

Abstract
Colorectal cancer has a relatively low sensitivity to paclitaxel. The purpose of this study was to investigate the role of connexin 43 (Cx43), which is a structural component of gap junctional communication (GJC), in paclitaxel cytotoxicity in colorectal cancer cells. Three colorectal cancer cell lines (HCT106, HCT116 and LoVo) were transfected with Cx43 and used to examine paclitaxel cytotoxicity. A western blot assay was used to confirm Cx43 expression in transfected cell lines as well as the expression of several proteins that are associated with paclitaxel cytotoxicity. A parachute dye-coupling assay was used to measure GJC function. An MTT assay was used to analyze the viability of paclitaxel-treated cells. Cx43 expression level and GJC function were significantly upregulated by the transfection (P<0.05). The viability of transfected cells was significantly inhibited compared with that of untransfected cells when treated with paclitaxel (20 or 80 nM) at high culture density but not at low culture density (P<0.05). Cx43 transfection significantly increased the mitotic arrest, tubulin polymerization and apoptosis effects of paclitaxel (P<0.05). It was also found that paclitaxel had an inhibitory effect on GJC function after 12 h of treatment in LoVo cells (P<0.05). These results indicate that Cx43 may serve as a target of paclitaxel chemotherapy for colorectal cancer.
Keywords: connexin 43, gap junctional communication, paclitaxel, colorectal cancer, cytotoxicity
Introduction
Colorectal cancer (CRC) is one of the most common types of malignancy worldwide (1). Although surgery is the most effective treatment option for CRC, chemotherapy is also used to control tumor growth and reduce recurrence (2). The application of novel antitumor drugs has improved the prognosis of CRC; however, intrinsic or acquired chemoresistance is frequently encountered during chemotherapy (3). Therefore, there it is necessary to understand the molecular mechanisms underlying drug resistance.
Paclitaxel, a member of the taxane class of agents, has been widely used as an antineoplastic agent for >20 years and is effective in the treatment of a number of types of cancer and other malignancies (4–6). Paclitaxel binds to tubulin and stabilizes microtubule filaments to induce mitotic arrest during the G2/M phase and promote apoptosis (7). However, as a single agent, paclitaxel has low cytotoxicity in adenocarcinoma of the colon or rectum (8). There are multiple known mechanisms of paclitaxel resistance, such as microtubule mutations (9), upregulation of survivin expression (10) and protein kinase B/extracellular signal-regulated kinase activation (11), and additional factors continue to be evaluated.
Previous studies have demonstrated that connexins, a group of tumor suppressor genes, are potential anti-oncogenic targets for chemotherapy, and the upregulation of connexin expression can increase sensitivity to antitumor drugs in many cancer types (12,13). Connexins form gap junctions, which are important intercellular channels. The downregulation of connexin expression and loss of gap junctional communication (GJC) are important events in carcinogenesis (14). Connexin 43 (Cx43) is a member of the gap junction protein family of connexins and is widely expressed in different tissues to provide GJC (15). Expression of Cx43 is reduced in many types of cancer; however, some cancer cell lines exhibit a substantial level of Cx43 expression, such as human mammary carcinoma (16) and CRC (17). Thus, it was speculated that Cx43 expression level may be associated with paclitaxel cytotoxicity in CRC.
In the present study, the influence of Cx43 expression on paclitaxel cytotoxicity was evaluated in three CRC cell lines. The results of this study may be relevant in the development of a new paclitaxel-based chemotherapy strategy for patients with CRC and a high level of Cx43 expression.
Materials and methods
Reagents and antibodies
All cell culture media, trypsin and antibiotics were purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA), and fetal bovine serum (FBS) was purchased from HyClone (GE Healthcare Life Sciences, Logan, UT, USA). Rabbit anti-Cx43 polyclonal antibody (C6219), rabbit anti-caspase-3 polyclonal antibody (C9598), rabbit anti-survivin polyclonal antibody (SAB3500269), rabbit anti-α-tubulin polyclonal antibody (SAB4500087), rabbit anti-cyclin B1 polyclonal antibody (C8831), rabbit anti-β-actin polyclonal antibody (SAB2100037), anti-rabbit IgG-peroxidase antibody (SAB3700870), MTT, neomycin (G418) and paclitaxel were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Rabbit anti-ATPase β3 (Na+/K+) polyclonal antibody (GTX114272) was purchased from GeneTex, Inc. (Irvine, CA, USA). ProteoExtract Native Membrane Protein Extraction kit and Immobilon membranes were purchased from EMD Millipore (Billerica, MA, USA). pTARGET Mammalian Expression Vector system was purchased from Promega Corporation (Madison, WI, USA). Lipofectamine reagent was purchased from Invitrogen (Thermo Fisher Scientific, Inc.). Electrochemical Luminescence (ECL) Plus substrate, bicinchoninic acid (BCA) reagents and radioimmunoprecipitation (RIPA) lysis buffer were purchased from CWBio (Beijing, China).
