

Abstract
Myosin heavy chain (MHC), the major component of myosin motor molecule, plays an essential role in force production during muscle contraction. However, a comprehensive analysis of MHC proteoforms arising from sequence variations and post-translational modifications (PTMs) remains challenging due to the difficulties in purifying MHC (~223 kDa) and achieving complete sequence coverage. Herein, we have established a strategy to effectively purify and comprehensively characterize MHC from heart tissue by combining size-exclusion chromatography (SEC) and middle-down mass spectrometry (MS). First, we have developed a MS-compatible SEC method for purifying MHC from heart tissue with high efficiency. Next, we have optimized the Glu-C, Asp-N and trypsin limited digestion conditions for middle-down MS. Subsequently, we have applied this strategy with optimized conditions to comprehensively characterize human MHC and identified β-MHC as the predominant isoform in human left ventricular tissue. Full sequence coverage based on highly accurate mass measurements has been achieved using middle-down MS combining 1 Glu-C, 1 Asp-N and 1 trypsin digestion. Three different PTMs: acetylation, methylation and trimethylation were identified in human β-MHC and the corresponding sites were localized to the N-terminal Gly, Lys34 and Lys129, respectively, by electron capture dissociation (ECD). Taken together, we have demonstrated this strategy is highly efficient for purification and characterization of MHC, which can be further applied to studies of the role of MHC proteoforms in muscle-related diseases. We also envision that this integrated SEC/middle-down MS strategy can be extended for the characterization of other large proteins over 200 kDa.
Graphical Abstract
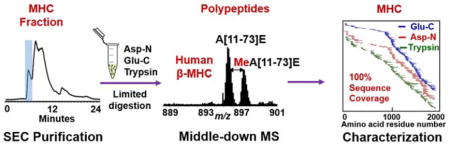
Introduction
Myofilaments, composed of thin and thick filaments, are responsible for muscle contraction and relaxation.1,2 The major component of the thick filament is myosin (also known as myosin II, conventional myosin), which comprises a family of molecular motors.3,4 Myosin interacts with actin on the thin filament to convert chemical energy from ATP hydrolysis to mechanical energy that powers muscle contraction.5 Myosin is a hexameric protein consisting of two myosin heavy chains (MHCs, ~223 kDa), two pairs of regulatory light chains (RLCs, ~19 kDa) and two pairs of essential light chains (ELCs, ~22 kDa) (Figure 1a).3,4 MHC can be generally divided into the globular head (also known as subfragment 1, or the S1 catalytic head domain) and coiled-coil tail (also referred to as subfragment 2, or the S2 filament-forming rod) domains.6 Each globular head can be further divided into the motor domain which contains the ATPase and actin-binding region, and the neck domain which is associated with ELC and RLC. In contrast to well characterized RLCs and ELCs including the sequence and post-translational modifications (PTMs),7–9 a comprehensive characterization of MHC remains challenging mainly due to its large molecular weight (MW) (~223 kDa), making it difficult to purify MHC and to achieve complete sequence coverage.
Figure 1.

(a) Schematic drawing of myosin consisting of myosin heavy chains (MHC) and light chains. (b) Purification of MHC by SEC. Representative UV chromatogram showing separation of human myofilament protein mixture by SEC. SEC column: PolyHydroxyethyl A, 200 × 9.4 mm, 5 μm, 500 Å. (c) SDS-PAGE analysis of the myofilament protein mixture and purified MHC by SEC.
Widely used methods for MHC purification typically require multiple precipitation-dissolution cycles and the use of high concentration of salts, which resulted in a prolonged purification process with potential problems in solubilizing proteins after precipitations.10–13 Recently, bottom-up mass spectrometry (MS) with trypsin digestion either in-gel or in-solution has been utilized for characterization of MHC.14–17 However, the sequence coverage is very low since only a small fraction of the MHC peptides have been recovered and detected by MS.14–17 In contrast to bottom-up MS, top-down MS analyzes intact proteins, which provides a bird’s eye view of all proteoforms18 arising from alternative splicing of mRNAs and PTMs.19–24 However the top-down approach still faces significant challenges for characterization of high MW proteins (> 100 kDa) mainly due to the decay of signal to noise ratio (S/N) as a function of increasing MW,25 as well as the difficulty in protein solubilization, ionization, and fragmentation.22,26 Thus, a hybrid middle-down MS approach27–32 which involves the digestion of proteins into polypeptides (3–20 kDa) appears to be an attractive alternative for characterization of large proteins (>100 kDa). These polypeptides are easier to separate and detect compared to larger intact proteins.26–28 Meanwhile, the middle-down approach significantly increases confidence in protein sequence verification and provides comprehensive PTM characterization.
In this study, we have developed a strategy combining size exclusion chromatography (SEC) and middle-down MS to efficiently purify and completely characterize MHC from human heart tissue. Compared to the existing methods for the purification of MHC which can take a day or longer,10–12 our method employed a fast extraction of myofilament proteins (~2 hr) followed by SEC separation of MHC (~10 min) in MS-friendly buffers. The protein fractions are collected automatically without additional steps needed for the removal of detergents or salts, which greatly simplifies the procedures and increases the purification efficiency. Unlike the small peptides (< 3 kDa) produced in bottom-up experiments, polypeptides ranging from 3 to 30 kDa have been obtained by limited proteolysis using Glu-C, Asp-N and trypsin in the middle-down MS approach. With only 1 Glu-C, 1 Asp-N and 1 trypsin digestions, we have achieved full sequence coverage for human MHC based on highly accurate mass measurements and completely characterized all PTMs.
Materials and methods
Chemical and Reagents
All reagents were purchased from Fisher Scientific (Fair Lawn, NJ, USA) and Sigma-Aldrich (St Louis, MO, USA) unless noted otherwise. Glu-C, Asp-N and trypsin endoproteinases were purchased from Promega (Madison, WI, USA). Standard MHC protein purified from porcine heart was purchased from Sigma-Aldrich (St Louis, MO, USA). All solutions were prepared with HPLC grade water (Fisher Scientific, Fair Lawn, NJ, USA).
