

Abstract
The synthesis of a series of neomycin derivatives carrying the 2-hydroxyethyl substituent on N6′ and/or N6‴ both alone and in combination with a 4′-O-ethyl group is described. By means of cell-free translation assays with wild-type bacterial ribosomes and their hybrids with eukaryotic decoding A sites, we investigate how individual substituents and their combinations affect activity and selectivity at the target level. In principle, and as shown by cell-free translation assays, modifications of the N6′ and N6‴ positions allow to enhance target selectivity without compromising antibacterial activity. As with the 6′OH paromomycin, the 4′-O-ethyl modification further affects the ribosomal activity, selectivity, and antibacterial profile of neomycin and its 6′-N-(2-hydroxyethyl) derivatives. The modified aminoglycosides show good antibacterial activity against model Gram-positive and Gram-negative microbes including the ESKAPE pathogens S aureus, K pneumoniae, E cloacae, and A baumannii.
Keywords: aminoglycosides, ototoxicity, multidrug-resistant infectious diseases, decoding A site, cell-free translation assay, synthesis
Graphical Abstract
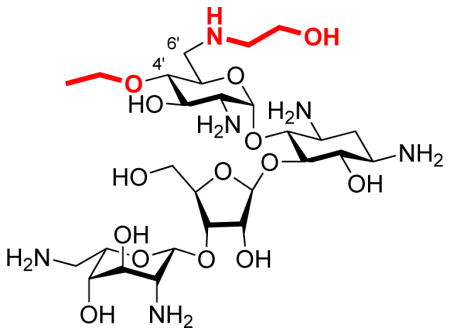
Introduction
There is currently much interest in the development of novel aminoglycoside antibiotics (AGAs) with which to combat the growing menace of multidrug resistant infectious diseases.1–9 Numerous strategies have been patented in recent years for overcoming AGA resistance determinants.10 These studies are exemplified by plazomicin 1, a semi-synthetic derivative of the natural 4,6-disubstituted 2-deoxystreptamine AGA sisomicin 2 (Figure 1), that was designed to overcome most common AGA resistance determinants and which is currently in phase III clinical trials.5,11,12 New generations of AGAs must also be engineered to overcome the typical AGA side effects of oto- and nephrotoxicity,5,13–15 even if the latter can be in part ameliorated in the clinic through the use of a once daily dosing regime.16 AGA antibacterial activity arises from binding to the decoding A site in helix 44 of the bacterial ribosome, as detailed by X-ray crystallographic and genetic studies,17–20 resulting in the disruption of bacterial protein synthesis. AGA ototoxicity is a consequence of AGA binding to the decoding A site of the mitochondrial ribosome and is particularly pernicious in patients with the A1555G mutant mitochondrial ribosome,21–23 known as the deafness allele.14,15,24,25 Improvement of AGA therapeutic indexes can be achieved by exploiting the phylogenetic differences in the nucleic acid bases forming the AGA-binding site. In practice, such AGA optimizations are enabled by the use of cell-free translation assays using a series of ribosomes and mutant hybrid ribosomes carrying either prokaryotic or eukaryotic decoding A sites.26 An alternative approach to the minimization of ototoxicity in AGAs exploits the limitations of the mechanotransducer channel responsible for AGA uptake into cochlea hair cells.27,28
Figure 1.

Some Natural and Semisynthetic Aminoglycoside Antibiotics
The observation that the unusual monosubstituted 2-deoxystreptamine AGA apramycin 3 causes little or no ototoxicity in animal models29,30 and avoids modifications by most aminoglycoside modifying enzymes (AMEs), thereby retaining excellent activity against many multidrug resistant (MDR) Gram-negative and Gram-positive organisms, encouraged us to design next generation AGAs by modification of the 4,5-disubstituted 2-deoxystreptamine AGA paromomycin 4.31,32 In particular we determined that functionalization at the paromomycin 4′-position with short alkyl chains, as in the 4′-O-ethyl derivative 5 (Figure 1), increases selectivity for the prokaryotic over the eukaryotic ribosomes, decreases oto- and nephrotoxicity, and overcomes several AME′s that act on the parent.31 In addition, like apramycin, these 4,5-AGA derivatives are little susceptible to inactivation by the 1405G ribosomal methyltransferases, which completely abrogate the activity of all 4,6-AGAs, including plazomicin.5,33–36
Having ascertained that comparable 4′-O-modifications do not result in increased ribosomal selectivity in the kanamycin class of 4,6-AGAs,37,38 we now turn our attention to the 4,5-AGA neomycin B 6. Neomycin B 6 differs from paromomycin 4 by the replacement of a single hydroxyl group at the 6′-position by an amino group, which gives rise to two problems. First, neomycin B shows significantly higher affinity for the human mitochondrial ribosome and its A1555G mutant.14,15,21,22,24,39 Second, the presence of the primary amino group at the 6′-site renders neomycin B susceptible to inactivation by the AAC(6′) class of aminoglycoside acetyltransferase (AAC) aminoglycoside modifying enzymes AMEs.5,34,40 Several studies have reported in recent years on the synthesis of improved neomycin derivatives with reduced susceptibility to AMEs, including derivatives of the 6′-amino group.8,41–49 As demonstrated originally by a Schering-Plough group45,46 and subsequently exploited by ISIS,41 and Achaogen50 6′-amino AGAs can be modified by alkylation of the 6′-amino group to overcome the effects of AAC(6′) AMEs. However, the effects of all these modifications on binding to the eukaryotic mitochondrial ribosomes and, consequently, on the ototoxicity of such derivatives are not known. We describe the synthesis of a number of neomycin B derivatives carrying the N-(2-hydroxyethyl) modification at either the 6′- and/or 6‴-positions, alone or in combination with the 4′-O-ethyl modification, and the influence of these modifications on antiribosomal and antibacterial activity. We find that the 6′-(2-hydroxyethyl) modification of neomycin B confers the advantage, beyond the anticipated suppression of the AAC(6′) AMEs, of increased ribosomal selectivity with little or no reduction of antibacterial activity. We also find that the 4′-O-ethyl substitution of neomycin B affects ribosomal selectivity in a manner comparable to that previously demonstrated for paromomycin.
Design
In targeting the neomycin B 6′-position for modification by N6′-alkylation we focused on incorporation of the 2-hydroxyethyl group, first introduced in the sisomicin series of 4,6-AGAs by workers at Schering,51,52 and a feature of plazomicin,11 as it is reportedly not detrimental to antibacterial activity. 1,2- and 1,3-Aminoalcohol modifications of the aminoglycosides are also suggested to enhance binding to phosphodiesters and nucleic acid bases, particularly to the Hoogsteen-face of guanosine.53,54 More importantly in view of the reduced basicity of 2-hydroxyalkyl amines as compared to the corresponding amines,55 we considered that the N-(2-hydroxyalkylation) would reduce the affinity of neomycin B for the mitochondrial ribosome and its deafness mutant. In view of the successful reduction in ototoxicity and nephrotoxicity wrought by modification of paromomycin at the 4′-position and the concomitant elimination of certain resistance modifications by this substitution, we targeted neomycin B derivatives modified by O-ethylation at the 4′-position and N-(2-hydroxyethylation) at the 6′-position. The synthetic schemes developed to provide access to these targeted derivatives also afforded facile access to neomycin derivatives modified at the 5″- and 6‴-positions, whose antiribosomal and antibacterial profiles were also studied.
