

Abstract
Occludin is a tight junction protein that forms the permeability barrier, which is typically disturbed in ischemic associated diseases. The aim of the present study was to determine whether somatostatin receptor 2 (SSTR2) in RF/6A cells is involved in the modulation of the downregulation of occludin induced by high glucose, and to evaluate the implicated molecules. RF/6A cells were maintained in Dulbecco's modified Eagle medium and treated with 0 or 30 mM D-glucose. SSTR2 agonist octreotide (OCT), OCT with SSTR2 antagonist cycle-somatostatin (c-SOM) and neuropilin 1 (NRP1) inhibitor ATWLPPR, respectively, were administered to RF/6A cells under high glucose conditions. Cell apoptosis was evaluated by terminal deoxynucleotidyl transferase dUTP nick-end labeling. Western blot analysis was used to detect the protein expression level of SSTR2, occludin, vascular endothelial growth factor (VEGF), protein kinase B (Akt), phosphorylated Akt (p-Akt), extracellular signal-related kinases (ERK) and p-ERK proteins. The amount of VEGF released was determined by ELISA. Notably, the level of occludin reduced significantly under high glucose conditions. The results indicated that the administration of OCT prevented the reduction of occludin induced by high glucose, and co-administration with c-SOM reversed the effect of OCT. Increased VEGF secretion and expression of VEGF, p-Akt and p-ERK in RF/6A cells induced by high glucose were inhibited by OCT. ATWLPPR also prevented the downregulation of occludin, but did not inhibit p-Akt and p-ERK levels under high glucose conditions. The current study concluded that the activation of SSTR2 prevents high glucose-induced occludin downregulation in RF/6A cells, and VEGF, NRP1, p-Akt and p-ERK were implicated in this process. The pharmacological effects of SSTR2 targeting to endothelium may be used to assess the role of resistance of permeability and anti-inflammation.
Keywords: somatostatin receptor 2, occludin, vascular endothelial growth factor, neuropilin 1, high glucose
Introduction
Diabetic retinopathy (DR) is one of the primary causes of blindness globally. More than 300 million people are estimated to have diabetes worldwide, and 40% have retinopathy to some extent, which includes ~8.2% with vision-threatening retinopathy (1,2). Primary pathological hallmarks of DR are retinal ischemia and proliferation, induced by microcirculatory disturbance and microangiopathy. The process includes oxidative stress, vascular endothelial growth factor (VEGF)-induced damage and inflammatory changes (3).
Previous studies have indicated that somatostatin (an endogenous peptide) or its analogue exhibit anti-inflammatory effects and neuroprotection against DR or retinal ischemia-reperfusion injury (4–6). Somatostatin receptors (SSTR1-5) are widely distributed in the retina (7,8). Activation of retinal SSTR2 may reduce VEGF release from damaged neurons and limit the VEGF response (9). It has been demonstrated in an oxygen-induced retinopathy model that activation of SSTR2 may downregulate VEGF and exert an anti-angiogenesis effect via the SHP-1/STAT3 pathway (10). VEGF combined with its receptors, VEGF receptor (VEGFR2) and neuropilin 1 (NRP1), has an important role in the disruption of the blood-retinal barrier (BRB), which is a core pathological change in retinal dysfunction of DR (11–13). Tight junctions of vascular endothelial cells are key components of BRB. Occludin, a transmembrane protein in the tight junctions, exerts its role by forming the permeability barrier. The high glucose conditions of DR may lead to VEGF release and occludin degradation in cultured human retinal endothelial cells (5).
A recent study indicated that SSTR2 was scarcely expressed in VEGF-containing cells, and SSTR2 or VEGF barely localized in retinal vessels (10). However, under hypoxic conditions, SSTR2 and VEGF immunoreactivity was noted in retinal capillaries (10). This is consistent with another study that demonstrated VEGF was released from damaged neurons and entered vessels under hypoxic conditions (9). Therefore, it is not clear whether SSTR2 exists in normal vascular endothelial cells, and whether activation of SSTR2 is able to regulate the degradation of occludin induced by high levels of glucose in vascular endothelial cells. The present study aimed to elucidate this and determine whether SSTR2 modulates the occludin degradation induced by high glucose via the VEGF/NRP1/protein kinase B (Akt) signaling pathway.
Materials and methods
Materials
RPMI-1640 culture medium and fetal bovine serum (FBS) were purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA). D-glucose, penicillin and streptomycin were obtained from DingGuo Biotech Co., Ltd., (Shanghai, China). Octreotide (OCT) and cyclo-somatostatin (c-SOM; both Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) were dissolved and diluted in 0.9% NaCl to 1 mM.