Cell lines
Human colorectal carcinoma cell lines HCT106, HCT116, LoVo were donated by Dr Shiwu Zhang, Department of Pathology, Tianjin Union Medical Center (Tianjin, China). All cell lines were cultured at 37°C in an atmosphere containing 5% CO2 in RPMI-1640 medium supplemented with 10% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin.
Cx43 transfection
Total RNA was extracted from LoVo cells at confluence by TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.). A total of 5 µg of total RNA was used for cDNA synthesis using BeyoRT cDNA synthesis kit (D7166; Beyotime Institute of Biotechnology, Haimen, China). The reverse transcription reaction mixture contained 5 µg total RNA, 1 µl random hexamer primer and diethyl pyrocarbonate -treated water to produce a final volume of 12 µl. After incubation in 70°C for 5 min, the mixture was cool down on ice, the following agents were added: 4 µl reaction buffer, 1 µl RNase inhibitor, 2 µl dNTP mix and 1 µl reverse transcriptase. The mixture was gently mixed and centrifuged transiently at 6,000 × g at 4°C. The supernatant was incubated at 25°C for 10 min and 42°C for 60 min. The reaction was terminated by incubation at 70°C for 10 min and the product was used for the following polymerase chain reaction (PCR). cDNA of the human Cx43 coding region was amplified by PCR using PCR kit with Taq (D7232; Beyotime Institute of Biotechnology) according to the manufacturer's instructions. The Cx43 primers used were as follows: forward, 5′-cacaattgagtggaatcttgatg-3′; reverse, 5′-caa cat ggg tga ctg gag c-3′. β-actin was used as the control. The β-actin primers used were as follows: forward, 5′-gtg ggg cgc ccc agg cac ca-3′; reverse, 5′-cttccttaatgtcacgcacgatttc-3′. The PCR reaction mixture contained 5 µl 10xPCR buffer, 4 µl 2.5 mM dNTP, 0.1 µg template, 2 µl primers, 0.25 µl Taq DNA polymerase, and was added to 50 µl of total volume by double distilled water. The PCR profile was 94°C for 30 sec, 55°C for 30 sec, and 72°C for 60 sec for 30 cycles, followed by 72°C for 10 min. The results were analyzed and quantified by Quantity One software (v4.62; Bio-Rad Laboratories, Inc., Hercules, CA, USA). A total of 45 ng purified PCR product was inserted into 140 ng pTARGET vector (A1410; Promega Corporation, Madison, WI, USA). Constructed expression vector (0.8 µg/106 cells) was transfected into the aforementioned three cell lines by lipofection using Lipofectamine reagent, according to the manufacturer's protocol. After transfection, cultures were selected with 400 µg/ml G418. Cells of subclones were diluted and seeded to obtain further clones.
Parachute dye-coupling assay
Functional GJC of transfected and paclitaxel-treated cells was evaluated as described by Wang et al (18). Briefly, cells for analysis were grown to confluence in 6-well plates. Two fluorescent dyes, CM-Dil and Calcein-AM, were used to analyze GJC function. CM-Dil is a membrane dye that is not able to spread to coupled cells. Calcein-AM can be converted intracellularly into the GJC-permeable dye calcein (18). Donor cells in one well were stained with fresh culture medium containing 10 µg/ml calcein-AM and 5 µg/ml CM-Dil for 30 min at 37°C. After this incubation, donor cells were washed with culture medium three times to remove unincorporated dye. Donor cells were then trypsinized and seeded onto a monolayer of receiver cells grown in another well. Receiver cells were cultured in a 6-wells plate at 37°C in an atmosphere containing 5% CO2 to confluence (106 cells/well) when donor cells were seeded. The ratio of donor to receiver was 1:150. Cells were cultured for 4 h at 37°C in order to allow GJC between donor and receiver cells. GJC function was then measured using a fluorescence microscope (Olympus CKX41; Olympus Corporation, Tokyo, Japan). Red fluorescence of CM-Dil was used to locate donor cells, and green fluorescence of calcein-AM was used to calculate the average number of fluorescent receiver cells around each donor cell. This number was used to represent the degree of GJC function. Five fields of each group were used to calculate.