Synthesis
4′-O-Ethyl neomycin B 11 was readily prepared from the paromomycin 4′,6′-diol 756 by selective sulfonylation of the primary alcohol with 2,4,6-tri-isopropylbenzenesulfonyl chloride to give 8, followed by displacement with azide to afford 9. Alkylation with sodium hydride and ethyl iodide then gave 10, and hydrogenolysis followed by filtration on Sephadex and lyophilization from aqueous acetic acid yielded 11 in the form of its acetate salt (Scheme 1).
Scheme 1.
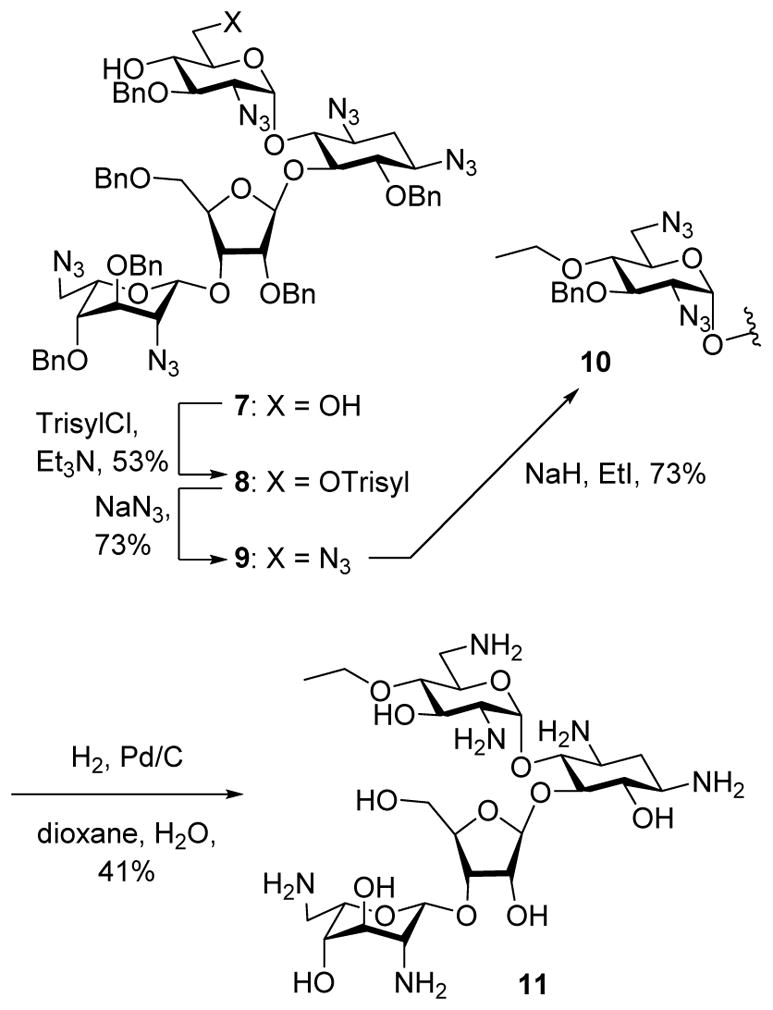
Synthesis of 4′-O-ethyl neomycin B.
Following a literature protocol,57 treatment of neomycin B with N-(benzyloxycarboxy)-succinimide and sodium carbonate in aqueous dioxane gave 32% of an inseparable 1:1 mixture of the N6′ and N6‴-benzyl carbamates 12 and 13, and 29% of the N6′,N6‴ bis(benzyl carbamate) 14 (Scheme 2). Treatment of the 12/13 mixture with imidazolesulfonyl azide58,59 and a catalytic amount of copper sulfate then afforded 53% of the combined penta-azides 15 and 16, from which the carbamate groups were removed by saponification in hot aqueous dioxane, giving a difficulty separable 1:1 mixture of 17 and 18 in 57% yield. Reductive amination of this mixture with benzyloxyacetaldehyde and sodium cyanoborohydride60 gave a 47% combined yield of the N6′ and N6‴-(2-benzyloxyethyl) derivatives 19 and 20, which were separated by preparative HPLC. The precise location of the 2′-benzyloxyethyl group on N6′ or N6‴ of 19 and 20 was determined by NMR methods, particularly Heteronuclear Multiple Bond Coherence (HMBC) correlations between the methylene groups either side of the alkylated N6′ or N6‴ (eg, C6′H2NCH2CH2OH). Finally, application of a two-step deprotection protocol, involving Staudinger reaction with trimethylphosphine and sodium hydroxide in hot aqueous THF,56 followed by hydrogenolysis gave the N6′ and N6‴ (2-hydroxyethyl) derivatives of neomycin B 21 and 22 in 23 and 26% yield (Scheme 2). Application of the same overall sequence of reactions to the N6′,N6‴ biscarbamate 14 gave the N6′,N6‴-bis(2-hydroxyethyl) neomycin derivative 26, via the intermediates 23–25 (Scheme 2).
Scheme 2.

Synthesis of the Neomycin Derivatives 21, 22, and 26.
Careful separation of the 17/18 mixture gave samples of the pure regioisomers 17 and 18. Treatment of 17 with acetic anhydride in methanol gave the acetamide 27, hydrogenolysis of which afforded an authentic sample of the known61,62 6′-N-acetylneomycin 28 (Scheme 3). Treatment of 18 with sodium nitrite in aqueous acetic acid57,63,64 resulted in 36% of the 6‴-deamino-6‴-hydroxyneomycin pentaazide 29, whose physical characteristics differed from those of the regioisomeric paromomycin pentaazide.65 Application of the Staudinger reaction to 29 then afforded the known57 N6‴-deamino-N6‴-hydroxy neomycin B derivative 30 (Scheme 3)
Scheme 3.

Synthesis of the Neomycin Derivatives 28 and 30.
Treatment of the 4′-O-ethyl paromomycin derivative 531 with imidazolesulfonyl azide and catalytic copper sulfate afforded the penta-azide 31 in 57% yield (Scheme 4). Reaction with toluenesulfonyl chloride in pyridine then gave an approximately 1:0.7 mixture of the regioisomeric tosylates 32 and 33 in 30% combined unoptimized yield and 10% of the ditosylate 34. Stirring the 32/33 mixture in ethanolamine at room temperature then gave 52% of a mixture of 35 and 36 which, after separation by preparative HPLC, were converted by Staudinger reaction to the neomycin derivative 37 and the paromomycin derivative 38 in 42 and 44% yield, respectively. Application of the same sequence of tosylate displacement by ethanolamine and Staudinger reaction to the bis(toluenesulfonate) 34 gave the neomycin derivative 40 via the intermediate 39.
