1042. doi: 10.1242/dev.087643. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias H, Hinck L. Mammary gland development. Wiley Interdiscip Rev Dev Biol. 2012;1:533–557. doi: 10.1002/wdev.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- Morroni M, Giordano A, Zingaretti MC, Boiani R, De Matteis R, Kahn BB, Nisoli E, Tonello C, Pisoschi C, Luchetti MM, et al. Reversible transdifferentiation ofsecretory epithelial cells into adipocytes in the mammary gland. Proc Natl Acad Sci U S A. 2004;101:16801–16806. doi: 10.1073/pnas.0407647101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimpf J, Schneider WJ. From cholesterol transport to signal transduction: low density lipoprotein receptor, very low density lipoprotein receptor, and apolipoprotein E receptor-2. Biochim Biophys Acta. 2000;1529:287–298. doi: 10.1016/s1388-1981(00)00155-4. [DOI] [PubMed] [Google Scholar]
- Oka K, Ishimura-Oka K, Chu MJ, Sullivan M, Krushkal J, Li WH, Chan L. Mouse very-low-density-lipoprotein receptor (VLDLR) cDNA cloning, tissue-specific expression and evolutionary relationship with the low-density-lipoprotein receptor. Eur J Biochem. 1994;224:975–982. doi: 10.1111/j.1432-1033.1994.00975.x. [DOI] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52:6–34. doi: 10.1194/jlr.R009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010;468:1100–1104. doi: 10.1038/nature09584. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Kawarabayasi Y, Nakai T, Sakai J, Yamamoto T. Rabbit Very Low-Density-Lipoprotein Receptor - a Low-Density-Lipoprotein Receptor-Like Protein with Distinct Ligand Specificity. P Natl Acad Sci USA. 1992;89:9252–9256. doi: 10.1073/pnas.89.19.9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiebel O, Oka K, Robinson K, Sullivan M, Martinez J, Nakamuta M, Ishimura-Oka K, Chan L. Mouse very low-density lipoprotein receptor (VLDLR): gene structure, tissue-specific expression and dietary and developmental regulation. Atherosclerosis. 1999;145:239–251. doi: 10.1016/s0021-9150(99)00068-4. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Walker A. Breast milk as the gold standard for protective nutrients. J Pediatr. 2010;156:S3–7. doi: 10.1016/j.jpeds.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007a;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- Wan Y. PPARgamma in bone homeostasis. Trends in endocrinology and metabolism: TEM. 2010;21:722–728. doi: 10.1016/j.tem.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007b;13:1496–1503. doi: 10.1038/nm1672. [DOI] [PubMed] [Google Scholar]
- Wan Y, Saghatelian A, Chong LW, Zhang CL, Cravatt BF, Evans RM. Maternal PPAR gamma protects nursing neonates by suppressing the production of nflammatory milk. Genes Dev. 2007c;21:1895–1908. doi: 10.1101/gad.1567207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JC, Patel DD, Jones MD, Knight BL, Soutar AK. Characterization and tissue-specific expression of the human ‘very low density lipoprotein (VLDL) receptor’ mRNA. Hum Mol Genet. 1994;3:531–537. doi: 10.1093/hmg/3.4.531. [DOI] [PubMed] [Google Scholar]
- Wei W, Schwaid AG, Wang X, Wang X, Chen S, Chu Q, Saghatelian A, Wan Y. Ligand Activation of ERRalpha by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab. 2016;23:479–491. doi: 10.1016/j.cmet.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Wang X, Yang M, Smith LC, Dechow PC, Sonoda J, Evans RM, Wan Y. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab. 2010;11:503–516. doi: 10.1016/j.cmet.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Zeve D, Suh JM, Wang X, Du Y, Zerwekh JE, Dechow PC, Graff JM, Wan Y. Biphasic and dosage-dependent regulation of osteoclastogenesis by beta-catenin. Mol Cell Biol. 2011;31:4706–4719. doi: 10.1128/MCB.05980-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagyu H, Kitamine T, Osuga J, Tozawa R, Chen Z, Kaji Y, Oka T, Perrey S, Tamura Y, Ohashi K, et al. Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J Biol Chem. 2000;275:21324–21330. doi: 10.1074/jbc.M002541200. [DOI] [PubMed] [Google Scholar]
- Yagyu H, Lutz EP, Kako Y, Marks S, Hu Y, Choi SY, Bensadoun A, Goldberg IJ. Very low density lipoprotein (VLDL) receptor-deficient mice have reduced lipoprotein lipase activity. Possible causes of hypertriglyceridemia and reduced body mass with VLDL receptor deficiency. J Biol Chem. 2002;277:10037–10043. doi: 10.1074/jbc.M109966200. [DOI] [PubMed] [Google Scholar]
- Zabalawi M, Bharadwaj M, Horton H, Cline M, Willingham M, Thomas MJ, Sorci-Thomas MG. Inflammation and skin cholesterol in LDLr-/-, apoA-I-/- mice: link between cholesterol homeostasis and self-tolerance? J Lipid Res. 2007;48:52–65. doi: 10.1194/jlr.M600370-JLR200. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.