

Abstract
Background
The serine protease HtrA is an important factor for regulating stress responses and protein quality control in bacteria. In recent studies, we have demonstrated that the gastric pathogen Helicobacter pylori can secrete HtrA into the extracellular environment, where it cleaves-off the ectodomain of the tumor suppressor and adherens junction protein E-cadherin on gastric epithelial cells.
Results
E-cadherin cleavage opens cell-to-cell junctions, allowing paracellular transmigration of the bacteria across polarized monolayers of MKN-28 and Caco-2 epithelial cells. However, rapid research progress on HtrA function is mainly hampered by the lack of ΔhtrA knockout mutants, suggesting that htrA may represent an essential gene in H. pylori. To circumvent this major handicap and to investigate the role of HtrA further, we overexpressed HtrA by introducing a second functional htrA gene copy in the chromosome and studied various virulence properties of the bacteria. The resulting data demonstrate that overexpression of HtrA in H. pylori gives rise to elevated rates of HtrA secretion, cleavage of E-cadherin, bacterial transmigration and delivery of the type IV secretion system (T4SS) effector protein CagA into polarized epithelial cells, but did not affect IL-8 chemokine production or the secretion of vacuolating cytotoxin VacA and γ-glutamyl-transpeptidase GGT.
Conclusions
These data provide for the first time genetic evidence in H. pylori that HtrA is a novel major virulence factor controlling multiple pathogenic activities of this important microbe.
Keywords: Adherens junction, Tight junction, E-cadherin, Helicobacter pylori, Protease, HtrA, Type IV secretion system, T4SS, CagA, EPIYA, Src, Abl, Integrin, Transwell
Background
Helicobacter pylori is a Gram-negative, flagellated pathogen, which persistently colonizes the human stomach [1, 2]. About 50% of the world population carries these bacteria, and infections are associated with chronic, often asymptomatic gastritis in all infected individuals. However, more severe gastric diseases such as peptic ulceration, mucosa-associated lymphoid tissue (MALT) lymphoma and gastric adenocarcinoma can arise in a subset of patients [3, 4]. The clinical outcome of H. pylori infection is regulated by several key elements including the genetic predisposition of the host, the bacterial genotype and environmental factors [5–7]. Dozens of bacterial determinants have been described to impact H. pylori pathogenicity. Two classical virulence factors are known, the vacuolating cytotoxin (VacA) and the cytotoxin-associated genes pathogenicity island (cagPAI). The cagPAI encodes a type IV secretion system (T4SS) for transport of the oncoprotein CagA across the bacterial membranes into host target cells [8, 9]. Upon delivery, CagA undergoes phosphorylation at C-terminal Glu-Pro-Ile-Tyr-Ala (EPIYA) sequence repeats by the c-Src and c-Abl family of tyrosine kinases [10–12]. Translocated CagA binds to and activates or inactivates a series of signaling factors in a phosphorylation-dependent and phosphorylation-independent fashion [13, 14]. The T4SS can also induce profound pro-inflammatory responses such as the release of chemokine interleukin-8 (IL-8) via transcription factor NF-κB, which proceeds widely independently of CagA delivery [15–17]. On the other hand, VacA is an autotransporter and secreted into the extracellular space, where it induces multiple responses including cell vacuolation, alteration of endo-lysosomal trafficking, immune cell inhibition and apoptosis [5, 18]. Other pathogenicity-associated processes comprise urease-triggered neutralization of acidic pH, flagella-mediated motility, expression of multiple adhesins (BabA/B, SabA, AlpA/B, HopQ, HopZ, OipA and others), inhibition of T cell proliferation by secreted γ-glutamyl-transpeptidase GGT, and secretion of proteases such as HtrA [3, 19–21].
High temperature requirement protein A (HtrA ) family members comprise a set of evolutionarily related serine proteases and chaperones, which are found in most prokaryotes and eukaryotes [22–24]. HtrA proteases are generally transported into the periplasm, where they form proteolytically active oligomers with important function in protein quality control [25, 26]. Its chief role is to remove damaged, misfolded or mislocalized proteins in the periplasm. HtrA proteins contain no regulatory components or ATP binding domains [22]. Thus, they are commonly referred to as ATP-independent chaperone-proteases. Bacterial HtrA proteases commonly comprise an N-terminal signal sequence, followed by a trypsin-like serine protease domain and one or two PDZ modules at the C-terminus, which permit protein–protein interactions [23, 27–29]. Inactivation of the htrA gene by mutation regularly results in high temperature sensitivity of many bacteria [30–35]. For a long time it was supposed that HtrA proteases are strictly functioning only inside the bacterial periplasm. However, we have previously introduced a new characteristic for the HtrAs of Campylobacter jejuni and H. pylori. These HtrA proteins can be actively secreted into the extracellular environment, where they cleave host cell factors [36–41]. It has been demonstrated that secreted HtrA from both species can open the adherens junctions in cultured polarized epithelial cells in vitro by cleaving the extracellular NTF (N-terminal fragment)-domain of E-cadherin, a well-known cell-to-cell adhesion factor [37, 39, 42]. Inactivation of C. jejuni htrA results in downregulated E-cadherin cleavage and bacterial transmigration across polarized cell monolayers in vitro [35, 39], and reduced apoptosis and immunopathology in the gut of infected mice in vivo [43, 44]. Similarly, HtrA is fundamental for the virulence of various other pathogens including Yersinia enterocolitica, Klebsiella pneumoniae, Chlamydia trachomatis, Salmonella enterica, Listeria monocytogenes, Legionella pneumophila, Shigella flexneri, Burkholderia cenocepacia and Borrelia burgdorferi [31, 32, 34, 45–50]. However, an htrA knockout strain in H. pylori is not yet available because the generation of mutants was unsuccessful in a broad collection of worldwide strains, suggesting that htrA may represent an essential gene in H. pylori [51, 52]. To study the role of HtrA further, we aimed to overexpress HtrA in H. pylori and examine various virulence properties of the bacteria. Our results show that overexpression of HtrA in H. pylori results in elevated secretion rates of the protease, cleavage of E-cadherin, bacterial transmigration and delivery of CagA into polarized epithelial cells.