Scheme 4.

Synthesis of the Neomycin Derivatives 37, 38, and 40.
Antiribosomal and Antibacterial Activity
Together with the parent neomycin 6 and the comparators paromomycin 4 and the 4′-O-ethyl paromomycin 5, compounds 11, 21, 22, 26, 28, 37, 38, and 40 (Figure 2) were screened for their ability to inhibit protein synthesis in cell-free translation assays employing wild type bacterial ribosomes and recombinant hybrid bacterial ribosomes26 carrying the complete decoding A site cassettes (Figure 3) of the human mitochondrial (Mit13), the A1555G mutant deafness allele of the human mitochondrial (A1555G), and the human cytoplasmic (Cyt14) ribosomes. The different activities of all synthetic AGAs and the comparators for the wild type bacterial ribosome versus the hybrid ribosomes are presented in Table 1. Such activities measured at the target level serve as an indicator of in vivo selectivity in the absence of significant differences in uptake between bacterial and eukaryotic cells. Compounds were also screened for antibacterial activity against methicillin resistant clinical isolates of the Gram-positive bacterium Staphylococcus aureus (MRSA) and against clinical isolates of the Gram-negative bacterium Escherichia coli (E coli) obtained from the Diagnostic Division of the Institute of Medical Microbiology (Table 2); in addition to methicillin resistance MRSA strains AG042 and AG044 are also resistant to gentamicin. No significant activity was observed against clinical isolates of the Gram-negative bacterium Pseudomonas aeruginosa (P aeruginosa). The more active compounds were also screened for activity against the ESKAPE pathogens Klebsiella pneumoniae (K pneumoniae), Enterobacter cloacae (E cloacae), and Acineto baumannii (A baumannii) (Table 3). Selected compounds were additionally screened for activity against a collection of wild type and recombinant E coli strains carrying defined resistance determinants29 in order to ascertain their susceptibility to common AMEs (Table 4).3,5,40,66
Figure 2.

Natural and Semisynthetic Aminoglycosides Screened for Antiribosomal and Antibacterial Activity and to Verify Engineered Strains of E coli.
Figure 3.

Decoding A sites of prokaryotic and eukaryotic ribosomes. The bacterial AGA binding pocket is boxed. The bacterial numbering scheme is illustrated for the AGA binding pocket. Changes from the bacterial ribosome binding pocket are coloured green. The A1555G mutant conferring hypersusceptibility to AGA ototoxicity is coloured red.
Table 1.
| Compound | IC50,
μg/mL (μM) |
Selectivity |
|||||
|---|---|---|---|---|---|---|---|
| Bacterial | Mit13 | A1555G | Cyt14 | Mit13 | A1555G | Cyt14 | |
| Paromomycin 4 | 0.02 (0.03) | 50.2 (81.8) | 5.39 (8.79) | 9.4 (15.3) | 2506 | 267 | 471 |
| Neomycin 6 | 0.01 (0.02) | 1.62 (2.62) | 0.22 (0.36) | 17.1 (27.9) | 162 | 22 | 1712 |
| 5 | 0.08 (0.12) | 99.3 (153.9) | 114.7 (177.8) | 102.9 (159.5) | 1240 | 1434 | 1286 |
| 11 | 0.01 (0.02) | 0.94 (1.47) | 2.76 (4.31) | 32.3 (50.4) | 94 | 276 | 3232 |
| 21 | 0.01 (0.02) | 13.1 (19.9) | 5.51 (8.37) | 38.3 (158.2) | 1310 | 551 | 3831 |
| 22 | 0.01 (0.02) | 9.72 (14.8) | 0.74 (1.12) | 21.5 (32.7) | 972 | 74 | 2151 |
| 30 | 0.01 (0.02) | 12.2 (19.8) | 1.13 (1.84) | 54.7 (89.2) | 1216 | 113 | 5471 |
| 37 | 0.07 (0.10) | 2.91 (4.25) | 20.3 (29.6) | 60.5 (88.4) | 42 | 289 | 865 |
| 26 | 0.04 (0.06) | 44.6 (63.3) | 11.7 (16.6) | 76.3 (108.4) | 1114 | 292 | 1908 |
| 28 | 0.16 (0.24) | 122 (185) | 606 (921) | 295 (448) | 776 | 3860 | 1879 |
| 40 | 0.64 (0.88) | 43.2 (59.1) | 47.5 (65.1) | 53.4 (73.1) | 67 | 74 | 83 |
| 38 | 1.90 (2.88) | 74.8 (109.2) | 95.8 (140) | 83.9 (122) | 39 | 50 | 44 |
IC50s are given in μg/mL and in parentheses in μM.
Selectivities are obtained by dividing the eukaryotic by the bacterial values
Table 2.
| Cmpd |
MRSA
|
E coli
|
|||||
|---|---|---|---|---|---|---|---|
| AG038 | AG039 | AG042 | AG044 | AG001 | AG055 | AG003 | |
|
|
|
||||||
| Parom 4 | 4 (7) | >256 (>417) | >256 (>417) | 4–8 (7–13) | 4 (7) | 2–4 (3–7) | 2–4 (3–7) |
| Neom 6 | 1–2 (2–3) | 128 (209) | 128 (209) | 1 (2) | 2 (3) | 1–2 (2–3) | 1 (2) |
| 5 | 8–16 (12–25) | 16 (25) | 8–16 (12–25) | 8–16 (12–25) | 64 (99) | 32–64 (50–99) | 32 (50) |
| 11 | 4 (6) | 2 (3) | 2 (3) | 1–2 (2–3) | 4 (6) | 2–4 (3–6) | 2–4 (3–6) |
| 21 | 4 (6) | >128 (>195) | >128 (>195) | 2 (3) | 4 (6) | 2–4 (3–6) | 4 (6) |
| 22 | 2 (3) | >128 (>195) | >128 (>195) | 2 (3) | 2–4 (3–6) | 4 (6) | 4 (6) |
| 30 | 4 (7) | >128 (>209) | >128 (>209) | 2 (3) | 4 (7) | 4 (7) | 4 (7) |
| 37 | 8–16 (12–23) | 8 (12) | 4–8 (6–12) | 4–8 (6–12) | 16 (23) | 8–16 (12–23) | 8–16 (12–23) |
| 26 | 8 (11) | >128 (>182) | >128 (>182) | 4 (6) | 16–32 (23–45) | 16 (23) | 16 (23) |
| 28 | 8 (12) | >64 (>97) | >64 (>97) | 8–16 (12–24) | 8 (12) | 8 (12) | 8 (12) |
| 40 | 16 (22) | 16–32 (22–44) | 16 (22) | 16 (22) | 32 (44) | 16–32 (22–44) | 16–32 (22–44) |
| 38 | 32–64 (47–93) | 32–64 (47–93) | 32 (47) | 64 (93) | 64–128 (93–187) | 64–128 (93–187) | 32–64 (93–187) |
All values were determined in duplicate using two dilution series.