Paclitaxel treatment and cell survival assay
Stock solutions of 1 µM paclitaxel in dimethyl sulfoxide were freshly prepared and added to wild type (WT) or transfected cell lines at a series of concentrations (0, 1, 5, 20 and 80 nM). Cells with two culture densities (3×104 or 1×102 cells/cm2) were treated with paclitaxel in a 37°C incubator for 48 h and then their viability was tested. Briefly, cells were seeded into 6-well dishes. For high density cultures, cells were seeded at 3×104 cells/cm2 density and exposed to paclitaxel when the cells achieved 80–100% confluency, where GJC formation was possible. For low density cultures, cells were seeded at 1×102 cells/cm2 density in 6-well dishes. Following substrate attachment (at 10 h), the cultures were treated with paclitaxel.
The inhibitory effects of paclitaxel on cell viability were evaluated using a cell survival assay. Briefly, 20 µl 5 mg/ml MTT solution was added to each well after exposure to paclitaxel for 24 h, and the cells were incubated for 4 h at 37°C. The cell medium was removed, and 100 µl DMSO was added to dissolve the purple formazan crystals. After 10 min of slow vibration, fluorescence was monitored at a wavelength of 490 nm. Cell viability was calculated as a percentage, where the absorption of cells not treated with paclitaxel (control group) was considered to be 100%. The experiment was repeated three times for each cell line.
Western blot analysis
Cx43 expression in the membrane of WT and transfected cells was analyzed by western blotting. The WT and transfected clones of the three cell lines were identified. Briefly, the membrane proteins of cells were extracted using a ProteoExtract Native Membrane Protein Extraction kit, according to the manufacturer's instructions, and subjected to western blot analysis of Cx43. Protein content was quantified using BCA reagent. Protein samples were suspended in SDS loading buffer (Beyotime Institute of Biotechnology). After boiling, 50 µg proteins were run on 12% SDS-PAGE gels, then transferred to Immobilon membranes by the semi-dry blot method. ATPase β3 was used as a loading control. The membranes were blocked by blocking reagent (P0023B; Beyotime Institute of Biotechnology) at room temperature for 1 h. The membranes were probed with anti-Cx43 antibody (1:4,000) and anti-ATPase β3 (1:4,000) antibody at room temperature for 1 h, then with anti-rabbit IgG-peroxidase (1:10,000) at room temperature for 1 h using standard techniques. The signals were visualized using ECL Plus and exposed film. Expression levels were quantified using Quantity One software (v4.62; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and normalized against ATPase β3.
For further analysis of other proteins, high density cultured cells were treated with 80 nM paclitaxel at 37°C in an atmosphere containing 5% CO2, and harvested after 12 h of treatment. For the survivin and cyclin B1 expression assays and detection of caspase-3 cleavage, harvested cells were lysed with RIPA lysis buffer supplemented with protease inhibitor mixture (Roche Applied Science, Penzberg, Germany). Cell lysate was used for the western blot assay method described below.
For the detection of α-tubulin polymerization levels, soluble and polymerized tubulins in cell lysate were separated according to a previously published method (19). In brief, the harvested cells (1×106 cells) were lysed at 37°C with 100 µl hypotonic buffer [1 mM MgCl2, 20 mM Tris-HCl (pH 6.8), 2 mM EGTA, 0.5% NP-40, protease inhibitor mixture] for 5 min in the dark. The cell lysate was treated with an additional 100 µl hypotonic buffer and vortexed briefly, then sonicated on ice. The cell lysate was centrifuged at 10,000 × g for 10 min at room temperature. The supernatant contained the soluble tubulin and the pellet fraction included polymerized tubulin. The pellets were resuspended in 200 ml of hypotonic buffer. Expression levels of soluble and polymerized α-tubulin were determined by analysis of the supernatant and resuspended pellets, respectively. The percentage of polymerized tubulin was determined by dividing the densitometric value for polymerized tubulin by the total tubulin (polymerized + soluble) densitometric value.
After quantification of protein concentration using BCA reagents, equal amounts of proteins (20 µg/lane) were loaded onto gel and separated by 12% SDS-PAGE. The gel was transferred to Immobilon membranes by the semi-dry blot method. The membranes were blocked using blocking reagent (P0023B; Beyotime Institute of Biotechnology) at room temperature for 1 h. The membranes were probed with anti-survivin (1:4,000), anti-cyclin B1 (1:4,000), anti-α-tubulin (1:4,000), anti-caspase-3 (1:4,000), anti-β-actin (1:3,000) primary antibodies at room temperature for 1 h and anti-rabbit IgG-peroxidase secondary antibody (1:10,000) at room temperature for 1 h. The signals were visualized by ECL. β-actin was used as a loading control. Expression levels were quantified using Quantity One software and normalized against β-actin. The percentage of cleaved caspase-3 was determined by dividing the densitometric value of cleaved caspase-3 by the total caspase-3 (cleaved + procaspase-3) densitometric value.