Cardiac tissue samples
Human donor hearts were collected at the University of Wisconsin Hospital and Clinics according to protocols approved by the Institutional Review Board of the University of Wisconsin-Madison. The donor hearts were preserved in ice-cold cardioplegic solution until dissection and the dissected tissues were flash frozen immediately in liquid nitrogen and stored at −80 °C.
Myofilament protein extraction and MHC purification
Myofilament proteins were extracted from approximately 100–500 mg human left ventricular (LV) tissue with 3 mL HEPES buffer containing protease and phosphatase inhibitors (25 mM HEPES pH 7.4, 50 mM NaF, 2.5 mM EDTA, 0.25 mM Na3VO4 and 0.25 mM PMSF) and further homogenized in 2.5 mL trifluoroacetic acid (TFA) extraction solution (0.1% TFA, 1mM TCEP). The resulting homogenate was centrifuged and the supernatant (myofilament protein mixture) was collected for MHC purification. The myofilament protein mixture was then separated by a SEC column (PolyHydroxyethyl A, 200 mm length × 9.4 mm id, 5 μm particle size, 500 Å pore size, PolyLC Inc., Columbia, MD, USA) on an Acquity H-class UPLC system (Waters, Milford, MA, USA) with the UV detector set to detect absorbance at 280 nm. SEC fractions containing MHC were collected automatically and further concentrated by 100 kDa molecular weight cutoff (MWCO) filters to remove co-eluting small proteins and to concentrate MHC. The purity of MHC was confirmed by SDS-PAGE using 8% polyacrylamide gels. The detailed procedures are included in the Supporting Information.
Middle-down mass spectrometry
Bradford protein assays were performed to determine the MHC concentration prior to enzymatic digestion. In Glu-C, Asp-N and trypsin proteolysis reactions, 50–100 μg purified MHC was digested in 25 mM NH4HCO3 buffer at pH 8 with an enzyme-to-protein ratio of either 1:100 (for Glu-C and Asp-N proteolysis) or 1: 200 (for trypsin proteolysis). The reactions were incubated at 37 °C for 30 min and then deactivated with 2 μL 98% formic acid (FA).
The desalted polypeptides were further analyzed using the LC/MS+ method.33,34 Polypeptide mixture was separated by a home-made PLRP reversed-phase column (200 mm length × 500 μm id, 10 μm particle size, 1,000 Å pore size). PLRP-S particles were purchased from Agilent Technologies (Santa Clara, CA, USA). The polypeptide fractions were analyzed using a 7T LTQ/Fourier transform ion cyclotron resonance (LTQ/FT-ICR) Ultra mass spectrometer (Thermo Scientific Inc., Bremen, Germany) as described previously.21,23,34,35 For MS/MS experiments using ECD36, an electron energy between 0.5% and 10% (0.2–9.7 eV based on the offset of 0.3 eV) and a duration time between 20 ms and 70 ms were used for the fragmentation of the polypeptides of interest. The workflow of the middle-down MS pipeline is delineated in Figure S1.
In-house developed software tools, MASH Suite and MASH Suite Pro with MS-Align+37 were used for the MS and MS/MS data analysis.38,39 The MHC sequence in the Swiss-Prot protein database (Unit-ProtKB/Swiss-Prot) was used to predict the mass values of the polypeptides from Glu-C, Asp-N and trypsin digestions and polypeptides were assigned to potential MHC sequences with high mass accuracy (<10 ppm) and verified by MS/MS. The sequence coverage was calculated based on the sequence mapping of these assigned polypeptides, which is not the bond cleavages between amino acids. Quantitative analysis of methylated/un-methylated polypeptides was performed using peak intensities from the high-resolution FT-ICR mass spectra similarly as described previously.23,34 More detailed procedures are in the Supporting Information.
Results and discussion
Purification of MHC by size exclusion chromatography
After a rapid extraction of myofilament proteins from human heart tissue, we employed a ultra-high pressure SEC method utilizing a MS-friendly volatile solvent system40 for fast, high-resolution separation of myofilament proteins. Specifically, SEC with 0.1% TFA/H2O was used as the mobile phase to separate MHC from the human myofilament protein mixture. SDS-PAGE analysis of all the SEC fractions was performed as shown in Figure S2. The SEC fractions from 7 min to 9 min containing predominantly MHC were collected automatically (Figure 1b) and 100 kDa MWCO ultracentrifugal filters were used to concentrate the large proteins and to remove co-eluting low MW ones in the fractions. The total time required for the separation and filtration was less than 60 min. SDS-PAGE analysis was applied to evaluate the purity of MHCs after SEC separation. The myofilament protein mixture is shown in the first lane of a 8% polyacrylamide gel while a pure band around 220 kDa in the first fraction is shown in the second lane (Figure 1c), which demonstrated the successful purification of MHC from the myofilament protein mixture. In terms of purification yield, approximately 10–20 μg of high-purity MHC was obtained from 10 mg tissue.