Cell culture and experimental grouping
Rhesus monkey retinal fovea vascular endothelial cells (RF/6A) were obtained from Wuhan Boster Biological Technology, Ltd., (Wuhan, China). The RF/6A endothelial cells were maintained in Dulbecco's odified Eagle medium (DMEM; Wuhan Boster Biological Technology, Ltd.) culture medium supplemented with 10% FBS, streptomycin (100 µg/ml) and penicillin (100 IU/ml) in a 5% CO2 incubator at 37°C. Cells were seeded in 6-well plates at a density of 1×105 under normal (5.6 mM) and high glucose (30 mM) conditions, respectively. Passages 4–8 of the cells were used in the present study. The experimental groups in the current study were as follows: i) Control (normal conditions); ii) high glucose (HG; 30 mM D-glucose); iii) HG + Octreotide (OCT; 1 µM); iv) HG + OCT + c-SOM (OCT, 1 µM; c-SOM, 1 µM); and v) HG + VEGF co-receptor NRP1 antagonist (ATWLPPR: GL Biochem, Ltd., Shanghai China; 0.1 mM). Cells were observed and photographed with an inverted microscope (CX40RF200; Olympus Corp., Tokyo, Japan).
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)
TUNEL assay was performed using an in situ cell apoptosis detection kit (MK1022; Boster Biological Technology, Pleasanton, CA, USA) based on the manufacturer's protocol. RF/6A cells grown in normal and high glucose medium for 5 days were fixed with 4% paraformaldehyde at room temperature for 30 min. Following washing (3 times for 2 min) with phosphate-buffered saline (PBS) and distilled water, Proteinase K (1:200) was added to cells and incubated at 37°C for 10 min. Cells were washed with PBS and incubated with Labeling Buffer mixed with terminal deoxynucleotidyl and digoxidenin-11-deoxyuridine triphosphate (both provided in the kit) in a moist chamber at 37°C for 2 h. Cells were then washed with PBS (3 times for 2 min) and incubated at room temperature for 30 min with anti-digoxigenin peroxidase (provided in the kit) prior to treatment with with SABC-AP and chromogenic substrate (BCIP/NBT; provided in the kit).
Culture media VEGF measurement
A sandwich mouse VEGF ELISA kit (EK0541; Boster Biological Technology) was used to determine the level of VEGF according to the protocol provided by the manufacturer. Briefly, following collection of the cell culture fluid, with a 5-min centrifugation at 2,000 × g (4°C), the supernatant was diluted with sample diluent (provided in the kit) to a proportion of 1:2. From each sample, 100 ml supernatant was transferred to wells, which were coated with VEGF monoclonal antibody (1:100; provided in the kit). Subsequently, the avidin-biotin-peroxidase complex, ABC and chromogenic substrate were added in sequence according to the manufacturer's protocol. VEGF levels were determined by the absorbance at 450 nm using a microplate reader (Varioskan Flash; Thermo Fisher Scientific, Inc.) and normalized to the standard recombinant VEGF.
Western blot analysis
The culture solution was discarded and the RF/6A cells were washed with PBS three times and lysed by radioimmunoprecipitation lysis buffer containing EDTA-Na2, sodium fluoride and phenylmethylsulfonyl fluoride (all 1 mM). Cell extracts were collected and centrifuged for 5 min at 20,000 × g and 4°C. The supernatant was transferred into a 1.5-ml Eppendorf tube. Protein concentrations were measured using a Bradford Protein Assay kit (Beyotime Institute of Biotechnology, Co., Ltd., Haimen, China). Each protein sample (50 µg) was separated by 10% SDS-PAGE and transferred onto nitrocellulose membranes (EMD Millipore, Billerica, MA, USA). Following washing in TBST (2 times for 1 min), the membranes were blocked in 5% non-fat milk in Tris-buffered saline with Tween-20 and incubated overnight at 4°C with the primary antibodies, as follows: SSTR2 (Boster Biological Technology; BA1406-2;1:400), Occludin (Santa Cruz Biotechnology, Inc., Dallas, TX, USA; sc-8144; 1:400), Akt (Boster Biological Technology; BA0631; 1:400), phosphorylated-Akt (p-AKT; Santa Cruz Biotechnology, Inc.; sc-33,437; 1:500), extracellular signaling-regulated kinase (ERK), and p-ERK (both CST Biological Reagents Company Ltd., Shanghai, China; 9102s and 9101s; both 1:1,000). The secondary antibody was horseradish peroxidase-goat anti-rabbit IgG (Boster Biological Technology; BA1080; 1:2,000).
Statistical analysis
Results were presented as the mean ± standard deviation. Statistical analyses were performed by using SigmaStat (version 3.2; Systat software, Inc., San Jose, CA, USA). Different groups were compared using one-way analysis of variance (Turkey test or Kruskal Wallis were used as post-hoc tests) or Student t-test. P<0.05 was considered to determine statistically significant differences.