Results and discussion
Introduction and expression of a second htrA gene copy in H. pylori
Helicobacter pylori htrA is an essential bifunctional gene with crucial intracellular and extracellular functions [51, 52]. In order to study the function of htrA in more detail, we aimed to overexpress the protein by introduction of a second htrA gene copy of strain 26695 (htrA 26695) in the chromosome of H. pylori. For this purpose, we placed htrA 26695 under an inducible isopropyl-β-d-thiogalactopyranoside (IPTG)-responsive promotor as described [53]. Expression of htrA 26695 was driven by the pTac promotor construct (Fig. 1a, top) derived from plasmid pILL2150 [53]. Promoter activity was described as tightly regulated for LacZ expression and suitable for the analysis of essential H. pylori genes [53]. We transformed this construct in two H. pylori wild-type strains, P12 or 26695 (called Hp wt) and the resulting transformants were designated Hp htrA 26695. We obtained similar results for both H. pylori strains and subsequently show the representative results for one set of experiments. Hp wt and Hp htrA 26695 were grown for 24 h in brain heart infusion (BHI) liquid broth medium containing 10% FCS in the presence or absence of 1 mM or 2 mM IPTG, respectively, and the resulting lysates were checked for expression of HtrA and other well-known H. pylori proteins using Western blotting. The results indicate that HtrA expression in Hp htrA 26695 in the presence of IPTG increased up to about 2.4-fold compared to the control without IPTG, but did not change in Hp wt (Fig. 1b, c). Control blots using α-CagA and α-FlaA antibodies showed that the expression of CagA and FlaA proteins remained stable over time and were unaffected by addition of IPTG to all strains (Fig. 1b). These results demonstrate that the IPTG-dependent expression system works well for htrA 26695 in two H. pylori strains and is very useful for further analyses.
Fig. 1.
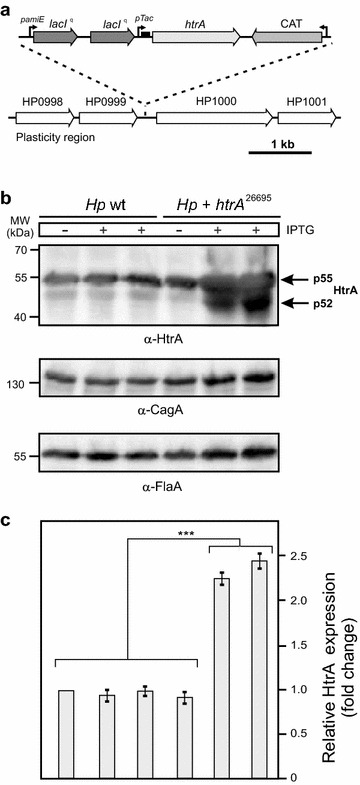
Chromosomal introduction of a second, inducible htrA gene copy in H. pylori. a Schematic presentation of the IPTG-inducible htrA 26695 expression construct with the chloramphenicol acetyl-transferase (CAT) cassette (top), which was cloned into the plasticity region of H. pylori between genes HP0999 and HP1000 (bottom). b This construct was transformed into H. pylori wild-type (Hp wt) strains 26695 and P12, and the expression of HtrA was investigated after 24 h growth in BHI medium in the presence or absence of 1 or 2 mM IPTG, respectively. The results for strain 26695 are shown. Western blotting using α-FlaA and α-CagA antibodies served as controls, indicating that equal amounts of proteins are present in the samples. c The band intensities of HtrA proteins were quantified densitometrically. The relative protein expression is given in “fold change”
Overexpression of HtrA in H. pylori enhances its proteolytic activity
Next, we aimed to analyse if Hp wt or Hp htrA 26695 can form proteolytically active HtrA oligomers in the absence or presence of IPTG. For this purpose, the samples generated for Fig. 1b were subjected to casein zymography. Bacterial pellets were loaded onto 0.1% casein containing gels and separated under non-reducing conditions and then renatured as described [38]. The results show that HtrA activity of Hp htrA 26695 increased up to ~2.5-fold in the presence of IPTG compared to Hp wt or the control without IPTG, giving rise to active HtrA oligomers with a molecular weight ranging from ~180 kDa to more than 200 kDa in the cell pellet (Fig. 2a, arrows). As further control, corresponding signals for proteolytically active HtrA were at a similar basal level in both strains in the absence of IPTG (Fig. 2a, b).