The SEC-based, MS-friendly method developed here significantly improves the purification efficiency as demonstrated by a significant reduction in the number of procedures involved and the time taken. The total time taken for the purification procedure including the myofilament extraction from heart tissue and SEC separation to obtain high purity MHC was less than 3 hrs. In contrast, previously developed methods10–13 for MHC purification generally involved successive precipitation-dissolution steps with or without ion exchange chromatography (i.e. DEAE), which are highly complex and could take a day or longer.12 In general, it is difficult to solubilize large myofilament proteins after multiple precipitations, thus requiring a large amount of starting tissue. Moreover, the use of high concentration of salts (e.g., 0.5 M KCl) makes it difficult to do subsequent MS analysis. Hence, only in-gel digestion from the gel band containing myosin or in-solution digestion from myosin-enriched pellets were used in bottom-up MS analysis, but with very limited recovery of MHC polypeptides.14–17 Our method uses low salt or salt-free buffers which is MS-friendly and the pure MHC collected can be used directly for MS analysis. Furthermore, the purity of MHC achieved using our method is much higher than the purity using the precipitation method which often co-precipitates with the low MW myosin light chains.12
Optimization of middle-down MS method
First, we sought to optimize the digestion conditions for middle-down MS analysis of MHC, including the digestion duration and enzyme-to-protein ratio, to minimize the amount of small peptides produced and to maximize the amount of polypeptides within the appropriate MW range (3–30 kDa). Here, Glu-C, Asp-N and trypsin were chosen for the limited proteolytic digestion of MHC. The middle-down MS method was optimized with standard MHC protein purified from porcine heart. Six different conditions for digestion were employed to assess the proteolytic results using Glu-C, Asp-N and trypsin. The reactions were incubated for 10, 30, or 60 min with 1:100 or 1:200 enzyme-to-protein ratios at 37 °C. The resulting polypeptides were then analyzed by 15% SDS polyacrylamide gels to evaluate the digestion results. Most polypeptides from Glu-C and Asp-N digestions were larger than 3 kDa in all the conditions assessed (Figure 2a). However, at 1:200 enzyme-to-protein ratio with either Glu-C or Asp-N, there was a significant amount of polypeptides greater than 50 kDa, which was not optimal for MS analysis. At a 1:100 enzyme-to-protein ratio, 30 min of digestion was optimal since this condition generates more polypeptides in the 3–30 kDa range than 10 min of digestion and fewer small peptides (<3 kDa) than 60 min of digestion (Figure 2a). For limited digestion reaction using trypsin, most polypeptides from the reactions in a 1:100 enzyme-to-protein ratio were smaller than 10 kDa (Figure 2a), and therefore, were not ideal for MS analysis. When using a 1:200 enzyme-to-protein ratio for digestion, even though there were more polypeptides produced, the small peptides remained abundant in solution, which would be expected to introduce significant suppression for the MS analysis of co-eluting polypeptides. Therefore, we mainly focused on the MS analysis of MHC polypeptides from 30-min Glu-C and Asp-N digestions at 37 °C with a 1:100 enzyme-to-protein ratio.
Figure 2.

Optimization of the middle-down MS method using purified MHC standard protein from pig heart tissue. (a) SDS-PAGE analysis of polypeptides after enzymatic digestion using Glu-C (i), Asp-N (ii) and trypsin (iii) under six different conditions: 10 min, 30 min, or 60 min incubation with 1:100 or 1:200 enzyme-to-protein ratio at 37 °C. M, molecular weight marker. (b) Identification of a very large polypeptide (~30 kDa) from pig β-MHC (MYH7_PIG, UniProtKB-P79293) digested by Glu-C. Left panel, mass spectrum of the precursor ion (M33+) and polypeptide sequence map showing bond cleavages. Right panel, representative fragment ions. Fragment assignments were made based on the predicted polypeptide sequence of the Glu-C digested pig β-MHC in the UniProt protein database. Expt’l, experimental monoisotopic mass based on data obtained from MS or MS/MS experiments. Calc’d, calculated monoisotopic mass based on amino acid sequences.
MHC polypeptides resulting from limited Glu-C digestion were separated by reversed-phase chromatography (RPC) and fractions were collected every 1 min for high-resolution MS and MS/MS analysis. These polypeptides were assigned to the sequence of the pig β-MHC (MYH7_Pig, UniProtKB-P79293) based on accurate mass measurements. The experimental mass values of ~70% polypeptides matched well with the theoretical polypeptide mass values of the pig β-MHC with high mass accuracy (< 10 ppm). Precursor ions of the selected polypeptides were subsequently isolated in the gas phase and subjected to MS/MS analysis to confirm the sequence assignments (Figure 2b). For example, a polypeptide of 30329.45 Da was assigned as the polypeptide R[1434–1696]E from pig β-MHC with a mass error of 5.6 ppm (Figure 2b). The MS/MS of this polypeptide contained 42 c ions and 18 z• ions, which matched well to the predicted ions from the polypeptide sequence (Figure 2b). Overall, MS and MS/MS analysis of the high abundance polypeptides resulting from standard protein digestion (Figure S3) demonstrated the feasibility of this middle-down MS method for the characterization of MHC sequences.
Sequence analysis of human cardiac MHC
Next, we applied our optimized middle-down MS method to analysis of purified human MHC by SEC. Sequence analysis of MHC from human LV tissue was carried out by limited proteolysis of MHC with Glu-C, Asp-N, and trypsin and the resulting polypeptides were further analyzed by high-resolution FT-ICR MS. Polypeptides were assigned to potential human cardiac MHC isoform sequences and the matches were evaluated based on high mass accuracy (< 10 ppm). The experimental masses of most polypeptides matched well with the theoretical masses of human β-MHC (MYH7_Human, UniProtKB-P12883) polypeptides (Table S1–S3). Subsequent MS/MS analysis further confirmed the expression of β-MHC isoform in human LV tissue with unique polypeptides from β-MHC in comparison to α-MHC (Figure S4): two representative unique polypeptides from Glu-C and Asp-N digestions were shown in Figure 3. The unique polypeptide from Glu-C digestion (Figure 3a) had an experimental mass of 7457.78 Da, which matched the theoretical mass of polypeptide L[1202–1264]E of human β-MHC. According to the tandem mass spectrum, 33 c ions and 38 z• ions from the polypeptide L[1202–1264]E were observed. Figure 3b showed a unique polypeptide from Asp-N digestion assigned as polypeptide D[218–381]A of human β-MHC with a molecular mass of 18560.24 Da. MS/MS analysis of this polypeptide showed 68 c ions and 37 z• ions, matching the predicted values from the sequence D[218–381]A. The tandem mass spectra of both polypeptides unambiguously confirmed the expected presence of β-MHC in human heart LV tissue.
Figure 3.

Identification of unique polypeptides from human β-MHC (MYH7_Human, UniProtKB-P12883) digested by Glu-C (a) and Asp-N (b). Left panel, mass spectra of the precursor ions and sequence maps showing bond cleavages. Right panel, representative fragment ions. Fragment assignments were made based on the predicted polypeptide sequences of the Glu-C/Asp-N digested human β-MHC in the UniProt protein database. These two polypeptides are also annotated in Figure 4.