Results
Observation of RF/6A cell appearance in high glucose conditions
Morphological observation demonstrated that the RF/6A cells exhibited a shuttle-like and polygonal appearance when they were not crowded in the well. As the concentration increased, a number of cells exhibited a cobblestone appearance. No difference in appearance was observed between normal and high glucose conditions (Fig. 1A). TUNEL assay was performed on day 5 to determine whether RF/6A cells were influenced by the high glucose conditions. The number of apoptosis-positive cells in high glucose conditions was higher than those exposed to normal conditions (Fig. 1B). Expression of SSTR2 in RF/6A cells. To evaluate the expression of SSTR2 in RF/6A cells, western blot analysis was performed on normal control cells and high glucose-treated cells. SSTR2 was expressed in the RF/6A cells both in normal and high glucose conditions. No significant difference in expression was observed in RF/6A cells treated with 30 mM D-glucose for 5 days, as compared with the untreated cells (Fig. 2).
Figure 1.

Observation of RF/6A cells in differing glucose concentrations. (A) Observation of RF/6A cells under normal, HG (30 nM) and HG + OCT conditions. Scale bar, 200 µm. (B) There was a marked increase in the number of TUNEL-positive cells in the HG group on day 5, as compared with the normal group. Red arrows indicate TUNEL-positive cells. Scale bar, 200 µm. HG, high glucose; OCT, octreotide; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling.
Figure 2.
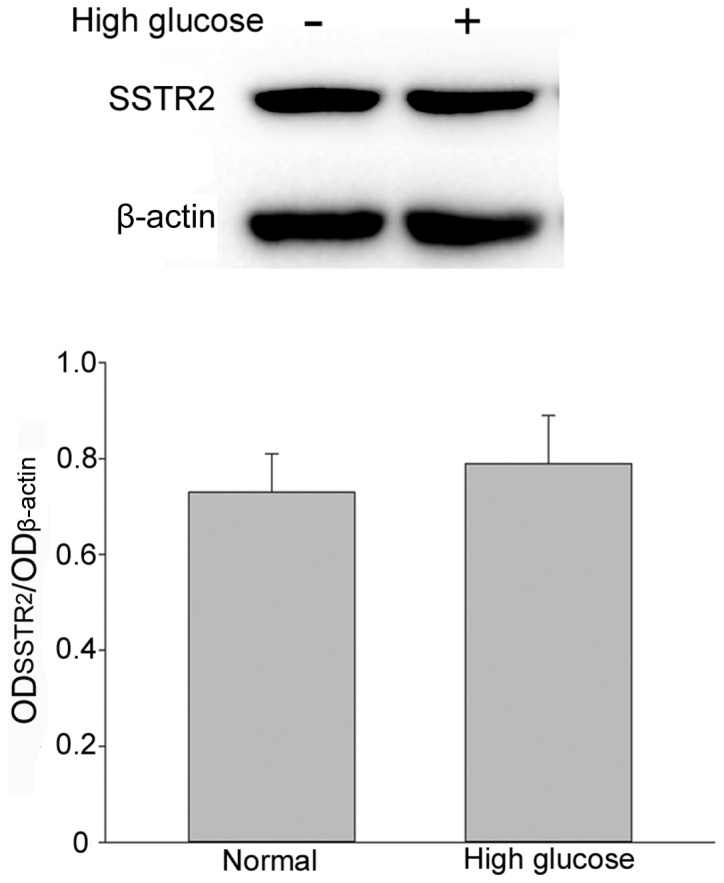
Evaluation of SSTR2 expression under normal and high glucose conditions using western blot analysis and β-actin was used as the loading control. Each bar represents the mean + standard deviation of OD data from three samples. SSTR2, somatostatin receptor 2; OD, optical density.
SSTR2 modulates the degradation of occludin induced by high glucose
Occludin is a transmembrane tight junction protein expressed in endothelial cells and is also a key factor in the formation of the permeability barrier. Under 30 mM D-glucose conditions, occludin expression was significantly reduced when compared with cells exposed to normal conditions (Fig. 3; P<0.05). For RF/6A cells under high glucose conditions, treatment with the SSTR2 agonist OCT (1 µM) restored the level of occluding, and this change was significant when compared with the high glucose group (P<0.05). Co-administration of SSTR2 antagonist c-SOM (1 µM) with octreotide significantly inhibited the effect of OCT (P<0.05). The results demonstrated that the specific SSTR2 was involved in modulation of occludin levels in the RF/6A cells under the high glucose conditions.
Figure 3.