Fig. 2.
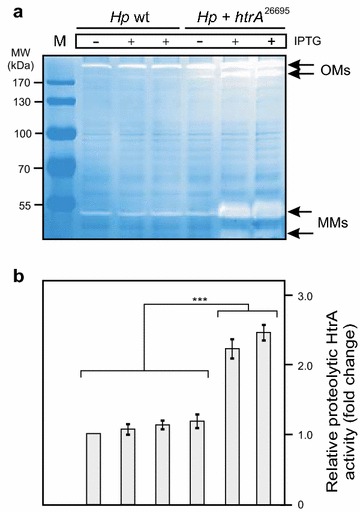
Expression of two htrA gene copies in H. pylori enhances the overall proteolytic activity of HtrA. a H. pylori wild-type strain 26695 (Hp wt) and 26695 transformed with htrA 26695 were grown for 24 h in BHI broth medium in the presence or absence of 1 or 2 mM IPTG, respectively. Bacterial pellets were prepared and subjected to investigation of protease activity by casein zymography. The position of proteolytically active HtrA monomers (MMs) and oligomers (OMs) is indicated with arrows. M, protein size marker. b The band intensities of proteolytically active HtrA proteins were quantified densitometrically. The relative HtrA activity is given in “fold change”
Induction of HtrA leads to higher secretion levels of HtrA, but not VacA and GGT
The next objective was to evaluate the level of secreted HtrA in the culture supernatants. After 24 h of growth, bacteria-free supernatants and cell pellets were prepared and the presence of secreted HtrA proteins in the supernatants was investigated by immunoblotting using α-HtrA antibodies (Fig. 3a). The results show that the bands for secreted HtrA in Hp htrA 26695 in the presence of IPTG increased up to ~1.8-fold compared to the strain without IPTG, but did not change significantly in Hp wt (Fig. 3a, b). As control, corresponding signals for secreted HtrA were at a similar basal level in both strains in the absence of IPTG (Fig. 3a, b). In further experiments, the supernatants were probed for two other well-known secreted H. pylori proteins, VacA and GGT. As shown in Fig. 3a, the band intensities for secreted VacA and GGT were constantly stable in both strains and did not change by adding IPTG (Fig. 3a, c). On the other hand, CagA is a well-known translocated T4SS effector protein, not secreted into the supernatant [54]. The α-CagA blots of the supernatants are devoid of CagA, indicating absence of lysed bacteria and cell debris in our samples as expected (Fig. 3a). Taken together, these experiments demonstrate that secretion of HtrA by Hp htrA 26695 is significantly enhanced after addition of IPTG compared to the Hp wt control, while the secretion levels of VacA and GGT remained unaffected.
Fig. 3.
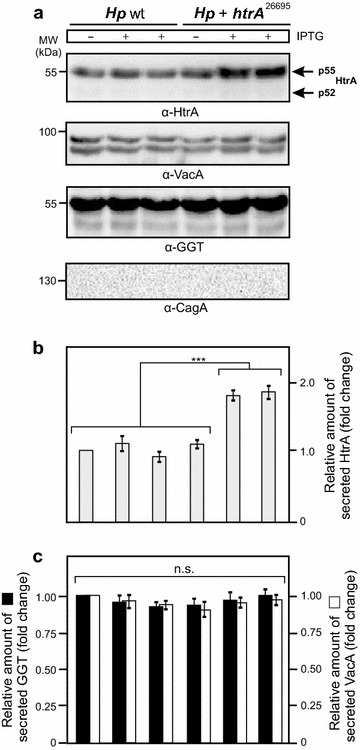
Elevated HtrA secretion levels in H. pylori does not affect the extracellular delivery of VacA and GGT. a H. pylori wild-type strain 26695 (Hp wt) and 26695 transformed with htrA 26695 were grown for 24 h in BHI broth medium with 1% β-cyclodextrin in the presence or absence of 1 or 2 mM IPTG, respectively. Bacteria-free supernatants were prepared and subjected to Western blotting using α-HtrA, α-VacA and α-GGT antibodies. The blots against α-CagA served as control. The band intensities of secreted HtrA (b) as well as VacA and GGT (c) were quantified densitometrically. The relative protein expression is given in “fold change”. All secretion assays were done in triplicates
Overexpression of HtrA does not affect host cell binding and IL-8 secretion by H. pylori
As next, we aimed to study the functional role of HtrA overexpression during infection of epithelial cells. For this purpose, monolayers of polarized MKN-28 cells were infected for 8 h with IPTG-induced or control Hp wt and Hp htrA 26695, respectively. To test if differential HtrA expression might affect host cell binding by H. pylori, we determined the CFU of bound bacteria by an established protocol [55]. The results show that the number of bound bacteria was similar between the samples and varied only between 10 and 16 CFU per MKN-28 cell (Fig. 4). In addition, we have analyzed the amount of chemokine IL-8 secreted into the supernatants. The levels of IL-8 were also at similar high level between the samples and varied only between ~12,000 and 17,000 pg/mL. These results suggest that overexpression of HtrA by IPTG induction does not affect the bacteria’s viability and host cell binding capabilities. The T4SS-dependent activity of H. pylori towards IL-8 secretion was also very high and similar between the samples, suggesting that T4SS functions are intact and remain unchanged with regard to the pro-inflammatory responses during infection with the different strains.