We were able to achieve 99% sequence coverage of human β-MHC based on polypeptides recovered from one Glu-C and one Asp-N digestions (Table S1 and S2). However, there was a short segment of human β-MHC (Q[475–483]E) that was not covered by any identifiable polypeptides. To obtain a full sequence coverage of human β-MHC, limited trypsin digestion was applied to the purified MHC as trypsin has cleavage sites different from Glu-C and Asp-N, and therefore was expected to give rise to a variety of polypeptides which could cover amino acids 475 to 483. Fortunately, we observed polypeptide Q[454–484]K of human β-MHC in the polypeptide mixture resulting from trypsin digestion (Table S3). Overall, a total of 730 polypeptides were mapped to the human β-MHC sequence and full sequence coverage was achieved based on accurate polypeptide masses (Figure 4), which demonstrated complete sequence coverage with polypeptides produced from limited proteolysis in middle-down MS. Among the three enzymes used for the digestion of MHC, Glu-C yielded the highest sequence coverage (99%) versus Asp-N (86%) and trypsin (95%) (Tables S1–S3). The overlaps between the polypeptide sequences improved the confidence of sequence mapping and protein identification.
Figure 4.

Schematic showing sequence coverage for human β-MHC from limited digestion experiments using Glu-C (blue), Asp-N (red) and trypsin (green). The mapping of polypeptides was based on accurate mass measurements and MS/MS experiments. Inset: A representative polypeptide of human β-MHC (D[554–627]A) from Asp-N digestion with accurate mass and confirmed sequence by MS/MS. Other two polypeptides, L[1203–1265]E and D[218–381]A, were the two polypeptides shown in Figure 3.
In addition to β-MHC, we detected several α-MHC (MYH6_HUMAN, UniProtKB-P13533) specific polypeptides with sequence assignments based on high mass accuracy (< 10 ppm error) (Table S4). Conceivably, the recovery of unique polypeptides from α-MHC was low due to the low abundance of α-MHC (< 10%),41 the high sequence similarity between α-MHC and β-MHC, and the increased complexity of the mixture after digestion. Therefore, it is difficult to comprehensively characterize α-MHC in human LV tissues using the current middle-down MS strategy. To improve the sensitivity of this method and enable the characterization of low abundant α-MHC specific polypeptides, the new generation instruments such as Bruker Solarix with absorption mode FT42,43 or Thermo Orbitrap with parallel ion detection and accumulation44 could be used. Alternatively, the characterization of human α-MHC could be conducted using human atrial tissue by the same SEC tandem middle-down MS strategy, as α-MHC is reported to have greater expression in cardiac atrial tissue (~90%).45
α-MHC and β-MHC are the two major isoforms resulting from two different genes, MYH6 and MYH7, respectively in mammalian cardiac muscle cells.5,46,47 The expression of α- and β-MHC is species- and chamber-dependent and is tightly controlled by developmental and hormonal factors.5,45,46 Even though these two MHC isoforms have a high degree (93%) of sequence homology, α- and β-MHC are functionally distinct.46,47 Myosin composed of α-MHC is known to have a higher ATPase activity than myosin composed of β-MHC.41 Alterations in the relative abundance of these two isoforms have a significant impact on the overall ATPase activity and contractile properties of the cardiac muscle.41,48 A previous study has shown a decrease in the relative abundance of α-MHC in human failing hearts compared to non-failing hearts, which was associated with cardiac dysfunction in heart failure.41 It has also been demonstrated that cardiac muscles comprised of a higher percentage of α-MHC isoforms produce greater power.47 Furthermore, an increase in β-MHC in rat LV tissue was observed in the investigation of MHC isoform transition during development of cardiac hypertrophy.49
Traditionally MHC isoforms can be separated by high-resolution SDS-PAGE electrophoretic methods.45,50–52 Recently, a new method using multi-color immunofluorescence has been developed for rapid determination of MHC isoform expression, which has been applied to rat, mouse and human skeletal muscles.53 Additionally, a RPC method was developed for the separation and quantification of MHC isoforms in purified myosin.54 Likely we could couple our SEC-based method with this RPC method for the comprehensive characterization of MHC together with isoform quantification.
Comprehensive characterization of MHC PTMs
Although the majority of polypeptides (~90%) detected in the MS analysis were mapped to human β-MHC, some polypeptides were not matched to any of the predicted polypeptides from proteolysis of human β-MHC. These unassigned polypeptides might represent those containing amino acid variants or PTMs. Thus, the unassigned polypeptides were further analyzed by MS/MS and the tandem mass spectra were used for the screening of potential unexpected sequence variants and PTMs by MASH Suite Pro38 with MS-Align+.37
We were able to observe post-translationally modified polypeptides with methylation at Lys34 methylation and trimethylation at Lys129 in the polypeptide mixture from Glu-C digestion of MHC proteins (Figure 5). For example, a polypeptide of 16396.28 Da was not assigned to any polypeptide sequences from the unmodified human β-MHC. The precursor ion of this polypeptide was isolated and fragmented using ECD. The polypeptide was subsequently identified as polypeptide D[74–217]E of β-MHC with a mass increase of 42.05 Da, which matched a trimethylation. Importantly, high-resolution MS obtained by LTQ-FT entails high mass accuracy and allows for the differentiation of trimethylation (42.05 Da) from acetylation (42.01 Da).55 The trimethylated polypeptide mass (3.7 ppm error) is more consistent with the experimental mass than is an acetylated polypeptide (6.1 ppm error). In total, 27 c ions and 51 z• ions were mapped to the trimethylated polypeptide D[74–217]E. Four fragment ions, c52, c57, z•87 and z•92, collectively indicated the localization of trimethylation to the “NPYKW” (Asn-Pro-Tyr-Lys-Trp) segment of the sequence (Figure 5a). After considering the fragmentation maps of two additional trimethylated polypeptides (Figure S5), the trimethylation site was assigned to Lys129. The absence of an unmodified polypeptide of D[74–217]E on the MS spectra (Figure S6), suggests that either MHC is fully trimethylated at Lys129 or the relative abundance of the unmodified polypeptide was too low to be detected. Another PTM of MHC, methylation at Lys34, was also characterized in multiple polypeptides using the same strategy (Figure 5b). A 14.01 Da increase in MW at Lys34 of polypeptide A[11–73]E from human β-MHC indicated methylation at Lys34. Four fragment ions shown in Figure 5b, c23, c24, z•39 and z•40 unambiguously confirmed the methylation site. Un-methylated polypeptide A[11–73]E of β-MHC was also observed on the mass spectrum with a mass difference of 14.01 Da (Figure S7a), which indicates that Lys34 is partially methylated with approximate 40% occupancy.