MICs are given in μg/mL and in parentheses in μM.
Table 3.
| Cmpd |
K pneumoniae
|
E cloacae
|
A baumannii.
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AG261 | AG262 | AG263 | AG290 | AG291 | AG292 | AG225 | AG226 | AG286 | |||
|
|
|
|
|||||||||
| Parom 4 | 1 (2) | 1 (2) | 1 (2) | 2 (3) | 1–2 (2–3) | 2 (3) | 4 (7) | 2–4 (3–7) | 1–2 (2–3) | ||
| Neom 6 | 0.5 (0.8) | 0.5 (0.8) | 1 (2) | 1 (2) | 1 (2) | 1 (2) | 4 (7) | 4 (7) | 0.5–1 (0.8–2) | ||
| 5 | 4 (6) | 8 (12) | 8 (12) | 8–16 (12–25) | 8–16 (12–25) | 8 (12) | 16 (25) | 16 (25) | 32 (25) | ||
| 11 | 0.5 (0.8) | 1 (2) | 1 (2) | 1 (2) | 1 (2) | 1 (2) | 4 (6) | 4 (6) | 1–2 (2–3) | ||
| 21 | 0.5–1 (1–2) | 1 (2) | 0.5 (1) | 1 (2) | 1 (2) | 1 (2) | 2 (3) | 2 (3) | 1 (2) | ||
| 22 | 0.5 (0.8) | 0.5 (0.8) | 1 (2) | 1 (2) | 0.5 (0.8) | 0.5 (0.8) | 2 (3) | 2 (3) | 0.5 (0.8) | ||
| 30 | 0.5 (0.8) | 0.5 (0.8) | 1 (2) | 1 (2) | 0.5–1 (0.8–2) | 2 (3) | 2 (3) | 2 (3) | 1 (2) | ||
| 37 | 2–4 (3–6) | 4 (6) | 4 (6) | 8 (12) | 4 (6) | 4 (6) | 8–16 (12–23) | 4–8 (6–12) | 8 (12) | ||
All values were determined in duplicate using two dilution series.
MICs are given in μg/mL and in parentheses in μM.
Table 4.
Antimicrobial Data Against Wild Type and Engineered Strains of E coli Carrying Specific Resistance Determinants (MIC, μg/mL, (μM))a,b.
| Strain | BM13 (AG006) | AG007 | AG105 | AG009 | AG036 | AG037 |
|---|---|---|---|---|---|---|
| Resistance Mechanism | Wild type | AAC(3) | AAC(2′) | AAC(6′) | ANT(4′, 4″) | APH(3′, 5″) |
| Gentamicinc | 0.5 (1) | 32 (67) | 32 (67) | -d | 0.5 (1) | 0.25 (0.5) |
| Kanamycin Ac | 1–2 (2–4) | -d | 1–2 (2–4) | 128 (265) | 16 (33) | >256 (>530) |
| Tobramycinc | 0.5 (1) | -d | -d | 64 (137) | 16 (34) | 1 (2) |
| Neomycin 6 | 1 (2) | 4 (7) | 2 (3) | 8 (13) | 32 (52) | >256 (>417) |
| 22 | 1 (2) | 2 (3) | 2 (3) | 16 (24) | 32 (49) | >256 (>389) |
| 21 | 1 (2) | 2 (3) | 2 (3) | 2 (3) | 64 (97) | >256 (>389) |
| 28 | 4–8 (6–12) | 16 (12) | 4 (6) | 16 (12) | >64 (>24) | >256 (>389) |
| 30 | 1 (2) | 2 (3) | 2–4 (3–7) | 64 (104) | 128 (207) | >256 (>414) |
| 37 | 2–4 (3–6) | 8–16 (12–23) | 8 (12) | 16 (23) | 4 (6) | >256 (>374) |
| 11 | 0.5 (0.8) | 2 (3) | 2 (3) | 32–64 (50–100) | 0.5 (0.8) | >256 (>399) |
| 26 | 2 (3) | 8 (11) | -d | 8–16 (11–23) | >64 (>93) | >256 (>364) |
| 40 | 4 (5) | 8 (11) | -d | 16–32 (22–44) | 8 (11) | >256 (>351) |
All values were determined in duplicate using two dilution series.
MICs are given in μg/mL and in parentheses in μM.
These AGAs were used to verify the resistance phenotype type of the recombinant strains.
Not determined
Discussion
Influence of Substituents on Antibacterioribosomal Activity
The inhibitory activity of neomycin against the bacterial ribosome is not affected by the introduction of an ethyl group onto the 4′-hydroxy group, or by a 2-hydroxyethyl substituent onto either the 6′- or the 6‴-amino group (11, 21 and 22, Table 1). Neither is it affected by the replacement of the 6‴-amino by a hydroxy group (30, Table 1), consistent with earlier observations on the antibacterial activity of 3057,67 and with more recent work using the 6‴-amino group of neomycin as a viable locus of derivatization through amide bond formation.68 Conversely, the analogous replacement of the 6‴-amino by a hydroxy group in paromomycin is reported to produce a significant loss of antibacterial activity.57,67 The lack of effect of these modifications on the antibacterioribosomal activity of neomycin contrasts with the effect of introducing a 4′-O-ethyl group onto paromomycin which results in a four-fold reduction of activity (5, Table 1). Presumably, in the neomycin series the presence of the 6′-amino group is able to compensate for any reduction in antiribosomal activity due to the modifications made at the 4′- and 6‴-positions in 11, 22, and 30, whereas the corresponding modifications in the paromomycin series, with the 6′-hydroxy group, cause a significant loss of activity. This difference between the neomycin and paromomycin series arises from the strength of the critical hydrogen bond between the 6′-substituent and N1 of A1408 in the decoding A site. In the neomycin series this H bond involves an ammonium ion as donor, and is estimated to be ≥3 kcal.mol−1 stronger than the 6′-OH-A1408 H bond in the paromomycin complex.69 It is noteworthy that the 6′-N-2-hydroxyethyl group in 21 does not impact the critical H bond to A1408 to an extent detectable in the antibacterioribosomal assay, despite the ~1 pKa unit reduction in basicity it imposes on the 6′-amine. In contrast to alkylation, acetylation of the 6′-amine in neomycin to give the amide 28 has a significant adverse effect on binding to the bacterial ribosome consistent with the AAC(6′) mechanism of aminoglycoside resistance.1–9
Although neither the 6′-N-2-hydroxyethyl, nor the 4′-O-ethyl, or the 6‴-N-2-hydroxyethyl modifications of neomycin are sufficient by themselves to bring about a measureable reduction in antibacterioribosomal activity, the combination of two such modifications in a single molecule, as in 37 and 26 does bring about a marked reduction in activity (Table 1). Necessarily, therefore, each of these individual modifications does have a small detrimental effect on binding, although this is not sufficient to affect IC50 activity. Indeed, comparison of the antibacterioribosomal (Table 1) and antibacterial (Table 2) activities of 37 and 5, both of which carry the 4′-O-ethyl group, suggests that the influence of the 6′-N-2-hydroxyethyl substituent on binding approaches that of the 6′-hydroxyl group.