Involvement of SSTR2 in modulation of the degradation of occludin induced by high glucose, as evaluated by western blot and densitometric analysis. A high concentration of glucose induced degradation of occluding, this was restored by administration of the SSTR2 agonist OCT, and the effect of octreotide was reversed by SSTR2 antagonist c-SOM. **P<0.01 vs. untreated normal condition; #P<0.05 vs. high glucose group; $P<0.05 vs. high glucose + OCT group. Each column represents the mean + standard deviation of OD data from three samples. β-actin was used as the loading control. SSTR2, somatostatin receptor 2; OCT, octreotide; c-SOM, cyclo-somatostatin.
OCT inhibits the VEGF expression and secretion involved in the activation of Akt and ERK in the high glucose condition
A sandwich ELISA assay was performed to evaluate the secretion level of VEGF from RF/6A cells. As presented in Fig. 4A, cells exposed to 30 mM glucose exhibited in a significant increase in VEGF levels from day1 (P<0.05). In cells subjected to high glucose, treatment with 1 mM OCT significantly inhibited the release of VEGF into the culture medium from day 3 (P<0.05; Fig. 4A). Expression levels of VEGF, Akt, p-Akt, ERK and p-ERK were determined by western blot analysis. As presented in Fig. 4B and C, high glucose conditions significantly up-regulated the expression of VEGF in RF/6A cells (P<0.05). At the same time, the expression of p-Akt and p-ERK also increased significantly compared with the cells under normal conditions (P<0.05). The increase in expression of VEGF, p-Akt and p-ERK was significantly inhibited by treatment with OCT (P<0.05). VEGF is able to activate its downstream signaling molecules (Akt and ERK) by binding to its membrane receptor. Thus, these results demonstrated that OCT inhibited VEGF expression and secretion and subsequently the autocrine response induced by a high concentration of glucose.
Figure 4.

Effect of HG on VEGF expression and secretion in RF/6A cells, and the VEGF autocrine response was inhibited by OCT. (A) HG conditions resulted in an increase of VEGF release in a time-dependent manner. The increase of VEGF release may be inhibited by administration of OCT. (B) Expression of VEGF, Akt, p-Akt, ERK and p-ERK in RF/6A was evaluated by western blot and densitometric analysis and subsequently (C) quantified. VEGF, p-Akt and p-ERK upregulation in HG conditions was inhibited by treatment with OCT. Each column represents the mean + standard deviation of OD data from three samples. β-actin was used as the loading control. *P<0.05 vs. normal control; #P<0.05 vs. high glucose group. VEGF, vascular endothelial growth factor; Akt, protein kinase B; ERK, extracellular signal-related kinase; p-Akt, phosphorylated Akt; p-ERK, phosphorylated ERK; HG, high glucose; OCT, octreotide; OD, optical density.
Involvement of NRP1 in the effects of high glucose-induced occludin downregulation
VEGF is secreted from RF/6A cells and bound to its receptors VEGFR2/NRP1, exerting its role of downstream signal transduction. Western blot analysis indicated that treatment with NRP1 (VEGFR2 co-receptor) inhibitor ATWLPPR (0.1 mM) significantly prevented occludin downregulation under high glucose conditions (P<0.05; Fig. 5). p-Akt and p-ERK increased significantly in high glucose conditions (P<0.05; Fig. 5B); however, this was not influenced by ATWLPPR. These results indicate that NRP1 may be involved in the modulation of the VEGF autocrine or paracrine responses, as demonstrated in Fig. 6.
Figure 5.

NRP1 inhibitor ATWLLPPR prevented HGe-induced occludin downregulation. This was assessed using (A) western blot analysis, which was (B) quantified. Each column represents the mean + standard deviation of OD data from three samples. β-actin was used as the loading control. *P<0.05 vs. normal control, #P<0.05 vs. high glucose group. NRP1, neuropilin 1; OD, optical density; Akt, protein kinase B; ERK, extracellular signal-related kinase; p-Akt, phosphorylated Akt; p-ERK, phosphorylated ERK; HG, high glucose.
Figure 6.

Schematic diagram of the pathway linking SSTR2 activation to the prevention of occludin decrease induced by high glucose. Solid arrows, activation; dashed arrows, inhibition; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor; NRP1, neuropilin 1; SSTR2, somatostatin receptor 2; Akt, protein kinase B; ERK, extracellular signal-related kinase.
Discussion
The results of the present study indicate that SSTR2 exists in RF/6A cells cultured in normal and high glucose medium. Furthermore, no significant difference in SSTR2 expression was observed using densitometric analysis between the two culture conditions. Activation of SSTR2 prevented occludin downregulation under high glucose conditions and this process is involved in the increased expression of p-Akt, p-ERK and VEGF. Administration of the NRP1 inhibitor, ATWLPPR, inhibited the decrease of occludin induced by high glucose.