Statistical analysis
All data in figures represent the mean ± standard deviation. Statistical analysis was performed by Student's t-test using SPSS software (ver 17.0; SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant result.
Results
Confirmation of transfection and GJC function
Membrane expression of Cx43 in WT and Cx43-transfected cells of the three cell lines was analyzed using western blot assays (Fig. 1A and B). HCT106-WT and HCT116-WT exhibited a lower level of Cx43 expression compared with LoVo-WT. All of the transfected cell lines exhibited a significantly higher level of Cx43 expression compared with their WT counterparts (P<0.05; Fig. 1B). The results of parachute dye-coupling assays also suggested an association between GJC function and Cx43 expression. All transfected cell lines exhibited a significantly higher level of GJC function compared with their WT counterparts (P<0.05; Fig. 1C-F). This evidence suggested that GJC in transfected cells was associated with Cx43 expression.
Figure 1.

Effect of Cx43 expression on GJC function in colorectal carcinoma cell lines. (A) Western blot analysis of Cx43 expression in LoVo, HCT106 and HCT116 cell lines transfected with Cx43. ATPase β3 subunit served as a loading control. (B) Cx43 expression level was analyzed using Quantity One software (v4.62) and normalized against ATPase β3 expression level. Parachute dye-coupling assay results of the (C) LoVo, (D) HCT106 and (E) HCT116 cell lines. Scale bars represent 10 µm. (F) Quantified results of parachute dye-coupling assays in the three cell lines. Columns show the mean ± standard deviation from five independent experiments. *P<0.05 vs. the respective WT group. WT, wild type; Cx43, connexin 43; GJC, gap junctional communication.
Cx43 enhances paclitaxel-induced apoptosis
MTT assays indicated that the cytotoxicity of paclitaxel differed between WT and transfected cells (Fig. 2). High culture density ensures GJC function (20), and all transfected cell lines with high culture density were significantly more sensitive to paclitaxel compared with their WT counterparts when they were treated with 20 or 80 nM paclitaxel (P<0.05). At lower concentrations of paclitaxel treatment (1 or 5 nM), the cell viability rates of HCT106 and HCT116 were also significantly decreased by transfection with Cx43 (P<0.05 vs. WT). No significant differences in viability rates existed between transfected and WT cells when cells were seeded at low density. Caspase-3 is a frequently activated death protease. The activation of caspase-3 occurs at an early stage of apoptosis (21). The analysis of caspase-3 revealed that the activated (cleaved) form of caspase-3 was significantly increased in paclitaxel-treated Cx43-transfected cells compared with paclitaxel-treated WT cells from all three cell lines (P<0.05; Figs. 3 and 4A-C). No significant differences in caspase-3 activation were observed between untreated WT and Cx43-transfected cells (Figs. 3 and 4A-C).
Figure 2.

MTT assay of the three cell lines under paclitaxel treatment. WT and Cx43-transfected cells were treated with paclitaxel (0, 1, 5, 20 or 80 nM) for 48 h, then an MTT assay was performed. Results are expressed as the percentage of cell growth relative to untreated control cells. Data represent the mean ± standard deviation of three experiments. *P<0.05 vs. the respective WT group. H, high culture density (3×104 cells/cm2); L, low culture density (1×102 cells/cm2); WT, wild type; Cx43, connexin 43.
Figure 3.

Western blot analysis of caspase 3 cleavage, α-tubulin polymerization, and cyclin B1 and survivin expression levels in WT and Cx43-transfected cells treated with paclitaxel. Procaspase-3 and simultaneous generation of immunoreactive low molecular mass fragments p20/p17 (cleaved caspase-3) were detected. S, soluble tubulin; P, polymerized tubulin; -, untreated cells; +, treated cells; WT, wild type; Cx43, connexin 43.
Figure 4.

Densitometry analysis of western blots in WT and Cx43-transfected cells treated with paclitaxel. (A-C) Percentage of cleaved caspase-3 was determined by dividing the densitometric value of cleaved caspase-3 by the total caspase-3 (cleaved + procaspase-3) densitometric value. (D-F) Percentage of polymerized tubulin was determined by dividing the densitometric value for polymerized tubulin by the total tubulin (polymerized + soluble) densitometric value. (G) Cyclin-B1 and (H) survivin expression levels. The values shown are the expression levels of the ‘+’ group normalized against the ‘−’ group. *P<0.05 vs. the respective WT group. WT, wild type; Cx43, connexin 43; -, untreated cells; +, treated cells.