Influence of Substituents on Ribosomal Selectivity
To facilitate comparison of the influence of each synthetic modification on ribosomal selectivity, the experimental data presented in Table 1 have been converted into selectivity quotients (SelQ) with respect to the parent neomycin as presented in Table 5. In this Table, compounds with single modifications are listed first (Table 5, entries 1–6) in order of decreasing SelQ over the mitochondrial ribosome, followed by compounds with two modifications (Table 5, entries 7–9) and those with three modifications (Table 5, entries 10 and 11).
Table 5.
Change in Antiribosomal Selectivity (Selectivity Quotient) with Respect to the Parent Neomycin.
| Entry | Compound | Selectivity Quotient
(SelQ)a
|
||
|---|---|---|---|---|
| Mit13 | A1555G | Cyt 14 | ||
| 1 | paromomycin 4 | 15.47 | 12.14 | 0.28 |
| 2 | 21 | 8.09 | 25.05 | 2.24 |
| 3 | 30 | 7.51 | 5.14 | 3.20 |
| 4 | 22 | 6.00 | 3.37 | 1.26 |
| 5 | 28 | 4.5 | 176 | 1.1 |
| 6 | 11 | 0.58 | 12.55 | 1.89 |
| 7 | 5 | 7.66 | 65.18 | 0.75 |
| 8 | 26 | 6.88 | 13.28 | 1.12 |
| 9 | 37 | 0.26 | 13.14 | 0.51 |
| 10 | 40 | 0.42 | 3.37 | 0.05 |
| 11 | 38 | 0.24 | 2.27 | 0.03 |
SelQ = selectivity derivative/selectivity parent.
Compared to neomycin, paromomycin exhibits a ten to fifteen-fold greater selectivity for the bacterial over the mitochondrial and mutant mitochondrial ribosomes, but reduced selectivity over the cytoplasmic ribosome (Table 5, entry 1). The stronger A1408 N1-6′-NH3+ H bond in the neomycin-target interaction in part compensates for the loss of activity due to the G1491A substitution (Figure 3) at the base of the binding pocket on going from the bacterial to the mitochondrial ribosomes, whereas the weaker A1408 N1-6′-OH H bond in the paromomycin complex is less adept at this.69 The reduced selectivity of paromomycin for the bacterial over the cytoplasmic decoding A site is due to the 6′-hydroxy group of paromomycin accepting a hydrogen bond from G1408 in the cytoplasmic binding site in contrast to the repulsive interaction between the protonated 6′-amino group of neomycin and G140819,70 The incorporation of a 6′-N-2-(hydroxyethyl) group on neomycin increases selectivity for the bacterial over the mutant mitochondrial ribosome to a greater extent than over the wild type mitochondrial ribosome (21, Table 5, entry 2). Possibly, the looser mitochondrial binding site with two consecutive non-cognate base pairs at the foot of the binding site better accommodates the 2-hydroxyethyl group on N6′ than the less flexible mutant mitochondrial binding with a single non-Watson Crick pair in the same location. The modest increase in selectivity of 21 for the bacterial over the cytoplasmic ribosome can be attributed to increased repulsion with the G1408 in the cytoplasmic binding site, already present in neomycin,19,70 presumably for steric reasons. Overall, the simple introduction of a 2-hydroxyethyl group onto N6′ of neomycin increases the selectivity mainly for the bacterial over the mitochondrial and mutant mitochondrial ribosomes. A moderate increase in selectivity over the mitochondrial wild type ribosome is observed with the 6′-acetamide 28 (Table 5, entry 5), which contrasts with the large increase in selectivity seen for the mutant mitochondrial ribosome. Evidently, the more rigid mutant mitochondrial ribosome is less able to adapt to the presence of the 6′acetamide than the wild type mitochondrial ribosome. Overall, the exploration of other substituents on N6′ of the neomycin class of 4,5-AGAs may be a fruitful avenue for future explorations both alone and in combination with other recently described modifications known to enhance antibacterial activity.8,41–49
Modifications to the 6‴-position of neomycin as in the 6‴-deamino-6‴-hydroxy and 6‴-N-2-hydroxyethyl derivatives 30 and 22, respectively, cause relatively modest changes to the ribosomal selectivity pattern (Table 5, entries 3 and 4) just as they do not significantly influence affinity for the bacterial ribosome itself (Table 1). This is consistent with ring IV of the 4,5-AGAs serving mainly as an electrostatic anchor to the decoding A site,38,71,72 rather than through any critical specific interactions, and with a single ammonium group in ring IV being sufficient in the neomycin, if not in the less aminated paromomycin.57,67
Ethylation of the 4′-hydroxy group of neomycin (11, Table 5, entry 6) causes little reduction in SelQ for the mitochondrial ribosome but an increase in SelQ for the mutant mitochondrial ribosomes. These changes in selectivity with respect to the parent qualitatively mirror those seen on 4′-O-ethylation of paromomycin 5. Unfortunately, the inherent selectivity of neomycin for the bacterial over the mutant mitochondrial ribosome (Table 1) is low, such that even the increase in selectivity affected by the introduction of the 4′-O-ethyl group will not reduce toxicity sufficiently for 11 to be an interesting compound in its own right. This stands in contrast to 5, the 4′-O-ethyl analogue of paromomycin, whose ribosomal selectivity profile is comparable to apramycin, and which does not cause significant ototoxicity in animal models.31
Turning to doubly modified derivatives, 5 can be considered as a neomycin analogue that incorporates both the 6′-hydroxy in place of the 6′-amino and the 4′-O-ethyl group. When viewed in this manner it is apparent that the effects of the two individual modifications (Table 5, entries 1 and 6) synergize effectively (Table 5, entry 7). In contrast, the combination of the 6′-N-2-(hydroxyethyl) and 6‴-N-2-(hydroxyethyl) modifications into a single molecule, 26, does not produce a synergistic improvement in the selectivity quotient (Table 5, entry 8), nor does the combination of the 6′-N-2-(hydroxyethyl) and 4′-O-ethyl modification in 37 (Table 5, entry 9).