A previous study demonstrated that the SSTR2 agonist, OCT, has anti-inflammation and anti-oxidation effects and alleviated retinal edema in retinas following ischemia-reperfusion (5). It has been indicated that OCT was associated with the integrity of blood vessel endothelium, as confirmed by previous studies (14–16). Damage of endothelial cells is often observed in the early stage of DR (3,17,18). Somatostatin and its receptors are widely distributed in the retina and serves important and complex physiological roles (7,19). The current study focused on an endothelial cell line, RF/6A, and determined the existence of SSTR2 initially. A previous study assessing hypoxic retinas indicated that SSTR2 immunostaining was increasingly noted in capillaries (20). It has also been indicated that SSTR2 and SSTR5 are expressed in proliferative endothelial cells but not in quiescent cells (21). Whether SSTR2 in the endothelium serves a downstream role and modulates the integrity of endothelium remains unknown. The results of the present study indicated that high glucose conditions did not change the expression level of SSTR2 in RF/6A cells. Tight junctions among the endothelium in capillary vessels are the functional base of BRB (3). Occludin is an important tight junction transmembrane protein, responsible for the permeability of the barrier (22). Tissues or cells exposed to ischemia, hypoxia and high glucose conditions may exhibit a reduction of tight junction proteins, such as occludin, and this process forms the primary pathological base of BRB injury as it is demonstrated by enhanced permeability (3,22–24). In the current study, the condition culture medium with 30 mM glucose led to the decreased expression of occludin in RF/6A cells and this reduction was regulated by SSTR2. A study on the intestinal epithelia indicated that activation of SSTR2 led to protective effects on the intestinal barrier by modulation of the expression of tight junction proteins (25). Accumulated evidence has indicated that the administration of somatostatin or SSTR2 agonist had anti-inflammatory and neuroprotective effects in ischemic retinopathy (4,5). However, whether SSTR2 acts directly on capillaries epithelium, and the mechanism involved, remains unclear.
It is well-known that high glucose conditions are able to increase VEGF expression (22,26). The current study also indicated that not only the release of, but also the expression of VEGF in RF/6A cells was enhanced by high glucose. VEGF is an important factor that contributes to the permeability and the integrity of blood vessels (24). In patients with diabetes, increased VEGF in the eyes has been implicated in elevated vascular permeability and breakdown of BRB (27,28). VEGF-induced permeability is mediated by the downregulation of tight junction proteins (occludin), and the molecular mechanisms may be associated with urokinase plasminogen activator, nitric oxide and protein kinase C (24,29–32). The results of the current study demonstrated that high glucose induced an increase in VEGF expression, which may be inhibited by OCT, a SSTR2 preferring agonist. It is therefore hypothesized that SSTR2 regulates occludin, potentially by controlling the expression and secretion of VEGF. The results of the present study were consistent with other research performed in a mouse model of retinopathy (33). Although there remains a lack of such reports in RF/6A cells, previous studies have demonstrated that octreotide prevents the upregulation of VEGF induced by hypoxia (10,33). VEGF, as a specific mitogen, has been demonstrated to have an effective role in autocrine regulation of endothelial cells (34,35). Thus, the VEGF autocrine response may contribute to the vessel permeability changes in DR (22).
Akt and ERK are important cellular signal molecules that serve their biological functions when activated by phosphorylation. Activation of Akt may inhibit apoptosis and promote cell survival and lumen formation (36). ERK is involved in regulating angiogenesis (37). A high concentration of glucose may induce oxidative stress, tumor necrosis factor-α upregulation and apoptosis in the endothelium (38). The results of the current study indicated that high glucose induced an increase of p-Akt and p-ERK in RF/6A cells. Treatment with OCT was then demonstrated to reduce the activation of Akt and ERK. The present study was not able to illustrate whether the effects of OCT were from the inhibition of VEGF or other pathways, leading to anti-oxidative stress. Numerous studies have demonstrated that Akt and ERK are common downstream signal molecules of VEGF and its receptors (13,24,39,40). Increased p-ERK and p-Akt were typically observed in retinal ischemia diseases and retinal neovascularization (41,42), and they were also required factors for the expression of VEGF (41,43).
NRP1 acts as a co-receptor for VEGF by forming a complex with VEGFR2. NRP1 may promote VEGF binding and potentiate VEGFR2 activation and intracellular signaling (44). To illustrate whether NRP1 was involved in the high glucose-induced VEGF autocrine response and occludin reduction, a NRP1 inhibitor, ATWLPPR was administered to RF/6A cells. The results indicated that ATWLPPR reduced the increase of occludin, but had no significant effects on the activation of Akt and ERK. This was consistent with a previous study that demonstrated NRP1 to be a required component for the regulation of vascular permeability, induced by VEGF (40). Evans et al (44) suggested that treatment of small interfering RNA on NRP1 in HUVEC cells did not affect the phosphorylation of AKT and ERK mediated by VEGF. This was in accordance with the results of the present study. Activation of Akt and ERK was unaffected by the inhibition of NRP1, this was potentially due to lower thresholds of the receptors activity.