Cx43 enhances the effect of tubulin polymerization caused by paclitaxel
Paclitaxel can bind to assembled tubulin and inhibit microtubule disassembly to lock microtubules in a polymerized state (22). As shown in Figs. 3 and 4D-F, western blotting results indicated that paclitaxel increased microtubule assembly. This increase was significantly higher in Cx43-transfected cells compared with their WT counterparts (P<0.05). In the cells that were not treated with paclitaxel, no significant differences in tubulin polymerization were observed between WT and Cx43-transfected cells (Figs. 3 and 4D-F).
Cx43 increases cyclin B1 and survivin expression
Arrest of the cell cycle at mitotic phase is a molecular mechanism of paclitaxel-induced cytotoxicity. Cyclin B1 is a regulatory protein involved in mitosis. It has a critical role in regulating cyclin-dependent kinase 1, which initiates progression from the G2 phase to mitosis (23). The quantity and activity of cyclin B1 increase through the cell cycle (24). As shown in Figs. 3 and 4G, the cyclin B1 expression levels in all cells increased markedly after paclitaxel treatment. The increase was significantly higher in transfected cells compared with their WT counterparts (P<0.04). These results suggest that Cx43 promotes the mitotic arrest caused by paclitaxel.
Survivin is an anti-apoptotic protein that is highly expressed in cancer. Paclitaxel-mediated mitotic arrest of cancer cells is associated with survivin induction, and survivin expression increases in the G2/M phase of cell cycle (25). As shown in Figs. 3 and 4H, survivin expression levels increased after paclitaxel treatment. This increase was significantly higher in transfected cells compared with their WT counterparts (P<0.05).
Effect of paclitaxel treatment on GJC function
The parachute dye-coupling assay was used to evaluate whether GJC function was inhibited by paclitaxel treatment. GJC function was represented by the mean number of coupled cells, and the results are shown in Fig. 5. No inhibitory effect on GJC function in HCT106 or HCT116 cells was observed within 12 h of paclitaxel treatment. LoVo exhibited a significant inhibition at 12 h but not earlier. Cells with longer treatment times were not suitable for the analysis of GJC function because they were reduced in size and lost intercellular contact.
Figure 5.
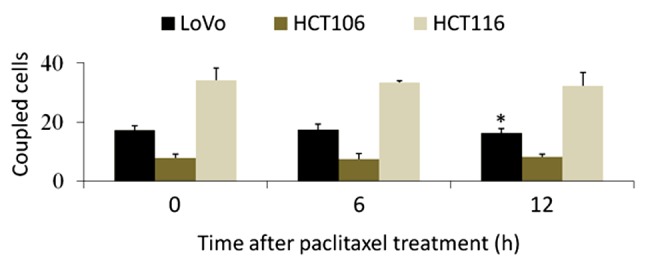
Parachute dye-coupling analysis of gap junctional communication after different durations of paclitaxel treatment. The group with no treatment (0 h) was used as the control. Columns show the mean ± standard deviation from five independent experiments. *P<0.05 vs. 0 h.
Discussion
Cx43 is a structural component of gap junctions, which permits the transfer of small water-soluble molecules (molecular weight <1 kDa) directly between cells without passing through the cell membrane (14). Paclitaxel and other small molecules involved in apoptosis can be transferred through GJC and the cytotoxic effect is enhanced in Cx43-transfected cells (26). The effect of Cx43 on cytotoxicity varies in different cell types and with different antitumor drugs. For example, Cx43 expression is upregulated in glioblastoma multiforme cells, which are resistant to a frontline antitumor drug, temozolomide (27). However, Cx43 enhances paclitaxel cytotoxicity in HeLa cells (28) and cisplatin cytotoxicity in mesothelioma cells (29). The mechanism of the effect of Cx43 on antitumor drugs is complex; a previous study reported that Cx43 sensitized tumor testicular cells in response to cisplatin but caused normal testicular cells to become resistant to cisplatin (20). Therefore, three CRC cell lines, LoVo, HCT106 and HCT116, were used in the present study in order to perform a comprehensive analysis of the role of Cx43 in CRC, and it was demonstrated that Cx43 enhanced paclitaxel cytotoxicity in all three of these cell lines.