Fig. 4.
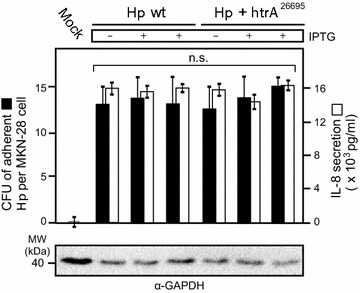
Higher expression of HtrA does not change host cell binding capabilities and IL-8 production by H. pylori. IPTG-induced or control H. pylori wild-type strain 26695 (Hp wt) and 26695 transformed with htrA 26695 were grown for 24 h in BHI broth medium with 10% FCS. Polarized MKN-28 cells were then co-incubated with H. pylori for 8 h using an MOI of 50. The numbers of adherent bacteria were determined by adhesion assays and counting the CFU on GC agar plates. The concentrations of secreted chemokine IL-8 were quantified by standard ELISA. All infection assays were done in triplicates
Overexpression of HtrA enhances disruption of cell-to-cell junctions by H. pylori
In the next set of experiments, confluent polarized Caco-2 cells were infected with the various IPTG-induced H. pylori strains for 24 h and subsequently fixed for immunofluorescence microscopy staining against the adherens junction protein E-cadherin and H. pylori. The results confirm that the signals of Hp wt or Hp htrA 26695 bacteria (red) attached to the host cells are similarly high between the samples. However, while the mock control cells exhibited typical E-cadherin signals between all neighbouring cells, H. pylori infection disrupted the E-cadherin staining significantly (Fig. 5b–d). Individual cells showing downregulated or dislocated E-cadherin signals are marked with blue and yellow arrowheads, respectively (Fig. 5b, c). The number of cells with changed E-cadherin patterns was more pronounced during infection with Hp htrA 26695 (Fig. 5c, d). Longer infection times up to 48 h, however, led to a complete disruption of the E-cadherin patterns by Hp htrA 26695 bacteria (data not shown). These data indicate that overexpression of HtrA is associated with enhanced damage of the cell-to-cell junctions over time.
Fig. 5.
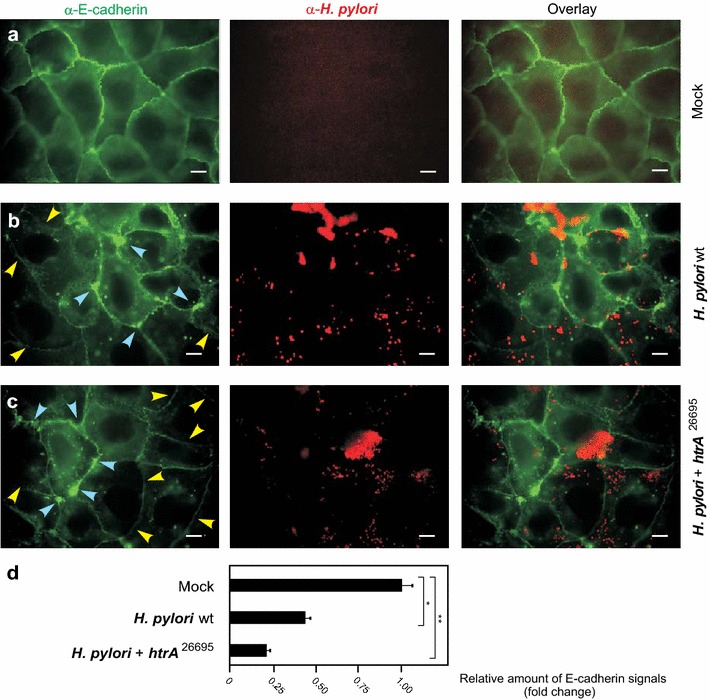
Enhanced disruption of cell-to-cell junctions by H. pylori expressing two htrA gene copies. Polarized Caco-2 cells were left untreated (mock) (a) or infected for 24 h with H. pylori wild-type (wt) (b) and H. pylori htrA 26695 (c). The cells were fixed with methanol and subjected to immunofluorescence using α-E-cadherin (green) and α-H. pylori (red) antibodies. Arrowheads mark cells showing significantly downregulated (blue) or disrupted (yellow) E-cadherin signals. Scale bars, 10 μm. d Quantification of overall fluorescence signals for E-cadherin stainings, which are given in “fold change”. The total E-cadherin signal in the uninfected mock control was set as “1”
Overexpression of HtrA enhances bacterial transmigration across polarized cells
In addition, we determined the transmigration rates by the different H. pylori strains. For this purpose, polarized Caco-2 and MKN-28 cells were grown in a transwell filter system for 14 days to reach confluent monolayers. The cells were infected with IPTG-induced or control Hp wt or Hp htrA 26695 for 24 h in the apical chamber. Transmigrated bacteria were harvested from the bottom chambers, grown on GC agar plates, and the CFUs were determined (Fig. 6a). The results show that the number of transmigrated Hp htrA 26695 bacteria in the presence of IPTG were about 420–520 × 103 CFU and increased up to ~2.2-fold compared to the corresponding IPTG-induced control Hp wt bacteria (Fig. 6). As a further control, the numbers of transmigrated bacteria of both strains in the absence of IPTG were at a similar basal level of approximately 200–265 × 103 H. pylori (Fig. 6a). In addition, we measured the transepithelial electrical resistance (TER) in the same experiments. In agreement with the results obtained above, the data show that the TER values dropped down significantly (Fig. 6b), correlating with enhanced cell damage (Fig. 5) and transmigration by Hp htrA 26695 bacteria in the presence of IPTG (Fig. 6a).