In conclusion, the results of the present study demonstrated that SSTR2 in RF/6A cells prevented the high glucose-induced decrease of occludin. NRP1, increased expression of VEGF, and activation of Akt and ERK were involved in this process. This may account for the regulating mechanisms of OCT on vascular permeability in diabetes or high glucose-mediated VEGF autocrine or paracrine response.
Acknowledgements
The present study was funded by the National Natural Science Foundation of China (grant no. 31300884) and Science and Technology Development Project of Henan Province (grant no. 162300410233 and 162300410036).
References
- 1.Ciulla TA, Amador AG, Zinman B. Diabetic retinopathy and diabetic macular edema: Pathophysiology, screening, and novel therapies. Diabetes Care. 2003;26:2653–2664. doi: 10.2337/diacare.26.9.2653. [DOI] [PubMed] [Google Scholar]
- 2.Klein R, Klein BE, Moss SE, Cruickshanks KJ. The wisconsin epidemiologic study of diabetic retinopathy: XVII. The 14-year incidence and progression of diabetic retinopathy and associated risk factors in type 1 diabetes. Ophthalmology. 1998;105:1801–1815. doi: 10.1016/S0161-6420(98)91020-X. [DOI] [PubMed] [Google Scholar]
- 3.Zhang C, Wang H, Nie J, Wang F. Protective factors in diabetic retinopathy: Focus on blood-retinal barrier. Discov Med. 2014;18:105–112. [PubMed] [Google Scholar]
- 4.Hernández C, García-Ramírez M, Corraliza L, Fernández-Carneado J, Farrera-Sinfreu J, Ponsati B, González-Rodríguez A, Valverde AM, Simó R. Topical administration of somatostatin prevents retinal neurodegeneration in experimental diabetes. Diabetes. 2013;62:2569–2578. doi: 10.2337/db12-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Sun Z, Shen J, Wu D, Liu F, Yang R, Ji S, Ji A, Li Y. Octreotide protects the mouse retina against ischemic reperfusion injury through regulation of antioxidation and activation of NF-κB. Oxid Med Cell Longev. 2015;2015:970156. doi: 10.1155/2015/970156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernández C, Simó-Servat O, Simó R. Somatostatin and diabetic retinopathy: Current concepts and new therapeutic perspectives. Endocrine. 2014;46:209–214. doi: 10.1007/s12020-014-0232-z. [DOI] [PubMed] [Google Scholar]
- 7.Cervia D, Casini G, Bagnoli P. Physiology and pathology of somatostatin in the mammalian retina: A current view. Mol Cell Endocrinol. 2008;286:112–122. doi: 10.1016/j.mce.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 8.Cristiani R, Petrucci C, Dal Monte M, Bagnoli P. Somatostatin (SRIF) and SRIF receptors in the mouse retina. Brain Res. 2002;936:1–14. doi: 10.1016/S0006-8993(02)02450-2. [DOI] [PubMed] [Google Scholar]
- 9.Cervia D, Catalani E, Dal Monte M, Casini G. Vascular endothelial growth factor in the ischemic retina and its regulation by somatostatin. J Neurochem. 2012;120:818–829. doi: 10.1111/j.1471-4159.2011.07622.x. [DOI] [PubMed] [Google Scholar]
- 10.Mei S, Cammalleri M, Azara D, Casini G, Bagnoli P, Dal Monte M. Mechanisms underlying somatostatin receptor 2 down-regulation of vascular endothelial growth factor expression in response to hypoxia in mouse retinal explants. J Pathol. 2012;226:519–533. doi: 10.1002/path.3006. [DOI] [PubMed] [Google Scholar]
- 11.Deissler HL, Lang GK, Lang GE. Capacity of aflibercept to counteract VEGF-stimulated abnormal behavior of retinal microvascular endothelial cells. Exp Eye Res. 2014;122:20–31. doi: 10.1016/j.exer.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 12.Gelfand MV, Hagan N, Tata A, Oh WJ, Lacoste B, Kang KT, Kopycinska J, Bischoff J, Wang JH, Gu C. Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding. Elife. 2014;3:e03720. doi: 10.7554/eLife.03720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herzog B, Pellet-Many C, Britton G, Hartzoulakis B, Zachary IC. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol Biol Cell. 2011;22:2766–2776. doi: 10.1091/mbc.E09-12-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Celiker U, Ilhan N, Ozercan I, Demir T, Celiker H. Octreotide reduces ischaemia-reperfusion injury in the retina. Acta Ophthalmol Scand. 2002;80:395–400. doi: 10.1034/j.1600-0420.2002.800409.x. [DOI] [PubMed] [Google Scholar]
