

Genome wide association studies (GWASs) have identified multiple coronary artery disease (CAD) risk loci,1 yet moving from association to mechanistic insights and therapeutic translation remains a major challenge.2 A number of GWASs have identified LIPA as a novel locus for CAD.3–5 LIPA encodes lysosomal acid lipase (LAL), the major lysosomal enzyme hydrolyzing cholesteryl esters (CE) and triglycerides (TG) derived from lipoproteins taken up by cells.6 Prior to its GWAS discovery for CAD, loss-of-function (LOF) mutations in LIPA were identified as the cause of rare Mendelian disorders, including Wolman disease, an infantile-onset disorder due to complete LOF mutations, as well as cholesterol ester storage disease (CESD), a later-onset disorder with residual LAL activity resulting in hepatosplenomegaly, hyperlipidemia, liver failure and premature atherosclerosis.7, 8 Although rare LIPA LOF alleles in CESD are linked to accelerated atherosclerosis and hyperlipidemia, surprisingly the common LIPA CAD risk alleles are not associated with altered plasma lipids,9 liver traits9 or reduced expression of LIPA in liver.10 Indeed, the CAD risk alleles have no expression quantitative trait locus (eQTL) in liver tissue10 yet do, however, have eQTLs for higher LIPA mRNA in monocytes3, 11 and macrophages.12
These paradoxical data raise important questions for the field, particularly as LAL enzyme replacement therapy is approved for clinical use in Wolman disease and CESD, but whether this will ameliorate premature atherosclerosis and affect cardiovascular outcomes remains to be determined.13, 14 First, what is the directionality of CAD causal variant(s) at the LIPA GWAS locus – are they LOF as might be anticipated based on the effects of rare Mendelian variants that cause CESD and atherosclerosis, or are these unexpectedly gain of function (GOF) variants that increase LIPA expression and function in monocytes/macrophages, as suggested by the eQTL studies? If GOF or LOF variants in monocytes/macrophages contribute to CAD in the general population, what is the underlying biological mechanism? Second, is the CAD-associated actions of the LIPA GWAS locus cell-specific, restricted to actions in monocytes/macrophages and not active in hepatocytes, as one might surmise based on cell-specific eQTL data? Third, what is the genetic mechanism of the CAD locus in the disease relevant cells – and is there one or many functional variants that contribute to the GWAS CAD signal?
In this issue, Morris et al15 have begun to tackle these intriguing questions in their study of the potential causal variant for CAD at the GWAS LIPA locus and the directionality of its actions. They report that rs1051338 (NM_000235.3: c.46A>C, p.Thr16Pro), a coding variant in high linkage disequilibrium with the GWAS lead single nucleotide polymorphisms (SNPs), may serve as the potential causal variant at the LIPA locus for CAD. By using in silico prediction, overexpression of LAL in COS7 cell lines, and comparing LAL expression and activity in primary macrophages from risk allele and non-risk allele carriers, the authors propose that the risk allele (C) at rs1051338, which encodes a non-synonymous threonine to proline change (Thr16Pro) within the signal peptide of LAL, may impair LAL protein translocation from the endoplasmic reticulum resulting in proteosomal degradation and reduced LAL protein and activity in macrophages. The results also showed that lysosomal LAL activity in the risk allele carriers was lower than that in the non-risk allele carriers and that this was associated with a trend toward reduced efflux after [3H]-cholesterol labeled acetylated-low density lipoprotein loading. Thus, they propose that the GWAS risk locus for CAD is indeed LOF and mediated by the rs1051338 LIPA variant.
Although these data are highly suggestive they are not yet definitive for rs1051338 being “the” causal variant at the LIPA GWAS locus. The effects of the variant on LAL protein degradation were determined by exogenous LAL overexpression in COS7 cell line, a line that is not a disease-relevant cell type and the results mainly relied on the use of pharmacological inhibitors. Some experiments were repeated using human monocyte-derived macrophages but only in four homozygous risk allele and non-risk allele carriers, which is a small sample size for detection of the modest effects expected of a common variant for a complex trait identified by GWAS. Indeed, this is revealed by the lack of difference in LIPA expression in the monocytes3, 11 and macrophages12 in the presented data despite the published eQTL for rs1051338 and other linked GWAS lead SNPs at the locus. Furthermore, the functional impact of the variant examined in this study focused only on the efflux capacity of [3H]-cholesterol labeled acetylated-low density lipoprotein and failed to show a statistically significant difference by allele groups. Other phenotypes relevant to the LOF of LIPA, such as lysosomal cholesteryl ester hydrolysis,16 autophagy17 and macrophage alternative activation18 were not studied.