Fig. 6.
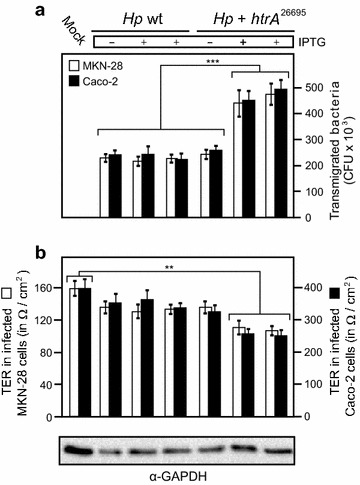
Upregulation of bacterial transmigration by H. pylori expressing two htrA genes. a Caco-2 and MKN-28 cells were cultured in a transwell filter system for 14 days to reach polarized monolayers, followed by apical infection with IPTG-induced or control Hp wt or Hp htrA 26695, respectively. After 24 h, transmigrated bacteria in the bottom chambers were quantified by counting CFUs. The results show that the number of transmigrated Hp htrA 26695 bacteria in the presence if IPTG increased ~2.5-fold compared to IPTG-induced control Hp wt. b Determination of the TER values in the same set of experiments. All experiments were done in triplicates
Overexpression of HtrA results in elevated E-cadherin cleavage and CagA phosphorylation
As next we investigated the cleavage of E-cadherin in infected vs. non-infected polarized epithelial cell lines by Western blotting after 24 h of incubation (Fig. 7). The results show that the intensity of cell-associated full-length E-cadherin signals dropped-down during infection with Hp htrA 26695 bacteria in the presence of IPTG and the corresponding cleaved-off NTF-domain, present in the supernatant, increased up to ~twofold compared to the corresponding IPTG-induced control Hp wt bacteria (Fig. 7a, c). As another control, the signals of the NTF-fragment produced by both strains in the absence of IPTG were at a similar basal level (Fig. 7a, c). Remarkably, the extent of E-cadherin cleavage induced by the different strains correlated with the intensity of signals obtained for translocated phospho-CagA in the same experiments (Fig. 7b, c). The strongest phospho-CagA signals were always observed for Hp htrA 26695 bacteria in the presence of IPTG, suggesting that increased HtrA expression and activity significantly positively regulate T4SS functions with regard to translocation and phosphorylation of CagA in polarized epithelial cells.
Fig. 7.