- 15.Rauca C, Schäfer K, Höllt V. Effects of somatostatin, octreotide and cortistatin on ischaemic neuronal damage following permanent middle cerebral artery occlusion in the rat. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:633–638. doi: 10.1007/s002109900136. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Wang L, Zhang X, Cui L, Xing Y, Dong L, Liu Z, Li Y, Zhang X, Wang C, et al. The protection by octreotide against experimental ischemic stroke: Up-regulated transcription factor Nrf2, HO-1 and down-regulated NF-κB expression. Brain Res. 2012;1475:80–87. doi: 10.1016/j.brainres.2012.07.052. [DOI] [PubMed] [Google Scholar]
- 17.Rojas M, Zhang W, Xu Z, Lemtalsi T, Chandler P, Toque HA, Caldwell RW, Caldwell RB. Requirement of NOX2 expression in both retina and bone marrow for diabetes-induced retinal vascular injury. PLoS One. 2013;8:e84357. doi: 10.1371/journal.pone.0084357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moran E, Ding L, Wang Z, Cheng R, Chen Q, Moore R, Takahashi Y, Ma JX. Protective and antioxidant effects of PPARα in the ischemic retina. Invest Ophthalmol Vis Sci. 2014;55:4568–4576. doi: 10.1167/iovs.13-13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cervia D, Casini G. The neuropeptide systems and their potential role in the treatment of mammalian retinal ischemia: A developing story. Curr Neuropharmacol. 2013;11:95–101. doi: 10.2174/157015913804999423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dal Monte M, Ristori C, Videau C, Loudes C, Martini D, Casini G, Epelbaum J, Bagnoli P. Expression, localization, and functional coupling of the somatostatin receptor subtype 2 in a mouse model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2010;51:1848–1856. doi: 10.1167/iovs.09-4472. [DOI] [PubMed] [Google Scholar]
- 21.Adams RL, Adams IP, Lindow SW, Zhong W, Atkin SL. Somatostatin receptors 2 and 5 are preferentially expressed in proliferating endothelium. Br J Cancer. 2005;92:1493–1498. doi: 10.1038/sj.bjc.6602503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spoerri PE, Afzal A, Li Calzi S, Shaw LC, Cai J, Pan H, Boulton M, Grant MB. Effects of VEGFR-1, VEGFR-2, and IGF-IR hammerhead ribozymes on glucose-mediated tight junction expression in cultured human retinal endothelial cells. Mol Vis. 2006;12:32–42. [PubMed] [Google Scholar]
- 23.Muthusamy A, Lin CM, Shanmugam S, Lindner HM, Abcouwer SF, Antonetti DA. Ischemia-reperfusion injury induces occludin phosphorylation/ubiquitination and retinal vascular permeability in a VEGFR-2-dependent manner. J Cereb Blood Flow Metab. 2014;34:522–531. doi: 10.1038/jcbfm.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harhaj NS, Felinski EA, Wolpert EB, Sundstrom JM, Gardner TW, Antonetti DA. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci. 2006;47:5106–5115. doi: 10.1167/iovs.06-0322. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Wang Q, Xu H, Tao L, Lu J, Cai L, Wang C. Somatostatin regulates tight junction proteins expression in colitis mice. Int J Clin Exp Pathol. 2014;7:2153–2162. [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao B, Cai J, Boulton M. Expression of placenta growth factor is regulated by both VEGF and hyperglycaemia via VEGFR-2. Microvasc Res. 2004;68:239–246. doi: 10.1016/j.mvr.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C, Mori F, Ciulla TA, Ways K, Jirousek M, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes. 1997;46:1473–1480. doi: 10.2337/diab.46.9.1473. [DOI] [PubMed] [Google Scholar]
- 28.Amin RH, Frank RN, Kennedy A, Eliott D, Puklin JE, Abrams GW. Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 1997;38:36–47. [PubMed] [Google Scholar]