Compounds 38 and 40 are triply modified neomycin derivatives that are byproducts of the synthetic schemes; they correspond to the doubly modified analogs 5 and 37 whose 5″-hydroxy groups have been replaced by ethanolamine residues linked via nitrogen. The most striking feature of these two compounds is the marked decrease in selectivity they display for the bacterial over the cytoplasmic ribosomes (Table 5, entries 11 and 10). This is due to an increase in affinity for the cytoplasmic ribosome that reflects that seen in 5″-amino-5″-deoxy derivatives of the 4,5-AGA ribostamycin by the Baasov group, and which forms the basis of the suggested application of such compounds for the read-through treatment of diseases such as cystic fibrosis and the treatment of infections by protozoal parasites such as Leishmaniosis.73–75
Antibacterial Activity
Most of the single modifications of neomycin displayed antibacterial activity comparable to that of the parent against MRSA and E coli (Table 2), and against the ESKAPE pathogens K pneumoniae, E cloacae, and A baumannii (Table 3). In addition, the 4′-O-ethyl derivative 11 shows strong activity against two clinical strains of MRSA that are resistant to neomycin and to paromomycin. In this respect 11 functions analogously to the 4′-O-ethyl derivative of paromomycin 5 and other 4′-O-substituted paromomycin derivatives,32 and also 4′-substituted kanamycin derivatives,38,76 by circumventing the activity of the AME responsible for the loss of activity in the parent.
Double and triple modifications of neomycin resulted in a greater loss of antibacterial activity than any single modification. Nevertheless, it is noteworthy that those compounds carrying the 4′-O-ethyl modification retain activity against neomycin-resistant strains of MRSA (Table 2), and in particular that 4′-O-ethyl-6′-N-(2-hydroxyethyl) neomycin 37 displays strong activity against the ESKAPE pathogens K pneumoniae, E cloacae, and A baumannii (Table 3).
Influence of Aminoglycoside Modifying Enzymes on Antibacterial Activity
Consistent with the precedent in the paromomycin and other series,31,32,38,76 derivatization of the 4′-hydroxy group as in 11 and 37 overcomes resistance due to the presence of the aminoglycoside nucleotidyltranferase ANT(4′,4″) (Table 4). Consistent with the lack of susceptibility of the parent, and the general lower susceptibility of the 4,5-AGAs than the 4,6-AGAs to AACs,35 none of the derivatives screened were significantly inactivated by the presence of either the AAC(3) or AAC(2′) resistance determinants. Unfortunately, none of the modifications enacted provided protection against the aminoglycoside phosphotransferase APH(3′,5″) mechanism of resistance.
With regard to the AAC(6′) resistance determinant, the antibacterial activity of the parent neomycin is reduced some eight fold in the presence of this AME (Table 4). This is consistent with acetylation of the 6′-position of neomycin not resulting in complete abrogation of activity as has been repeatedly reported for enzymatically-derived material, and as substantiated here with an authentic sample of the 6′-acetamide 28.61,62,77 A greater loss of activity in the presence of the AAC(6′) AME is seen for derivatives 11, 22 and 30 consistent with the notion that, while 6′-N-acetylation of neomycin is tolerated to some extent, the incorporation of a second modification causes a more significant loss of activity.77 This pattern fits the general picture discussed above according to which double modifications of the neomycin framework abrogate ribosomal binding and antibacterial activity to a greater extent than single modifications. When the 6′-position is rendered inaccessible to modification by AAC(6′) by introduction of the 6′-(2-hydroxyethyl) ethyl group (21, 26, 37 and 40) susceptibility toward the engineered strain approaches that toward the wild type (Table 3), in contrast to the more substantial loss of activity seen with compounds retaining the 6′-primary amino group.
Conclusion
Introduction of a 2-aminoethyl substituent on neomycin B N6′ suppresses the action of the AAC(6′) resistance determinant, has no significant effect on antibacterial activity, and leads to enhanced selectivity for the bacterial ribosomal decoding A site over mitochondrial and mutant mitochondrial decoding A sites, indicating that this modification has multiple beneficial effects. Ethylation of the 4′-hydroxy group of neomycin B induces an increase in selectivity for prokaryotic over eukaryotic ribosomes, that is comparable to effect of the identical modification of paromomcyin, and has little influence on antibacterial activity against model Gram-negative and Gram-positive microbes. The 4′-O-ethyl modification of neomycin B has the further advantage of conferring neomycin resistance to the ANT(4′,4″) class of AMEs. Double and triple modifications of the neomycin B framework, introduced with a view to further enhancing ribosomal selectivity, offer few advantages as they generally result in reduced antibacterial activity.
Supplementary Material
Acknowledgments
We thank the NIH (AI123352), the University of Zurich and Wayne State University for support and Professor Patrice Courvalin of the Institut Pasteur for recombinant strains of E coli. We acknowledge support from the NSF (MRI-084043) for the purchase of the 600 MHz NMR spectrometer in the Lumigen Instrument Center at Wayne State University.
Footnotes
Supporting Information Available. Full experimental details and copies of 1H and 13C NMR spectra for the new AGAs (37–39, 42, 44, 50, and 51). This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Vakulenko SB, Mobashery S. Clin Microbiol Rev. 2003;16:430. doi: 10.1128/CMR.16.3.430-450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang J, Chang C-WT. In: Aminoglycoside Antibiotics. Arya DP, editor. Wiley; Hobeken: 2007. p. 141. [Google Scholar]
- 3.Houghton JL, Green KD, Chen W, Garneau-Tsodikova S. ChemBioChem. 2010;11:880. doi: 10.1002/cbic.200900779. [DOI] [PubMed] [Google Scholar]
- 4.Becker B, Cooper MA. ACS Chem Biol. 2013;8:105. doi: 10.1021/cb3005116. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong ES, Kostrub CF, Cass RT, Moser HE, Serio AW, Miller GH. In: Antibiotic Discovery and Development. Dougherty TJ, Pucci MJ, editors. Springer Science+Business Media; 2012. p. 229. [Google Scholar]
- 6.Fosso MY, Li Y, Garneau-Tsodikova S. Med Chem Commun. 2014;5:1075. doi: 10.1039/C4MD00163J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright GD. ACS Infect Dis. 2015;1:80. doi: 10.1021/id500052s. [DOI] [PubMed] [Google Scholar]
- 8.Maianti JP, Hanessian S. Med Chem Commun. 2016;7:170. [Google Scholar]
- 9.Labby KJ, Garneau-Tsodikova S. Fut Med Chem. 2013;5:1285. doi: 10.4155/fmc.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandrika NT, Garneau-Tsodikova S. MedChemCommun. 2016;7:50. doi: 10.1039/C5MD00453E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aggen JB, Armstrong ES, Goldblum AA, Dozzo P, Linsell MS, Gliedt MJ, Hildebrandt DJ, Feeney LA, Kubo A, Matias RD, Lopez S, Gomez M, Wlasichuk KB, Diokno R, Miller GH, Moser HE. Antimicrob Agent Chemother. 2010;54:4636. doi: 10.1128/AAC.00572-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhanel GG, Lawson CD, Zelenitsky S, Findlay B, Schweizer F, Adam H, Walkty A, Rubinstein E, Gin AS, Hoban DJ, Lynch JP, Karlowsky JA. Expert Rev Anti-infect Ther. 2012;10:459. doi: 10.1586/eri.12.25. [DOI] [PubMed] [Google Scholar]