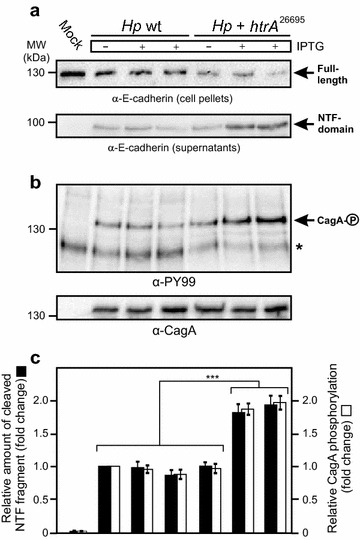
Elevated E-cadherin cleavage and CagA translocation in host cells by overexpression of HtrA. Polarized MKN-28 cells were infected with IPTG-induced or control Hp wt or Hp htrA 26695 for 24 h. a Cell pellets and supernatants were prepared and subjected to α-E-cadherin Western blotting. The full-length E-cadherin signals (130 kDa) in the cell pellets and cleaved-off NTF-domain (90 kDa) in the culture supernatants are marked with arrows. b Translocation and phosphorylation of CagA (arrow) were analyzed using α-PY-99 and α-CagA antibodies, respectively. The asterisk marks a phosphorylated 125 kDa host cell protein migrating below CagA. c The band intensities of cleaved E-cadherin NTF-fragments and phospho-CagA signals were quantified densitometrically. The relative amounts of NTF-fragment and phosphorylated CagA are given in “fold change”. All experiments were done in triplicates
Conclusions
Diverse pathogens encode proteases with crucial functions during infection, but knowledge on secreted proteases and their activities in H. pylori is very limited. In many bacteria, HtrA is a well-recognized factor in the periplasm, which contains chaperone and proteolytic functions with important roles in protein quality control involved in stress tolerance and bacterial survival [23, 25–29]. In addition, it was demonstrated that HtrA has a significant impact on the virulence of multiple bacterial pathogens including Borrelia, Burkholderia, Campylobacter, Chlamydia, Klebsiella, Legionella, Listeria, Salmonella, Shigella and Yersinia species. Interestingly, htrA does not appear as an essential gene in each of these bacteria because ΔhtrA knockout mutation has been described [31, 32, 34, 43–50]. In contrast, inactivation of the htrA gene in H. pylori has been unsuccessful in more than one hundred worldwide isolates, but the reasons for this failure are still unclear [37, 51, 52]. Remarkably, it was also demonstrated that pharmacological inhibition of HtrA protease activity effectively killed H. pylori, while it did not affect the growth and viability of other Gram-negative pathogens including Salmonella and Shigella [52].
Research progress on H. pylori HtrA is mainly hampered by the lack of ΔhtrA knockout mutants. Thus, other genetic manipulation strategies are required to study HtrA function during the infection process. Here we developed a genetic approach to overexpress HtrA in two clinical isolates, P12 and 26695. For this purpose, a second htrA gene copy was introduced into the H. pylori chromosome and placed under an IPTG-inducible promotor [53]. Once the HtrA proteins are translated by the bacteria they are delivered into the periplasm and subsequently secreted into the extracellular environment. This important new aspect seems to be conserved among a wide range of worldwide H. pylori isolates [52]. We could show here that overexpression of HtrA enhanced not only its proteolytic activity by up to ~2.5-fold, but also the secretion of the protease by ~1.8-fold. Interestingly, the secretion of other well-known bacterial virulence determinants, VacA and GGT, was not affected by HtrA overexpression, suggesting that the secretion of these factors follow different, non-linked pathways. In addition, we could demonstrate that various virulence-associated properties of H. pylori were also not affected including bacterial attachment to the epithelial cells and induction of pro-inflammatory responses such as the secretion of IL-8. In contrast, the transepithelial migration of H. pylori overexpressing HtrA increasing significantly up to ~2.2-fold compared to the control bacteria. This phenotype was accompanied by significantly enhanced damage to the adherens junction protein E-cadherin. Our Western blotting data demonstrated that HtrA-mediated cleavage of full-length E-cadherin was enhanced, leading to elevated levels of the 90 kDa E-cadherin NTF-fragment in the supernatants of infected cells. Immunofluorescence microscopy confirmed these observations and showed that the cell-to-cell junctions of infected Caco-2 cells were significantly more disrupted after 24 h compared to the wild-type control infection, explaining why higher numbers of bacteria can cross the epithelial barrier and reach basolateral compartments. Finally, we observed that the levels of CagA translocation and phosphorylation increased up to ~twofold in HtrA-overexpressing H. pylori compared to the control bacteria. These observations can be explained by reports showing that CagA delivery into host cells requires a receptor, which was identified as the basolateral integrin member α5β1 [56–62]. Integrins are well-known mammalian cell adhesion receptors, which facilitate anchoring of host cells to the extracellular matrix and which are absent at apical surfaces [63, 64]. These findings let us to suggest a novel mechanism how the T4SS of H. pylori works in polarized epithelial cells by cooperating with the secreted serine protease HtrA, which opens cell-to-cell junctions. Using an inducible genetic system to overexpress HtrA, we could enhance the proteolytic activity of HtrA, necessary for elevated paracellular transmigration of H. pylori across the polarized epithelial cells to reach basolateral membranes and inject CagA in an integrin-dependent fashion. Extensive research has shown in recent years that the above discussed features basically resemble a phenotype, called epithelial-mesenchymal transition (EMT). Gastric cancerogenesis is known for its aggressiveness and tendency to metastasize. EMT is the initial step in metastasis, orchestrated by various cellular factors [65]. We proposed that the activity of secreted HtrA is maybe the initial step in a signaling cascade, followed by CagA and probably others, that triggers EMT in gastric epithelial cells. Translocated CagA can then deregulate cell polarity and scattering, by various pathways including the interaction with partioning kinase Par1b changing cell polarity [66] and by stabilizing Snail, a transcriptional repressor of E-cadherin expression [67]. Taken together, these data provide for the first time genetic evidence that HtrA is a major novel virulence factor of H. pylori, controlling multiple pathogenic activities of this important microbe.