Figure 5.

Identification of the post-translationally modified polypeptides from Glu-C digestion of human β-MHC. Mass spectra of the precursor ions, sequence maps and representative fragment ions of the selective polypeptides show trimethylation at Lys129 (a) and methylation at Lys34 (b).
To the best of our knowledge, this is the first time that methylation and trimethylation on β-MHC have been identified in human heart tissue. Previously, these two modifications were found in rabbit skeletal muscle MHC-2b (MYH4_RABIT, UniProtKB-Q28641)56 and adult chicken pectoralis muscle MHC (MYSS_CHICK, UniProtKB - P13538)57 using cyanogen bromide (CNBr) and trypsin digestion, respectively. Since these two modifications are in the myosin motor domain, it is highly possible that they are associated with differences in ATPase activity and the kinetics of actin-myosin interactions. It is likely that the amino acid sequence surrounding Lys129 is unusually hydrophobic and carries a charge at neutral pH, which suggests that it could form a hydrophobic pocket with a positive charge on trimethylated lysine.56 This structure may enable the binding of the adenine moiety to MHC and thus the interaction between positively charged lysine and the ATP phosphate group. Considering the high sequence homology between human β-MHC and rabbit MHC-2b (Figure S8) surrounding Lys129, the trimethylation at Lys129 may have the same function here as it does in rabbit MHC-2b.
For rabbit MHC-2b, a 60:40 relative abundance of monomethylated lysine relative to lysine at Lys34 was previously reported based on amino acid analysis.56 Here we have observed a lower relative abundance (~40%) of monomethylated polypeptide in several Lys34 methylated polypeptides of different length (Figure S7). In contrast to the partial methylation at Lys34, Lys129 is fully occupied by trimethylation in rabbit MHC-2b (referred to as Lys130).56 This is consistent with with our results from human MHC, where only the trimethylated polypeptide peak was detected while no unmodified polypeptide peak was observed (Figure S6), suggesting that that Lys129 in human β-MHC is also fully trimethylated. Further study needs to be done to address the significance of partial methylation of Lys34 versus possibly complete trimethylation of Lys129 of β-MHC. The observation of multiple polypeptides containing methylated Lys34 also demonstrate the high reliability of this middle-down MS method for the characterization of PTMs of large proteins (Figure S7).
In experiments using Asp-N for limited digestion, in addition to the trimethylation of Lys129 (Figure S9a), the removal of N-terminal methionine and acetylation of Gly2 was also confirmed by a small peptide of 2566.28 Da (Figure S9b). The modified peptide G[2–24]L from human β-MHC was confirmed by MS/MS with 9 c ions and 16 z• ions (Figure S9b). The removal of methionine and acetylation at the N-terminus, which is common in eukaryotic proteins, was also observed in rabbit MHC-2b56 and chicken skeletal MHC.57,58 A previous study has suggested that N-terminal residues are important for protein activities and the removal of N-terminal methionine is required for viability.58 For example, N-terminal acetylation of actin along with the removal of methionine strengthens the weak interaction between actin and myosin.59,60 Also, N-terminal acetylation on α-tropomyosin is essential for its binding to actin.61 Although the roles of methionine removal and N-terminal acetylation of MHC have yet to be defined, it is possible that they are involved in the regulation of MHC stability or the interaction of MHC with other proteins.
In terms of other PTMs, phosphorylation at Ser1510 is predicted in the UniProt database for human β-MHC (MYH7_HUMAN, UniProtKB-P12883) based on similarity analysis of rat β-MHC (MYH7_RAT, UniProtKB-P02564) in a large-scale proteomics study of rat tissues. We did observe a polypeptide containing unphosphorylated Ser1510; however, no corresponding phosphorylated polypeptide with a mass shift of 79.96 Da was observed (Figure S10). Overall, there was no phosphorylated polypeptide detected in human β-MHC in this study. It is likely that phosphorylation in MHC is species- and muscle-type dependent. Previously, MHC phosphorylation has been reported in a number of smooth muscle and non-muscle MHC of which the phosphorylation is known to regulate ATPase activity and filament assembly.3 Nevertheless, the presence and the role of phosphorylation of MHC in striated muscles (i.e., cardiac and skeletal muscles) remain obscure.
Moreover, age-specific changes in MHC PTMs, including carbonylation, methylation and deamidation, have been found in the MHC motor domain and tail region using a bottom-up approach in skeletal muscle tissues from the tibialis and vastus lateralis muscles.14 Protein carbonylation is generally considered to be an irreversible PTM and a major hallmark of oxidative stress-related disorders in pathological conditions.62 Since the human heart tissue samples used in this study are from healthy donors, it is conceivable that no carbonylation is present. Regarding deamidation, we have not observed appreciable amount of deamidation in this study based on both MS and MS/MS data. Enzymatic deamidation is reported to be associated with age-related diseases whereas non-enzymatic deamidation could occur in proteomic sample preparation.63 The key causative factors for the non-enzymatic deamidation were prolonged incubation time (8–24 h) and the mildly alkaline pH.63 Given the very short digestion (30 min) employed here for the middle-down experiments, conceivably the non-enzymatic deamidation will be very low, likely below the detection limit.