- 13.Talaska AE, Schacht J. In: Aminoglycoside Antibiotics: From Chemical Biology to Drug Discovery. Arya DP, editor. Wiley; Hoboken: 2007. p. 255. [Google Scholar]
- 14.Böttger EC, Schacht J. Hearing Research. 2013;303:12. doi: 10.1016/j.heares.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huth ME, Ricci AJ, Cheng AG. Int J Otolaryngol. 2011:937861. doi: 10.1155/2011/937861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drusano GL, Ambrose PG, Bhavnani SM, Bertino JS, Nafziger AN, Louie A. Clin Infect Dis. 2007;45:753. doi: 10.1086/520991. [DOI] [PubMed] [Google Scholar]
- 17.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Nature. 2000;407:340. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 18.Wasserman MR, Pulk A, Zhou Z, Altman RB, Zinder JC, Green KD, Garneau-Tsodikova S, Doudna Cate JH, Blanchard SC. Nat Commun. 2015;6:7896. doi: 10.1038/ncomms8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vicens Q, Westhof E. Biopolymers. 2003;70:42. doi: 10.1002/bip.10414. [DOI] [PubMed] [Google Scholar]
- 20.Hobbie SN, Pfister P, Bruell C, Sander P, François B, Westhof E, Böttger EC. Antimicrob Agents Chemother. 2006;50:1489. doi: 10.1128/AAC.50.4.1489-1496.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prezant TR, Agapian JV, Bohlman MC, Bu X, Öztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, Shohat M, Fischel-Ghodsian N. Nat Genetics. 1993;4:289. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- 22.Hobbie SN, Akshay S, Kalapala SK, Bruell C, Shcherbakov D, Böttger EC. Proc Natl Acad Sci, USA. 2008;105:20888. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hobbie SN, Bruell CM, Akshay S, Kalapala SK, Shcherbakov D, Böttger EC. Proc Natl Acad Sci, USA. 2008;105:3244. doi: 10.1073/pnas.0707265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akbergenov R, Shcherbakov D, Matt T, Duscha S, Meyer M, Perez-Fernandez D, Pathak R, Harish S, Kudyba I, Dubbaka SR, Silva S, Ruiz Ruiz M, Salian S, Vasella A, Böttger EC. In: Ribosomes: Structure, Function, and Dynamics. Rodnina MV, Wintermeyer W, Green R, editors. Springer-Verlag; Vienna: 2011. p. 249. [Google Scholar]
- 25.Shulman E, Belakhov V, Wei G, Kendall A, Meyron-Holtz EG, Ben-Shachar D, Schacht J, Baasov T. J Biol Chem. 2014;289:2318. doi: 10.1074/jbc.M113.533588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hobbie SN, Kalapala SK, Akshay S, Bruell C, Schmidt S, Dabow S, Vasella A, Sander P, Böttger EC. Nucl Acids Res. 2007;35:6086. doi: 10.1093/nar/gkm658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alharazneh A, Luk L, Huth M, Monfared A, Steyger PS, Cheng AG, Ricci AJ. PLoS One. 2011;6(7):e22347. doi: 10.1371/journal.pone.0022347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huth ME, Han KH, Sotoudeh K, Hsieh YJ, Effertz T, Vu AA, Verhoeven S, Hsieh MH, Greenhouse R, Cheng AG, Ricci AJ. J Clin Invest. 2015;125:583. doi: 10.1172/JCI77424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matt T, Ng CL, Lang K, Sha SH, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Xie J, Dubbaka SR, Perez-Fernandez D, Vasella A, Ramakrishnan V, Schacht J, Böttger EC. Proc Natl Acad Sci, USA. 2012;109:10984. doi: 10.1073/pnas.1204073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyer M, Freihofer P, Scherman M, Teague J, Lenaerts A, Böttger EC. Antimicrob Agent Chemother. 2014;54:6938. doi: 10.1128/AAC.03239-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duscha S, Boukari H, Shcherbakov D, Salian S, Silva S, Kendall A, Kato T, Akbergenov R, Perez-Fernandez D, Bernet B, Vaddi S, Thommes P, Schacht J, Crich D, Vasella A, Böttger EC. mBio. 2014;5 doi: 10.1128/mBio.01827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsushita T, Chen W, Juskeviciene R, Teo Y, Shcherbakov D, Vasella A, Böttger EC, Crich D. J Am Chem Soc. 2015;137:7706. doi: 10.1021/jacs.5b02248. [DOI] [PubMed] [Google Scholar]
- 33.Livermore DM, Mushtaq S, Warner M, Zhang JC, Maharjan S, Doumith M, Woodford N. J Antimicrob Chemother. 2011;66:48. doi: 10.1093/jac/dkq408. [DOI] [PubMed] [Google Scholar]
- 34.Garneau-Tsodikova S, Labby KJ. Med Chem Commun. 2016;7:11. doi: 10.1039/C5MD00344J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bacot-Davis VR, Bassenden AV, Berghuis IM. Med Chem Commun. 2016;7:103. [Google Scholar]
- 36.Bassenden AV, Rodionov D, Shi K, Berghuis AM. ACS Chem Biol. 2016;11:1339. doi: 10.1021/acschembio.5b01070. [DOI] [PubMed] [Google Scholar]
- 37.Salian S, Matt T, Akbergenov R, Harish S, Meyer M, Duscha S, Shcherbakov D, Bernet BB, Vasella A, Westhof E, Böttger EC. Antimicrob Agent Chemother. 2012;56:6104. doi: 10.1128/AAC.01326-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato T, Yang G, Teo Y, Juskeviciene R, Perez-Fernandez D, Shinde HM, Salien S, Bernet B, Vasella A, Böttger EC, Crich D. ACS Infect Dis. 2015;1:479. doi: 10.1021/acsinfecdis.5b00069. [DOI] [PubMed] [Google Scholar]
- 39.Qian Y, Guan MX. Antimicrob Agent Chem. 2009;53:4612. doi: 10.1128/AAC.00965-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magnet S, Blanchard JS. Chem Rev. 2005;105:477. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 41.Swayze EE, Hanessian S, Szychowski J, Adhikari SS, Pachamuthu K, Wang X, Migawa MT, Griffey RH. 2007064954 (A2). WO. doi: 10.1002/anie.200462092. [DOI] [PubMed]
- 42.Hainrichson M, Pokrovskaya V, Shallom-Shezifi D, Fridman M, Belakhov V, Shachar D, Yaron S, Baasov T. Bioorg Med Chem. 2005;13:5797. doi: 10.1016/j.bmc.2005.05.058. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Chiang FI, Wu L, Czyryca PG, Li D, Chang CWT. J Med Chem. 2008;51:7563. doi: 10.1021/jm800997s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J, Keller K, Takemoto JY, Bensaci M, Litke A, Czyryca PG, Chang CWT. J Antibiotics. 2009;62:539. doi: 10.1038/ja.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanessian S, Saavedra OM, Vilchis-Reyes MA, Maianti JP, Kanazawa H, Dozzo P, Matias RD, Serio A, Kondo J. Chem Sci. 2014;5:4621. [Google Scholar]
- 46.Maianti JP, Kanazawa H, Dozzo P, Matias RD, Feeney LA, Armstrong ES, Hildebrandt DJ, Kane TR, Gliedt MJ, Goldblum AA, Linsell MS, Aggen JB, Kondo J, Hanessian S. ACS Chem Biol. 2014;9:2067. doi: 10.1021/cb5003416. [DOI] [PubMed] [Google Scholar]
- 47.Watkins D, Kumar S, Green KD, Arya DP, Garneau-Tsodikova S. Antimicrob Agents Chem. 2015;59:3899. doi: 10.1128/AAC.00861-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin Y, Watkins D, Degtyareva NN, Green KD, Spano MN, Garneau-Tsodikova S, Arya DP. MedChemCommun. 2016 doi: 10.1039/C5MD00427F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanessian S, Giguere A, Grzyb J, Maianti JP, Saavedra, Aggen JB, Linsell MS, Goldblum AA, Hildebrandt DJ, Kane TR, Dozzo P, Gliedt MJ, Matias RD, Feeney LA, Armstrong ES. ACS Med Chem Lett. 2011;2:924. doi: 10.1021/ml200202y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldblum AA, Dozzo P, Kane TR, Aggen JB, Linsell MS, Hildebrandt DJ, Gliedt MJ. 2012/0122809 A1. US.