In addition to rs1051338, there are other linked SNPs at the LIPA locus, including those of similar allele frequency yet showing stronger association with increased risk of CAD and also with higher mRNA expression in eQTLs.3, 11 Some of these SNPs overlie open chromatin marks and other epigenetic features suggesting regulatory actions that might increase LIPA expression and contribute to CAD. The current study does not exclude this alternative hypothesis, one that is supported indirectly by data showing that increasing free cholesterol levels in lysosomes inhibit lysosome acidification and function and subsequent hydrolysis of lipoprotein CE, 19, 20 and that extracellular lysosomal synapse can degrade aggregated low density lipoprotein and contribute to foam cell formation.21–23 The functional effects, or lack thereof, of the linked SNPs in the region on LIPA transcription and mRNA expression in both monocytes and macrophages remain to be studied. This is particular important, as the current study did not address the published and surprising eQTL data of higher LIPA mRNA levels if the rs1051338 coding variant is indeed causal for CAD and encodes LOF of LAL activity.
Studying cell lines with endogenous LIPA expression on an isogenic background when the only genetic difference is each individual SNP will provide more reliable data than studying endogenous cells where the effects of individual variants in high linkage disequilibrium at the locus cannot be separated – as is the case for the current studies by Morris et al.15 Gene editing of human induced pluripotent stem cells (hiPSC) to introduce separately each risk-allele or to correct each risk allele, ideally differentiating the hiPSC lines to macrophages for functional studies,24 is ultimately required to provide definitive data in support of causal effects of any individual or combination of SNP variants.
In summary, Morris et al15 present important data suggesting that the rs1051338 Thr16Pro variant, in the LAL signal peptide, may be a causal LOF variant at the LIPA GWAS locus. Yet these studies do not address fully the LIPA paradox. Ultimately gene editing25 of isogenic hiPSC, with differentiation to macrophages (and other LIPA and CAD relevant cells) coupled to study of primary macrophages of much larger numbers of risk and non-risk allele carriers is warranted to parse the individual effects of each of the linked variants at the LIPA GWAS locus. Targeted mouse models with knock-in of human LIPA CAD alleles will also help to reveal the in vivo effects of specific variants at this GWAS locus on atherosclerosis. Further mechanistic study of macrophage LIPA in CAD risk will shed light on the potential for benefit and risk in therapeutic targeting of LIPA in CAD particularly in the context of the availability of LAL replacement therapy currently approved for use in CESD patients.
Figure 1. The LIPA paradox.
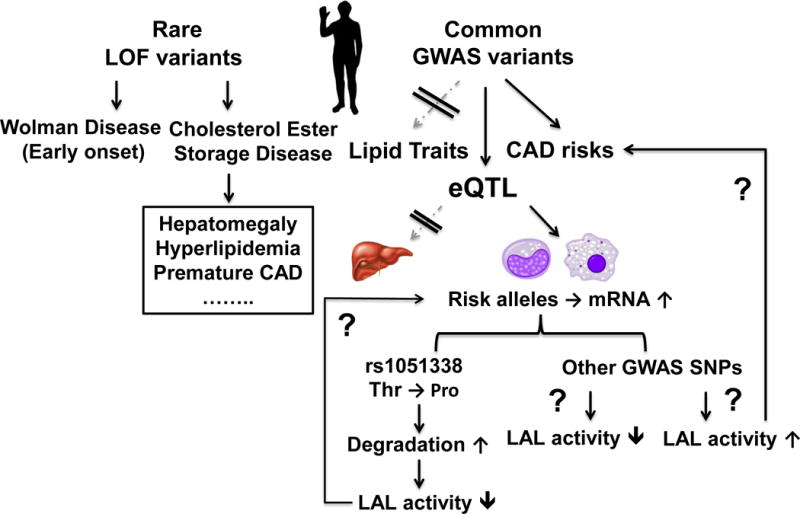
Genome wide association studies (GWAS) have identified LIPA as a novel locus for coronary artery disease (CAD). Rare loss-of-function (LOF) mutations of LIPA result in Wolman disease and cholesteryl ester storage disease with hepatomegaly, hyperlipidemia, and premature atherosclerosis. Surprisingly, the common LIPA CAD-GWAS risk alleles do not associate with altered plasma lipids or hepatic LIPA mRNA levels but relate to higher LIPA mRNA in monocytes and macrophages. The study by Morris et al15 suggests that rs1051338, a coding variant in high linkage disequilibrium with the GWAS lead single nucleotide polymorphisms (SNPs), causing a threonine to proline missense mutation within the signal peptide of LAL, leads to increased degradation and therefore reduced LAL activity. However, the directionality of LIPA CAD variants - whether LOF as suggested by Morris et al15 and based on the rare Mendelian variants or unexpected gain of function in monocytes/macrophages as suggested by expression quantitative trait loci (eQTL) analysis warrants further studies. The causal variant(s) and the functional impact and mechanisms of the variants on macrophage phenotypes remain to be fully defined.
Acknowledgments
Sources of funding
This work is supported by NIH grants R01-HL-113147 and K24-HL-107643 (to MPR), and partially supported by American Heart Association Postdoctoral Fellowship 15POST25620017 and NIH grant K99-HL-130574 (to HZ). MPR is also supported by R01-HL-111694.
Footnotes
Disclosures
None.