The expression levels of connexins differ in various types of CRC. Immunohistochemical studies have reported that Cx26, Cx32 and Cx43 are downregulated or relocalized in CRC (30,31). These findings suggest that dysregulation of connexins and loss of GJC may be early events in CRC development. However, other studies have reported increased connexin expression in CRC. For example, upregulated Cx43 expression was exhibited in advanced stages of CRC (32), and high levels of Cx26 were also identified in some colorectal patients with lung metastases (33). Therefore, connexins may be prognostic markers in CRC and are considered to be potential targets for cancer chemoprevention and chemotherapy.
Previous studies have indicated that Cx43 is able to sensitize cells to apoptosis in response to chemotherapeutic drugs through GJC (18,28); however, another study reported that the role of Cx43 in chemotherapy-induced apoptosis is independent of GJC function (12). The results of the MTT assay in the current study indicated a role for Cx43 in mediating the apoptosis of CRC cell lines. Cx43-transfected cells exhibited a lower viability rate when treated with paclitaxel (20 or 80 nM) than did their WT counterparts when they were cultured at high density. When cells were sparsely seeded, they were not in contact and not able to form GJC, and there was no significant difference in apoptotic rate between Cx43-transfected cells and their WT counterparts. This evidence indicates that upregulating Cx43 expression level alone does not sensitize CRC cells to paclitaxel, but GJC function increased by Cx43 can increase sensitivity to paclitaxel when the cells are in contact with each other.
Paclitaxel has long been recognized to induce mitotic arrest and tubulin polymerization, both of which lead to cell death (7). The current study indicates that Cx43 promotes these effects in CRC cells. Despite WT LoVo cells exhibiting considerable Cx43 expression levels, the GJC function of these cells was enhanced by Cx43 transfection, and paclitaxel displayed higher cytotoxicity on the transfected LoVo cell line compared with its WT counterpart.
The current results suggest that paclitaxel cytotoxicity is enhanced by Cx43 expression; however, it has previously been reported that paclitaxel inhibits GJC in certain cells, such as epithelial and cervical cancer cells (28,34). In a previous study, researchers analyzed the effect of paclitaxel and another agent in the taxane family, docetaxel, on GJC function in HeLa cells. The results revealed that paclitaxel had a stronger inhibitory effect on GJC compared with docetaxel, which resulted in paclitaxel having reduced cytotoxicity compared with docetaxel (28). The ability of paclitaxel to inhibit GJC in CRC cells was investigated in the present study, and no marked inhibitory effect occurred within 12 h of paclitaxel treatment. Western blotting results also suggested that cells transfected with Cx43 were more sensitive to paclitaxel than were their WT counterparts at 12 h after treatment. These results indicate that the cytotoxicity of paclitaxel was not notably decreased by paclitaxel-induced GJC inhibition in CRC cells. However, measurements of GJC function for longer times after paclitaxel treatment may be inaccurate due to cell shrinkage, which decreases intercellular contact.
In summary, the present study demonstrated the role of Cx43 in paclitaxel-induced cytotoxicity in CRC cells. GJC function in Cx43-transfected cells was upregulated compared with their WT counterparts. The enhancement effect of Cx43 on paclitaxel-induced cytotoxicity is dependent on GJC function. In addition, Cx43 promotes other cellular responses to paclitaxel, such as caspase-3 maturation, mitotic arrest and tubulin polymerization. The current study offers a possible approach for further in vitro and in vivo investigations concerning the effects of Cx43 expression on paclitaxel-treated CRC cells.
Acknowledgements
This study was supported by the Foundation of Tianjin Health Bureau (grant nos. 2014KY27, 2014KY28 and 2014KR14), the Foundation of the Committee on Science and Technology of Tianjin (grant nos. 15JCQNJC42200 and 13JCYBJC42700), the National Basic Research Program of China (grant no. 2016YFC0102404) and the National Natural Science Foundation of China (grant nos. 81503127 and 81472729).