Methods
MKN-28 and Caco-2 cell culture and H. pylori infection
Human MKN-28 cells (JCRB, #0253) were originally isolated from gastric adenocarcinoma. The Caco-2 cells (ATCC HTB-37) were obtained from a human colon adenocarcinoma. Both cell lines have been extensively used over the last twenty years as models for studying the gastrointestinal barrier. Cells were cultured in 6-well plates with RPMI1640 or DMEM medium, respectively, containing 4 mM glutamine (Invitrogen, Karlsruhe/Germany), and 10% FCS (Invitrogen, Karlsruhe/Germany) [68]. H. pylori strains 26695, P12 and their mutants were grown on horse serum GC agar plates supplemented with nystatin (1 μg/mL), vancomycin (10 μg/mL) and trimethoprim (5 μg/mL), and if necessary with 4 μg/chloramphenicol per mL. Growth was performed for 2 days at 37 °C in anaerobic chambers containing a CampyGen gas mix (Oxoid, Wesel/Germany) at 37 °C [69]. H. pylori was harvested and resuspended in phosphate buffered saline (PBS, pH 7.4) using sterile cotton swabs (Carl Roth, Karlsruhe/Germany). The bacterial concentration was measured in a spectrophotometer as optical density (OD) at 600 nm (Eppendorf, Hamburg/Germany). Infections were carried out at a multiplicity of infection (MOI) of 50 [70]. All infection assays were done in triplicates.
H. pylori mutagenesis
To introduce a second htrA gene copy in the H. pylori chromosome, we made use of the previously generated IPTG-inducible LacIq pTac system for lacZ gene expression as cloned in vector pILL2150 [53]. In this system, the promoters were engineered to be under the control of H. pylori RNA polymerase. The amiE gene promoter of H. pylori was taken to constitutively express the LacIq repressor, which is present in two copies (Fig. 1a, top). Expression of the lacZ reporter gene was driven by the pTac promotor as described [53]. We replaced the lacZ gene of pILL2150 by the htrA gene of strain 26695 using the NdeI and BamHI restriction sites. Then the complete cassette shown in Fig. 1a (top) was introduced in the chromosomal plasticity region of H. pylori strains P12 and 26695 (between ORFs HP0999 and HP1000) as shown in Fig. 1a (bottom) using established transformation methods [71, 72]. At the 3′ end, a chloramphenicol resistance gene cassette (CAT) was added to select clones. The correct integration and expression of the htrA gene was verified by PCR and Western blotting, respectively.
HtrA, VacA and GGT secretion assays
Wild-type and mutant H. pylori strains were suspended in BHI medium supplemented with 1% β-cyclodextrin (Sigma Aldrich) [73]. The optical density was determined and adjusted to OD600 = 0.2. To allow bacterial protein secretion in the culture supernatant, H. pylori was grown for 24 h under shaking at 160 rpm in the presence or absence of IPTG (Sigma Aldrich). The cell pellets and the supernatants were prepared by centrifugation at 4000 rpm. The supernatants were transferred through 0.21 μm sterile filters (Sigma Aldrich) to remove remnant bacterial cells. Lack of live bacteria in the supernatant was verified by the absence of bacterial growth after 5 days of incubation on GC agar plates. The resulting bacterial pellets and supernatants were then analysed by Western blotting as described below.
Transwell infection studies
MKN-28 and Caco-2 cells were cultured on 0.33 cm2 cell culture inserts with 3 μm pore size (Corning Life Sciences, Schiphol/Netherlands). The cells were grown to confluent monolayers, and then incubated for another 14 days to allow cell polarization [39]. TER was measured with an Electrical Resistance System (ERS) (World Precision Instruments, Berlin/Germany). Maximal TER values indicated that the monolayers reached maximal cell polarity [39]. The cells were infected in the apical compartment at MOI of 50 and the numbers of transmigrated bacteria were quantified in aliquots taken from the basal chambers and counting colony forming units (CFU) on GC agar plates after 5 days of incubation [35].
Cell binding assay
Infection of MKN-28 and Caco-2 cell monolayers was performed at a density of 3.5 × 105 cells in 6-well plates as described previously [55]. After infection, infected cells were washed three times with 1 mL of pre-warmed culture medium per well to remove non-adherent bacteria. To determine the total CFU corresponding to cell-associated bacteria, the infected monolayers were incubated with 1 mL of 0.1% saponin in PBS at 37 °C for 15 min. The resulting suspensions were diluted and plated on GC agar plates. The CFUs were counted after 5 days of incubation.
Casein zymography
Undiluted aliquots of the bacteria were loaded onto 10% SDS–PAGE gels containing 0.1% casein (Carl Roth, Karlsruhe/Germany) and separated by electrophoresis under non-reducing conditions. After protein separation, the gel was renatured in 2.5% Triton X-100 solution at room temperature for 60 min with gentle agitation, equilibrated in developing buffer (50 mM Tris–HCl, pH 7.4, 200 mM NaCl, 5 mM CaCl2, 0.02% Brij35) at room temperature for 30 min with gentle agitation, and incubated overnight in fresh developing buffer at 37 °C. Transparent HtrA bands with caseinolytic activity were visualized by staining with 0.5% Coomassie Blue R250 as described [35, 39].