- 29.Behzadian MA, Windsor LJ, Ghaly N, Liou G, Tsai NT, Caldwell RB. VEGF-induced paracellular permeability in cultured endothelial cells involves urokinase and its receptor. FASEB J. 2003;17:752–754. doi: 10.1096/fj.02-0484fje. [DOI] [PubMed] [Google Scholar]
- 30.Qaum T, Xu Q, Joussen AM, Clemens MW, Qin W, Miyamoto K, Hassessian H, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci. 2001;42:2408–2413. [PubMed] [Google Scholar]
- 31.Wu HM, Yuan Y, Zawieja DC, Tinsley J, Granger HJ. Role of phospholipase C protein kinase C, and calcium in VEGF-induced venular hyperpermeability. Am J Physiol. 1999;276:H535–H542. doi: 10.1152/ajpheart.1999.276.2.H535. [DOI] [PubMed] [Google Scholar]
- 32.Joussen IS, Poulaki V, Tsujikawa A, Qin W, Qaum T, Xu Q, Moromizato Y, Bursell SE, Wiegand SJ, Rudge J, et al. Suppression of diabetic retinopathy with angiopoietin-1. Am J Pathol. 2002;160:1683–1693. doi: 10.1016/S0002-9440(10)61115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dal Monte M, Ristori C, Cammalleri M, Bagnoli P. Effects of somatostatin analogues on retinal angiogenesis in a mouse model of oxygen-induced retinopathy: Involvement of the somatostatin receptor subtype 2. Invest Ophthalmol Vis Sci. 2009;50:3596–3606. doi: 10.1167/iovs.09-3412. [DOI] [PubMed] [Google Scholar]
- 34.Imaizumi T, Itaya H, Nasu S, Yoshida H, Matsubara Y, Fujimoto K, Matsumiya T, Kimura H, Satoh K. Expression of vascular endothelial growth factor in human umbilical vein endothelial cells stimulated with interleukin-1alpha-an autocrine regulation of angiogenesis and inflammatory reactions. Thromb Haemost. 2000;83:949–955. [PubMed] [Google Scholar]
- 35.Simorre-Pinatel V, Guerrin M, Chollet P, Penary M, Clamens S, Malecaze F, Plouet J. Vasculotropin-VEGF stimulates retinal capillary endothelial cells through an autocrine pathway. Invest Ophthalmol Vis Sci. 1994;35:3393–3400. [PubMed] [Google Scholar]
- 36.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha'afi RI, Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–312. doi: 10.1016/S0092-8674(00)81661-X. [DOI] [PubMed] [Google Scholar]
- 37.Amin MA, Volpert OV, Woods JM, Kumar P, Harlow LA, Koch AE. Migration inhibitory factor mediates angiogenesis via mitogen-activated protein kinase and phosphatidylinositol kinase. Circ Res. 2003;93:321–329. doi: 10.1161/01.RES.0000087641.56024.DA. [DOI] [PubMed] [Google Scholar]
- 38.Nacci C, Tarquinio M, Montagnani M. Molecular and clinical aspects of endothelial dysfunction in diabetes. Intern Emerg Med. 2009;4:107–116. doi: 10.1007/s11739-009-0234-7. [DOI] [PubMed] [Google Scholar]
- 39.Breslin JW, Pappas PJ, Cerveira JJ, Hobson RW, II, Durán WN. VEGF increases endothelial permeability by separate signaling pathways involving ERK-1/2 and nitric oxide. Am J Physiol Heart Circ Physiol. 2003;284:H92–H100. doi: 10.1152/ajpheart.00330.2002. [DOI] [PubMed] [Google Scholar]
- 40.Becker PM, Waltenberger J, Yachechko R, Mirzapoiazova T, Sham JS, Lee CG, Elias JA, Verin AD. Neuropilin-1 regulates vascular endothelial growth factor-mediated endothelial permeability. Circ Res. 2005;96:1257–1265. doi: 10.1161/01.RES.0000171756.13554.49. [DOI] [PubMed] [Google Scholar]
- 41.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bullard LE, Qi X, Penn JS. Role for extracellular signal-responsive kinase-1 and −2 in retinal angiogenesis. Invest Ophthalmol Vis Sci. 2003;44:1722–1731. doi: 10.1167/iovs.01-1193. [DOI] [PubMed] [Google Scholar]
- 43.Jin J, Yuan F, Shen MQ, Feng YF, He QL. Vascular endothelial growth factor regulates primate choroid-retinal endothelial cell proliferation and tube formation through PI3K/Akt and MEK/ERK dependent signaling. Mol Cell Biochem. 2013;381:267–272. doi: 10.1007/s11010-013-1710-y. [DOI] [PubMed] [Google Scholar]
- 44.Evans IM, Yamaji M, Britton G, Pellet-Many C, Lockie C, Zachary IC, Frankel P. Neuropilin-1 signaling through p130Cas tyrosine phosphorylation is essential for growth factor-dependent migration of glioma and endothelial cells. Mol Cell Biol. 2011;31:1174–1185. doi: 10.1128/MCB.00903-10. [DOI] [PMC free article] [PubMed] [Google Scholar]