References
- 1.Zhang QQ, Wu XJ, Tang T, Zhu SW, Yao Q, Gao BZ, Yuan XC. Quantitative analysis of rectal cancer by spectral domain optical coherence tomography. Phys Med Biol. 2012;57:5235–5244. doi: 10.1088/0031-9155/57/16/5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ades S. Adjuvant chemotherapy for colon cancer in the elderly: Moving from evidence to practice. Oncology (Williston Park) 2009;23:162–167. [PubMed] [Google Scholar]
- 3.Jensen NF, Stenvang J, Beck MK, Hanáková B, Belling KC, Do KN, Viuff B, Nygård SB, Gupta R, Rasmussen MH, et al. Establishment and characterization of models of chemotherapy resistance in colorectal cancer: Towards a predictive signature of chemoresistance. Mol Oncol. 2015;9:1169–1185. doi: 10.1016/j.molonc.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, Clarke-Pearson DL, Davidson M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with Stage III and Stage IV Ovarian Cancer. N Engl J Med. 1996;334:1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- 5.Holmes FA, Walters RS, Theriault RL, Forman AD, Newton LK, Raber MN, Buzdar AU, Frye DK, Hortobagyi GN. Phase II Trial of taxol, an active drug in the treatment of metastatic breast cancer. J Natl Cancer Inst. 1991;83:1797–1805. doi: 10.1093/jnci/83.24.1797-a. [DOI] [PubMed] [Google Scholar]
- 6.Murphy WK, Fossella FV, Winn RJ, Shin DM, Hynes HE, Gross HM, Davilla E, Leimert J, Dhingra H, Raber MN, et al. Phase II study of taxol in patients with untreated advanced non-small-cell lung cancer. J Natl Cancer Inst. 1993;85:384–388. doi: 10.1093/jnci/85.5.384. [DOI] [PubMed] [Google Scholar]
- 7.Rowinsky EK, Donehower RC. Paclitaxel (taxol) N Engl J Med. 1995;332:1004–1014. doi: 10.1056/NEJM199504133321507. [DOI] [PubMed] [Google Scholar]
- 8.Einzig AI, Neuberg D, Wiernik PH, Grochow LB, Ramirez G, O'Dwyer PJ, Petrelli NJ. Phase II trial of paclitaxel in patients with advanced colon cancer previously untreated with cytotoxic chemotherapy: An eastern cooperative oncology group trial (PA286) Am J Ther. 1996;3:750–754. doi: 10.1097/00045391-199611000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Orr GA, Verdier-Pinard P, Mcdaid H, Horwitz SB. Mechanisms of taxol resistance related to microtubules. Oncogene. 2003;22:7280–7295. doi: 10.1038/sj.onc.1206934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai HC, Huang CY, Su HL, Tang CH. CTGF increases drug resistance to paclitaxel by upregulating survivin expression in human osteosarcoma cells. Biochim Biophys Acta. 2014;1843:846–854. doi: 10.1016/j.bbamcr.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Wu G, Qin XQ, Guo JJ, Li TY, Chen JH. AKT/ERK activation is associated with gastric cancer cell resistance to paclitaxel. Int J Clin Exp Pathol. 2014;7:1449–1458. [PMC free article] [PubMed] [Google Scholar]
- 12.Du G, Yang Y, Zhang Y, Sun T, Liu W, Wang Y, Li J, Zhang H. Thrombocytosis and immunohistochemical expression of connexin 43 at diagnosis predict survival in advanced non-small-cell lung cancer treated with cisplatin-based chemotherapy. Cancer Chemother Pharmacol. 2013;71:893–904. doi: 10.1007/s00280-013-2080-6. [DOI] [PubMed] [Google Scholar]
- 13.Yu BB, Dong SY, Yu ML, Jiang GJ, Ji J, Tong XH. Total flavonoids of litsea coreana enhance the cytotoxicity of oxaliplatin by increasing gap junction intercellular communication. Biol Pharm Bull. 2014;37:1315–1322. doi: 10.1248/bpb.b14-00193. [DOI] [PubMed] [Google Scholar]
- 14.King TJ, Bertram JS. Connexins as targets for cancer chemoprevention and chemotherapy. Biochim Biophys Acta. 2005;1719:146–160. doi: 10.1016/j.bbamem.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Yamasaki H, Naus CC. Role of connexin genes in growth control. Carcinogenesis. 1996;17:1199–1213. doi: 10.1093/carcin/17.6.1199. [DOI] [PubMed] [Google Scholar]
- 16.Park JM, Munoz JL, Wona BW, Bliss SA, Greco SJ, Patel SA, Kandouz M, Rameshwar P. Exogenous CXCL12 activates protein kinase C to phosphorylate connexin 43 for gap junctional intercellular communication among confluent breast cancer cells. Cancer Lett. 2013;331:84–91. doi: 10.1016/j.canlet.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Sirnes S, Bruun J, Kolberg M, Kjenseth A, Lind GE, Svindland A, Brech A, Nesbakken A, Lothe RA, Leithe E, Rivedal E. Connexin43 acts as a colorectal cancer tumor suppressor and predicts disease outcome. Int J Cancer. 2012;131:570–581. doi: 10.1002/ijc.26392. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Zhang S, Zhang C, Zhao Z, Zheng X, Xue L, Liu J, Yuan XC. Investigation of an SPR biosensor for determining the influence of connexin 43 expression on the cytotoxicity of cisplatin. Analyst. 2016;141:3411–3420. doi: 10.1039/C6AN00264A. [DOI] [PubMed] [Google Scholar]