The promise of middle-down MS for complete characterization of large proteins (>200 kDa)
In this study, we have demonstrated the high promise of complete sequence characterization of large proteins over 200 kDa using middle-down MS. Previously Simpson et al. has coupled SEC with RPC as 2D separation strategies for protein profiling followed by bottom-up analyses,64 whereas Hummel et al. used SEC to purify Mustard 2S albumin allergen for bottom-up, middle-down and top-down proteomics.65 Nevertheless, this is the first time that the integrated strategy of SEC purification and middle-down MS has been applied to the characterization of proteins larger than 200 kDa. After optimization of limited proteolysis conditions, we achieved full sequence coverage for β-MHC (223 kDa) with middle-down MS using 1 Glu-C, 1 Asp-N, and 1 trypsin limited proteolysis (Figure 4). Specifically, 99% sequence coverage was achieved using a single middle-down MS experiment with limited Glu-C proteolysis (Table S1); whereas 86% or 95% sequence coverage was obtained using a single middle-down MS with limited Asp-N (Table S2) or trypsin (Table S3) proteolysis, respectively. Polypeptides in the mass range of 10 to 30 kDa have been detected in all three middle-down MS experiments with limited proteolysis using Glu-C (64 polypeptides), Asp-N (46 polypeptides), and trypsin (43 polypeptides). The detection of these polypeptides (10–30 kDa) not only increased the confidence of sequence verification but also allowed the reliable mapping of the PTMs and preserved the connectivity of multiple PTMs within the sequence covered by such polypeptides. Note that most of the proteins detected in previous large-scale top-down MS-based proteomics studies are between 10 and 30 kDa.20,22,24,26,66
In contrast, mainly small peptides (< 3 kDa) are produced by complete trypsin digestion in the typical bottom-up experiment. Despite its robustness and high throughput, the intrinsic limitation of the bottom-up approach is the loss of the connectivity among the small peptides and the potential missing PTMs due to the partial recovery of the digested peptides.67 On the other hand, top-down MS is the method of choice for characterization of complex proteoforms18 arising from alternative splicing of mRNAs and PTMs.19–24 However, it remains an unsolved challenge to analyze large proteins, especially those over 100 kDa, using a top-down MS approach, mainly due to the decay of S/N as a function of increasing MW25 together with difficulties in protein solubilization, ionization, and fragmentation.22,26 Compton et al. generated a quantitative model integrating the factors of charge states, isotope peaks and interfering species present in a typical mass spectrum, which demonstrated the significantly reduced ability to detect proteins over 25 kDa.25 Hence, the detection of proteins over 100 kDa is extremely challenging, particularly in a large-scale proteomics study.
With the advancement of the high-resolution mass spectrometer, the middle-down MS approach provides an attractive alternative for analysis of large proteins.26–30,68 In the middle-down MS approach, proteins are digested into relatively polypeptides (i.e., 3–20 kDa) which are amendable for high-throughput and highly sensitive MS analysis. Wu et al. developed a sensitive LC-MS-based middle-down MS strategy termed as Extended Range Proteomic Analysis (ERPA), which could be performed under similar chromatographic conditions but with high sequence coverage as demonstrated for β-casein and epidermal growth factor receptor.28 Cannon et al. used a capillary LC-LTQ orbitrap system to provide automated middle-down MS analysis of the proteolytic polypeptides in the mass range of 3–10 kDa.27 In addition, Heck and co-workers analyzed the glycoproteins68 and histone isoforms69 by combining RPC and middle-down MS. Overall, the advantages of middle-down MS include high-performance separation and high-sensitivity detection of polypeptides; high-confidence protein identification by sequencing polypeptides; high sequence coverage for comprehensive sequence characterization; and capability for identifying long-range combinatorial PTM patterns.26,30
Conclusion
In summary, we have developed a method combining MS-friendly SEC with middle-down MS for the efficient purification and complete characterization of MHC in human LV tissue without the use of gel electrophoresis and specific antibodies. We were able to obtain human cardiac MHC with high purity via SEC purification. Full sequence coverage was achieved by middle-down MS with high mass accuracy from 1 Glu-C, 1 Asp-N, and 1 trypsin digestion. PTMs including acetylation of N-terminal Gly, methylation of Lys34, and trimethylation of Lys129 in human β-MHC were observed and characterized by MS/MS. These results demonstrate that the integrated strategy of coupling SEC with middle-down MS is efficient for characterization of MHC, which holds great potential for the study of MHC sequence variants and PTM changes in muscle-related diseases. Furthermore, it is expected that this integrated SEC/middle-down MS strategy may be applicable to the characterization of other proteins larger than 200 kDa with appropriate modification of the protein extraction and separation steps.
Supplementary Material
Acknowledgments
We would like to thank Lichen Xiu for his help with protein purification and Dr. Andrew Alpert for providing the SEC columns. Financial support was kindly provided by NIH R01 HL109810 and HL096971 (to Y.G.). YG would like to acknowledge NIH R01GM117058 and NIH high-end instrument grant S10OD018475. We would also like to thank Wisconsin Partnership Funds for the establishment of UW Human Proteomics Program for the mass spectrometry facility.