References
- 1.Khera AV, Kathiresan S. Genetics of coronary artery disease: Discovery, biology and clinical translation. Nature reviews Genetics. 2017 doi: 10.1038/nrg.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nurnberg ST, Zhang H, Hand NJ, Bauer RC, Saleheen D, Reilly MP, Rader DJ. From loci to biology: Functional genomics of genome-wide association for coronary disease. Circulation research. 2016;118:586–606. doi: 10.1161/CIRCRESAHA.115.306464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wild PS, Zeller T, Schillert A, et al. A genome-wide association study identifies lipa as a susceptibility gene for coronary artery disease. Circulation. Cardiovascular genetics. 2011;4:403–412. doi: 10.1161/CIRCGENETICS.110.958728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Consortium CAD. Deloukas P, Kanoni S, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nature genetics. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coronary Artery Disease Genetics C. A genome-wide association study in europeans and south asians identifies five new loci for coronary artery disease. Nature genetics. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 6.Dubland JA, Francis GA. Lysosomal acid lipase: At the crossroads of normal and atherogenic cholesterol metabolism. Frontiers in cell and developmental biology. 2015;3:3. doi: 10.3389/fcell.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernstein DL, Hulkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. Journal of hepatology. 2013;58:1230–1243. doi: 10.1016/j.jhep.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Du H, Schiavi S, Levine M, Mishra J, Heur M, Grabowski GA. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Human molecular genetics. 2001;10:1639–1648. doi: 10.1093/hmg/10.16.1639. [DOI] [PubMed] [Google Scholar]
- 9.Global Lipids Genetics C. Willer CJ, Schmidt EM, et al. Discovery and refinement of loci associated with lipid levels. Nature genetics. 2013;45:1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Human genomics. The genotype-tissue expression (gtex) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS genetics. 2011;7:e1002260. doi: 10.1371/journal.pgen.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nedelec Y, Sanz J, Baharian G, et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell. 2016;167:657–669.e621. doi: 10.1016/j.cell.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 13.Burton BK, Balwani M, Feillet F, et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. The New England journal of medicine. 2015;373:1010–1020. doi: 10.1056/NEJMoa1501365. [DOI] [PubMed] [Google Scholar]
- 14.Jones SA, Rojas-Caro S, Quinn AG, et al. Survival in infants treated with sebelipase alfa for lysosomal acid lipase deficiency: An open-label, multicenter, dose-escalation study. Orphanet journal of rare diseases. 2017;12:25. doi: 10.1186/s13023-017-0587-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris GE, Braund PS, Moore JS, Samani NJ, Codd V, Webb TR. Coronary artery disease-associated lipa coding variant rs1051338 reduces lysosomal acid lipase levels and activity in lysosomes. Arteriosclerosis, thrombosis, and vascular biology. 2017 doi: 10.1161/ATVBAHA.116.308734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. The Journal of biological chemistry. 1975;250:8487–8495. [PubMed] [Google Scholar]
- 17.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell metabolism. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang SC, Everts B, Ivanova Y, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nature immunology. 2014;15:846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox BE, Griffin EE, Ullery JC, Jerome WG. Effects of cellular cholesterol loading on macrophage foam cell lysosome acidification. Journal of lipid research. 2007;48:1012–1021. doi: 10.1194/jlr.M600390-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Jerome WG, Cox BE, Griffin EE, Ullery JC. Lysosomal cholesterol accumulation inhibits subsequent hydrolysis of lipoprotein cholesteryl ester. Microscopy and microanalysis : the official journal of Microscopy Society of America, Microbeam Analysis Society, Microscopical Society of Canada. 2008;14:138–149. doi: 10.1017/S1431927608080069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh RK, Barbosa-Lorenzi VC, Lund FW, Grosheva I, Maxfield FR, Haka AS. Degradation of aggregated ldl occurs in complex extracellular sub-compartments of the lysosomal synapse. Journal of cell science. 2016;129:1072–1082. doi: 10.1242/jcs.181743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haka AS, Singh RK, Grosheva I, Hoffner H, Capetillo-Zarate E, Chin HF, Anandasabapathy N, Maxfield FR. Monocyte-derived dendritic cells upregulate extracellular catabolism of aggregated low-density lipoprotein on maturation, leading to foam cell formation. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:2092–2103. doi: 10.1161/ATVBAHA.115.305843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haka AS, Grosheva I, Chiang E, Buxbaum AR, Baird BA, Pierini LM, Maxfield FR. Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Molecular biology of the cell. 2009;20:4932–4940. doi: 10.1091/mbc.E09-07-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Xue C, Shah R, et al. Functional analysis and transcriptomic profiling of ipsc-derived macrophages and their application in modeling mendelian disease. Circulation research. 2015;117:17–28. doi: 10.1161/CIRCRESAHA.117.305860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musunuru K. Genome editing of human pluripotent stem cells to generate human cellular disease models. Disease models & mechanisms. 2013;6:896–904. doi: 10.1242/dmm.012054. [DOI] [PMC free article] [PubMed] [Google Scholar]