- 51.Nagabhushan TL. 3,997,524. US.
- 52.Davies DH, Mallams AK. J Med Chem. 1978;21:189. doi: 10.1021/jm00200a009. [DOI] [PubMed] [Google Scholar]
- 53.Hendrix M, Alper PB, Priestley ES, Wong CH. Angew Chem Int Ed. 1997;36:95. [Google Scholar]
- 54.Greenberg WA, Priestley ES, Sears PS, Alper PB, Rosenbohm C, Hendrix M, Hung SC, Wong CH. J Am Chem Soc. 1999;121:6527. [Google Scholar]
- 55.Morgenthaler M, Schweizer E, Hoffmann-Roder A, Benini F, Martin RE, Jaeschke G, Wagner B, Fischer H, Bendels S, Zimmerli D, Schneider J, Diederich F, Kansy M, Muller K. ChemMedChem. 2007;2:1100. doi: 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- 56.Pathak R, Perez-Fernandez D, Nandurdikar R, Kalapala SK, Böttger EC, Vasella A. Helv Chim Acta. 2008;91:1533. [Google Scholar]
- 57.Toda S, Nakagawa S, Naito T, Kawaguchi H. J Antibiotics. 1983;36:87. doi: 10.7164/antibiotics.36.87. [DOI] [PubMed] [Google Scholar]
- 58.Goddard-Borger ED, Stick RV. Org Lett. 2007;9:3797. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 59.Potter GT, Jayson GC, Miller GJ, Gardiner JM. J Org Chem. 2016;81:3443. doi: 10.1021/acs.joc.6b00177. [DOI] [PubMed] [Google Scholar]
- 60.Borch RF, Bernstein MD, Durst HD. J Am Chem Soc. 1971;93:2897. [Google Scholar]
- 61.Benveniste R, Davies J. Biochemistry. 1971;10:1787. doi: 10.1021/bi00786a009. [DOI] [PubMed] [Google Scholar]
- 62.Zhu CB, Sunada A, Ishikawa J, Ikeda Y, Kondo S, Hotta K. J Antibiotics. 1999;52:889. doi: 10.7164/antibiotics.52.889. [DOI] [PubMed] [Google Scholar]
- 63.Cassinelli G, Julita P, Arcamone F. J Antibiotics. 1978;31:382. doi: 10.7164/antibiotics.31.382. [DOI] [PubMed] [Google Scholar]
- 64.Williams JM. Adv Carbohydr Chem Biochem. 1975;31:9. [Google Scholar]
- 65.Pathak R, Böttger EC, Vasella A. Helv Chim Acta. 2005;88:2967. [Google Scholar]
- 66.Ramirez MS, Tolmasky ME. Drug Resist Updates. 2010;13:151. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Autissier D, BarthelemyY P, Mazieres N, Peyre M, Penasse L. J Antibiotics. 1981;34:536. doi: 10.7164/antibiotics.34.536. [DOI] [PubMed] [Google Scholar]
- 68.Shaul P, Green KD, Rutenberg R, Kramer M, Berkov-Zrihen Y, Breiner-Goldstein E, Garneau-Tsodikova S, Fridman M. Org Biomol Chem. 2011;9:4057. doi: 10.1039/c0ob01133a. [DOI] [PubMed] [Google Scholar]
- 69.Pfister P, Hobbie S, Brüll C, Corti N, Vasella A, Westhof E, Böttger EC. J Mol Biol. 2005;346:467. doi: 10.1016/j.jmb.2004.11.073. [DOI] [PubMed] [Google Scholar]
- 70.Perez-Fernandez D, Shcherbakov D, Matt T, Leong NC, Kudyba I, Duscha S, Boukari H, Patak R, Dubbaka SR, Lang K, Meyer M, Akbergenov R, Freihofer P, Vaddi S, Thommes P, Ramakrishnan V, Vasella A, Böttger EC. Nature Commun. 2014;5:4112/1. doi: 10.1038/ncomms4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hermann T, Westhof E. J Med Chem. 1999;42:1250. doi: 10.1021/jm981108g. [DOI] [PubMed] [Google Scholar]
- 72.Alper PB, Hendrix M, Sears P, Wong CH. J Am Chem Soc. 1998;120:1965. [Google Scholar]
- 73.Kandasamy J, Atia-Glikin D, Shulman E, Shapira K, Shavit M, Belakhov V, Baasov T. J Med Chem. 2012;55:10630. doi: 10.1021/jm3012992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shalev M, Baasov T. Med Chem Commun. 2014;5:1092. doi: 10.1039/C4MD00081A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shalev M, Rozenberg H, Smolkin B, Nasereddin A, Kopelyanskiy D, Belakhov V, Schrepfer T, Schacht J, Jaffe CL, Adir N, Baasov T. Nucl Acid Res. 2015;43:8601. doi: 10.1093/nar/gkv821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yan RB, Yuan M, Wu Y, You X, Ye XS. Bioorg Med Chem. 2011;19:30. doi: 10.1016/j.bmc.2010.11.065. [DOI] [PubMed] [Google Scholar]
- 77.Green KD, Chen W, Houghton JL, Fridman M, Garneau-Tsodikova S. ChemBioChem. 2010;11:119. doi: 10.1002/cbic.200900584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.