Footnotes
Supporting Information Available:
Additional information as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Moss RL, Diffee GM, Greaser ML. Rev Physiol Bioch P. 1995;126:1–63. doi: 10.1007/BFb0049775. [DOI] [PubMed] [Google Scholar]
- 2.Schiaffino S, Reggiani C. Physiol Rev. 1996;76:371–423. doi: 10.1152/physrev.1996.76.2.371. [DOI] [PubMed] [Google Scholar]
- 3.Warrick HM, Spudich JA. Ann Rev Cell Biol. 1987;3:379–421. doi: 10.1146/annurev.cb.03.110187.002115. [DOI] [PubMed] [Google Scholar]
- 4.Rayment I, Rypniewski W, Schmidt-Base K, Smith R, Tomchick D, Benning M, Winkelmann D, Wesenberg G, Holden H. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 5.Gupta MP. J Mol Cell Cardiol. 2007;43:388–403. doi: 10.1016/j.yjmcc.2007.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore JR, Leinwand L, Warshaw DM. Circ Res. 2012;111:375–385. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arrell DK, Neverova I, Fraser H, Marban E, Van Eyk JE. Circ Res. 2001;89:480–487. doi: 10.1161/hh1801.097240. [DOI] [PubMed] [Google Scholar]
- 8.Scruggs SB, Reisdorph R, Armstrong ML, Warren CM, Reisdorph N, Solaro RJ, Buttrick PM. Mol Cell Proteomics. 2010;9:1804–1818. doi: 10.1074/mcp.M110.000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregorich ZR, Peng Y, Cai WX, Jin YT, Wei LM, Chen AJ, McKiernan SH, Aiken JM, Moss RL, Diffee GM, Ge Y. J Proteome Res. 2016;15:2706–2716. doi: 10.1021/acs.jproteome.6b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murakami U, Uchida K, Hiratsuka T. J Biochem. 1976;80:611–619. doi: 10.1093/oxfordjournals.jbchem.a131316. [DOI] [PubMed] [Google Scholar]
- 11.Richards EG, Chung CS, Menzel DB, Olcott HS. Biochemistry. 1967;6:528-&. doi: 10.1021/bi00854a022. [DOI] [PubMed] [Google Scholar]
- 12.Shiverick KT, Thomas LL, Alpert NR. Biochim Biophys Acta. 1975;393:124–133. doi: 10.1016/0005-2795(75)90222-6. [DOI] [PubMed] [Google Scholar]
- 13.Wikmanco J, Zelis R, Fenner C, Mason DT. Biochem Bioph Res Co. 1973;51:1097–1104. doi: 10.1016/0006-291x(73)90040-5. [DOI] [PubMed] [Google Scholar]
- 14.Li MS, Ogilvie H, Ochala J, Artemenko K, Iwamoto H, Yagi N, Bergquist J, Larsson L. Aging Cell. 2015;14:228–235. doi: 10.1111/acel.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burniston JG, Connolly JB. Int J Cardiol. 2010;140:363–366. doi: 10.1016/j.ijcard.2008.11.058. [DOI] [PubMed] [Google Scholar]
- 16.Helmke SM, Yen CY, Cios KJ, Nunley K, Bristow MR, Duncan MW, Perryman MB. Anal Chem. 2004;76:1683–1689. doi: 10.1021/ac035144l. [DOI] [PubMed] [Google Scholar]
- 17.Zurmanova J, Malacova D, Puta F, Novak P, Ricny J, Soukup T. Phys Res. 2007;56:659–662. doi: 10.33549/physiolres.931280. [DOI] [PubMed] [Google Scholar]
- 18.Smith LM, Kelleher NL, Proteomics CTD. Nat Methods. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siuti N, Kelleher NL. Nat Methods. 2007;4:817–821. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Nature. 2011;480:254–U141. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge Y, Rybakova IN, Xu Q, Moss RL. Proc Natl Acad Sci U S A. 2009;106:12658–12663. doi: 10.1073/pnas.0813369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregorich ZR, Ge Y. Proteomics. 2014;14:1195–1210. doi: 10.1002/pmic.201300432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng Y, Gregorich ZR, Valeja SG, Zhang H, Cai W, Chen YC, Guner H, Chen AJ, Schwahn DJ, Hacker TA, Liu X, Ge Y. Mol Cell Proteomics. 2014;13:2752–2764. doi: 10.1074/mcp.M114.040675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ansong C, Wu S, Meng D, Liu X, Brewer HM, Deatherage Kaiser BL, Nakayasu ES, Cort JR, Pevzner P, Smith RD, Heffron F, Adkins JN, Pasa-Tolic L. Proc Natl Acad Sci U S A. 2013;110:10153–10158. doi: 10.1073/pnas.1221210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Compton PD, Zamdborg L, Thomas PM, Kelleher NL. Anal Chem. 2011;83:6868–6874. doi: 10.1021/ac2010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia BA. J Am Soc Mass Spectr. 2010;21:193–202. doi: 10.1016/j.jasms.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 27.Cannon J, Lohnes K, Wynne C, Wang Y, Edwards N, Fenselau C. J Proteome Res. 2010;9:3886–3890. doi: 10.1021/pr1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu SL, Kim J, Hancock WS, Karger B. J Proteome Res. 2005;4:1155–1170. doi: 10.1021/pr050113n. [DOI] [PubMed] [Google Scholar]
- 29.Fornelli L, Ayoub D, Aizikov K, Beck A, Tsybin YO. Anal Chem. 2014;86:3005–3012. doi: 10.1021/ac4036857. [DOI] [PubMed] [Google Scholar]
- 30.Moradian A, Kalli A, Sweredoski MJ, Hess S. Proteomics. 2014;14:489–497. doi: 10.1002/pmic.201300256. [DOI] [PubMed] [Google Scholar]
- 31.Valkevich EM, Sanchez NA, Ge Y, Strieter ER. Biochemistry. 2014;53:4979–4989. doi: 10.1021/bi5006305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu C, Tran JC, Zamdborg L, Durbin KR, Li M, Ahlf DR, Early BP, Thomas PM, Sweedler JV, Kelleher NL. Nat Methods. 2012;9:822–824. doi: 10.1038/nmeth.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitelegge JP, Zhang HM, Aguilera R, Taylor RM, Cramer WA. Mol Cell Proteomics. 2002;1:816–827. doi: 10.1074/mcp.m200045-mcp200. [DOI] [PubMed] [Google Scholar]