- 19.Shimomura M, Yaoi T, Itoh K, Kato D, Terauchi K, Shimada J, Fushiki S. Drug resistance to paclitaxel is not only associated with ABCB1 mRNA expression but also with drug accumulation in intracellular compartments in human lung cancer cell lines. Int J Oncol. 2012;40:995–1004. doi: 10.3892/ijo.2011.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong X, Wang Q, Yang Y, Zheng S, Tong X, Zhang S, Tao L, Harris AL. Gap junctions propagate opposite effects in normal and tumor testicular cells in response to cisplatin. Cancer Lett. 2012;317:165–171. doi: 10.1016/j.canlet.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 21.Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 22.Jordan MA, Kamath K. How do microtubule-targeted drugs work? An overview. Curr Cancer Drug Targets. 2007;7:730–742. doi: 10.2174/156800907783220417. [DOI] [PubMed] [Google Scholar]
- 23.Wolf F, Wandke C, Isenberg N, Geley S. Dose-dependent effects of stable cyclin B1 on progression through mitosis in human cells. EMBO J. 2006;25:2802–2813. doi: 10.1038/sj.emboj.7601163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito M. Factors controlling cyclin B expression. Plant Mol Biol. 2000;43:677–690. doi: 10.1023/A:1006336005587. [DOI] [PubMed] [Google Scholar]
- 25.Li F, Altieri DC. The cancer antiapoptosis mouse survivin gene: Characterization of locus and transcriptional requirements of basal and cell cycle-dependent expression. Cancer Res. 1999;59:3143–3151. [PubMed] [Google Scholar]
- 26.Mesnil M, Piccoli C, Tiraby G, Willecke K, Yamasaki H. Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins; Proc Natl Acad Sci USA; 1996; pp. 1831–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munoz JL, Rodriguez-Cruz V, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P. Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis. 2014;5:e1145. doi: 10.1038/cddis.2014.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang N, Wang Q, Wu D, Zhang S, Zhang Y, Tao L. Differential effects of paclitaxel and docetaxel on gap junctions affects their cytotoxicities in transfected HeLa cells. Mol Med Rep. 2013;8:638–644. doi: 10.3892/mmr.2013.1546. [DOI] [PubMed] [Google Scholar]
- 29.Sato H, Iwata H, Takano Y, Yamada R, Okuzawa H, Nagashima Y, Yamaura K, Ueno K, Yano T. Enhanced effect of connexin 43 on cisplatin-induced cytotoxicity in mesothelioma cells. J Pharmacol Sci. 2009;110:466–475. doi: 10.1254/jphs.08327FP. [DOI] [PubMed] [Google Scholar]
- 30.Hong R, Lim SC. Pathological significance of connexin 26 expression in colorectal adenocarcinoma. Oncol Rep. 2008;19:913–919. [PubMed] [Google Scholar]
- 31.Kanczuga-Koda L, Koda M, Sulkowski S, Wincewicz A, Zalewski B, Sulkowska M. Gradual loss of functional gap junction within progression of colorectal cancer-a shift from membranous CX32 and CX43 expression to cytoplasmic pattern during colorectal carcinogenesis. In Vivo. 2010;24:101–107. [PubMed] [Google Scholar]
- 32.Han Y, Zhang PJ, Chen T, Yum SW, Pasha T, Furth EE. Connexin43 expression increases in the epithelium and stroma along the colonic neoplastic progression pathway: Implications for its oncogenic role. Gastroenterol Res Pract. 2011;2011:561719. doi: 10.1155/2011/561719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ezumi K, Yamamoto H, Murata K, Higashiyama M, Damdinsuren B, Nakamura Y, Kyo N, Okami J, Ngan CY, Takemasa I. Aberrant expression of connexin 26 is associated with lung metastasis of colorectal cancer. Clin Cancer Res. 2008;14:677–684. doi: 10.1158/1078-0432.CCR-07-1184. [DOI] [PubMed] [Google Scholar]
- 34.Giessmann D, Theiss C, Breipohl W, Meller K. Decreased gap junctional communication in neurobiotin microinjected lens epithelial cells after taxol treatment. Anat Embryol (Berl) 2005;209:391–400. doi: 10.1007/s00429-005-0456-z. [DOI] [PubMed] [Google Scholar]