- 34.Chen YC, Ayaz-Guner S, Peng Y, Lane NM, Locher MR, Kohmoto T, Larsson L, Moss RL, Ge Y. Anal Chem. 2015;87:8399–8406. doi: 10.1021/acs.analchem.5b01745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng Y, Chen X, Zhang H, Xu Q, Hacker TA, Ge Y. J Proteome Res. 2013;12:187–198. doi: 10.1021/pr301054n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zubarev RA, Kelleher NL, McLafferty FW. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 37.Liu XW, Sirotkin Y, Shen YF, Anderson G, Tsai YS, Ting YS, Goodlett DR, Smith RD, Bafna V, Pevzner PA. Mol Cell Proteomics. 2012;11:M111.008524. doi: 10.1074/mcp.M111.008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai WX, Guner H, Gregorich ZR, Chen AJ, Ayaz-Guner S, Peng Y, Valeja SG, Liu XW, Ge Y. Mol Cell Proteomics. 2016;15:703–714. doi: 10.1074/mcp.O115.054387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guner H, Close PL, Cai W, Zhang H, Peng Y, Gregorich ZR, Ge Y. J Am Soc Mass Spectrom. 2014;25:464–470. doi: 10.1007/s13361-013-0789-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen X, Ge Y. Proteomics. 2013;13:2563–2566. doi: 10.1002/pmic.201200594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyata S, Minobe W, Bristow MR, Leinwand LA. Circ Res. 2000;86:386–390. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]
- 42.Qi YL, Barrow MP, Li HL, Meier JE, Van Orden SL, Thompson CJ, O’Connor PB. Anal Chem. 2012;84:2923–2929. doi: 10.1021/ac3000122. [DOI] [PubMed] [Google Scholar]
- 43.Kilgour DPA, Nagornov KO, Kozhinov AN, Zhurov KO, Tsybin YO. Rapid Commun Mass Sp. 2015;29:1087–1093. doi: 10.1002/rcm.7200. [DOI] [PubMed] [Google Scholar]
- 44.Michalski A, Damoc E, Hauschild JP, Lange O, Wieghaus A, Makarov A, Nagaraj N, Cox J, Mann M, Horning S. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reiser PJ, Portman MA, Ning XH, Moravec CS. Am J Physiol-Heart Circ. 2001;280:H1814–H1820. doi: 10.1152/ajpheart.2001.280.4.H1814. [DOI] [PubMed] [Google Scholar]
- 46.Krenz M, Robbins J. J Am Coll Cardiol. 2004;44:2390–2397. doi: 10.1016/j.jacc.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki T, Palmer BM, James J, Wang Y, Chen ZY, VanBuren P, Maughan DW, Robbins J, LeWinter MM. Circ-Heart Fail. 2009;2:334–341. doi: 10.1161/CIRCHEARTFAILURE.108.802298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.James J, Martin L, Krenz M, Quatman C, Jones F, Klevitsky R, Gulick J, Robbins J. Circulation. 2005;111:2339–2346. doi: 10.1161/01.CIR.0000164233.09448.B1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Izumo S, Lompre AM, Matsuoka R, Koren G, Schwartz K, Nadalginard B, Mahdavi V. J Clin Invest. 1987;79:970–977. doi: 10.1172/JCI112908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talmadge RJ, Roy RR. J Appl Physiol. 1993;75:2337–2340. doi: 10.1152/jappl.1993.75.5.2337. [DOI] [PubMed] [Google Scholar]
- 51.Warren CM, Greaser ML. Anal Biochem. 2003;320:149–151. doi: 10.1016/s0003-2697(03)00350-6. [DOI] [PubMed] [Google Scholar]
- 52.Sant’Ana Pereira JAA, Greaser M, Moss RL. Anal Biochem. 2001;291:229–236. doi: 10.1006/abio.2001.5018. [DOI] [PubMed] [Google Scholar]
- 53.Bloemberg D, Quadrilatero J. Plos One. 2012;7:e35273. doi: 10.1371/journal.pone.0035273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lemon DD, Papst PJ, Joly K, Plato CF, McKinsey TA. Anal Biochem. 2011;408:132–135. doi: 10.1016/j.ab.2010.08.041. [DOI] [PubMed] [Google Scholar]
- 55.Garcia BA, Hake SB, Diaz RL, Kauer M, Morris SA, Recht J, Shabanowitz J, Mishra N, Strahl BD, Allis CD, Hunt DF. J Biol Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 56.Tong SW, Elzinga M. J Biol Chem. 1983;258:3100–3110. [PubMed] [Google Scholar]
- 57.Hayashida M, Maita T, Matsuda G. J Biochem. 1991;110:54–59. doi: 10.1093/oxfordjournals.jbchem.a123543. [DOI] [PubMed] [Google Scholar]
- 58.Polevoda B, Sherman F. J Biol Chem. 2000;275:36479–36482. doi: 10.1074/jbc.R000023200. [DOI] [PubMed] [Google Scholar]
- 59.Abe A, Saeki K, Yasunaga T, Wakabayashi T. Biochem Bioph Res Co. 2000;268:14–19. doi: 10.1006/bbrc.1999.2069. [DOI] [PubMed] [Google Scholar]
- 60.Hansen JE, Marner J, Pavlov D, Rubenstein PA, Reisler E. Biochemistry. 2000;39:1792–1799. doi: 10.1021/bi991873c. [DOI] [PubMed] [Google Scholar]
- 61.Urbancikova M, Hitchcockdegregori SE. J Biol Chem. 1994;269:24310–24315. [PubMed] [Google Scholar]
- 62.Fedorova M, Bollineni RC, Hoffmann R. Mass Spectrom Rev. 2014;33:79–97. doi: 10.1002/mas.21381. [DOI] [PubMed] [Google Scholar]
- 63.Hao PL, Ren Y, Alpert AJ, Sze SK. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.O111.009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Simpson DC, Ahn S, Pasa-Tolic L, Bogdanov B, Mottaz HM, Vilkov AN, Anderson GA, Lipton MS, Smith RD. Electrophoresis. 2006;27:2722–2733. doi: 10.1002/elps.200600037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hummel M, Wigger T, Brockmeyer J. J Proteome Res. 2015;14:1547–1556. doi: 10.1021/pr5012262. [DOI] [PubMed] [Google Scholar]
- 66.Valeja SG, Xiu LC, Gregorich ZR, Guner H, Jin S, Ge Y. Anal Chem. 2015;87:5363–5371. doi: 10.1021/acs.analchem.5b00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chait BT. Science. 2006;314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- 68.Yang Y, Liu F, Franc V, Halim LA, Schellekens H, Heck AJR. Nat Commun. 2016;7 doi: 10.1038/ncomms13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benevento M, Tonge PA, Puri MC, Nagy A, Heck AJR, Munoz J. Proteomics. 2015;15:3219–3231. doi: 10.1002/pmic.201500031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.