Antibodies
The following antibodies were purchased: rabbit polyclonal α-CagA antibody (Austral Biologicals, San Ramon, CA/USA), monoclonal pan-phosphotyrosine α-PY99 (Santa Cruz, Santa Cruz, CA/USA), rabbit α-GAPDH (Santa Cruz), rabbit α-H. pylori (Dako, Glostrup/Denmark) and two monoclonal antibodies directed against the extracellular domain of E-cadherin, H-108 (Santa Cruz) and CD324 (BD Biosciences, San Jose, CA/USA). HtrA proteins were detected by rabbit polyclonal α-HtrA antiserum raised against purified recombinant HtrA (Biogenes, Berlin/Germany). Rabbit polyclonal α-FlaA and α-GGT antibodies were described previously [74, 75]. The α-VacA antibody (#123) was kindly provided by Timothy Cover (Nashville, TN/USA).
Immunofluorescence staining and microscopy
Immunofluorescence staining with different antibodies as shown in each experiment was performed as described [76]. Briefly, cell samples were fixed with methanol at −20 °C for 10 min followed by permeabilization with 0.5% Triton-X100 for 1 min and blocking with 1% BSA, 0.1% Tween-20 in PBS for 30 min. Proteins were stained with the above mentioned α-E-cadherin (BD) and α-H. pylori antibodies. As secondary antibodies, we used TRITC (tetramethylrhodamine isothiocyanate)-conjugated goat anti-rabbit and FITC (fluorescein isothiocyanate)-conjugated goat anti-mouse (Thermo Fisher Scientific, Darmstadt/Germany). Samples were analysed using a Leica DMI4000B fluorescence microscope and different lasers (Leica Microsystems, Wetzlar/Germany). Images were obtained via LAS AF computer software (Leica Microsystems) and E-cadherin staining was quantified as “fold change” using the ImageJ Software (version 2.0). The mock control was set as “1”.
SDS–PAGE, dot blots and immunoblotting
Bacterial pellets, cell-free supernatants or infected cells were mixed with equal amounts of 2× SDS–PAGE buffer and boiled for 5 min. Proteins were separated by SDS–PAGE on 8% polyacrylamide gels and blotted onto PVDF membranes (Immobilon-P, Merck Millipore) as described [77]. Before addition of the antibodies, membranes were blocked in TBST buffer (140 mM NaCl, 25 mM Tris–HCl pH 7.4, 0.1% Tween-20) with 3% BSA or 5% skim milk for 1 h at room temperature [78]. As secondary antibodies, horseradish peroxidase-conjugated α-mouse or α-rabbit polyvalent rabbit and pig immunoglobulin, respectively, were used (Life Technologies, Darmstadt/Germany). Antibody detection was performed with the ECL Plus chemiluminescence Western Blot kit (GE Healthcare Life Sciences, Munich/Germany) [79].
Quantification of IL-8 chemokines by ELISA
MKN-28 cells were incubated for 8 h with H. pylori, and mock cells with medium served as negative control. The culture supernatants were collected and stored at −80 °C until assayed. IL-8 concentrations in the supernatants were determined by standard ELISA according to manufacturer’s procedures (Becton–Dickinson, Heidelberg/Germany) [80].
Quantification of band intensities in Western blots and casein gels
Quantification of band signals on immunoblots was performed by densitometric analysis using the Image Lab software (BioRad, Munich/Germany) and indicated the “fold change” of protein expression or phosphorylation level per sample. As shown in the corresponding figures, the control band on each gel was set as “1”.
Statistics
All data were evaluated via Student’s t test with SigmaPlot statistical software (version 13.0). Statistical significance was defined by p ≤ 0.05 (*), p ≤ 0.01 (**) and p ≤ 0.001 (***).
Authors’ contributions
AH, MB, SB and NT designed and performed the experiments. NT, the senior/corresponding author, supervised the experiments and wrote the manuscript together with SB. All authors read and approved the final manuscript.
Acknowledgements
We thank Drs. Ivo Boneca (Institute Pasteur, Paris, France) for providing plasmid pILL2150, Benjamin Hoy/Silja Wessler (University of Salzburg, Austria) for cloning of the htrA gene, Timothy Cover (Vanderbilt University Nashville, TN/USA) for the α-VacA antibody and Benjamin Schmid (OICE Erlangen, Germany) for help with the IFM signal quantification. We also thank Wilhelm Brill and Vanessa Schmidt for excellent technical assistance. We acknowledge support by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) within the funding pogramme Open Access Publishing.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by the German Science Foundation to NT (project TE776/3-1), to MB (project BO4724/1-1) and SB (project B10 in CRC-796 and A04 in CRC-1181).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Aileen Harrer, Email: Aileen.Harrer@fau.de.
Manja Boehm, Email: Manja.Boehm@fau.de.
Steffen Backert, Email: Steffen.Backert@fau.de.
Nicole Tegtmeyer, Email: Nicole.Tegtmeyer@fau.de.
References
- 1.Robinson K, Letley DP, Kaneko K. The human stomach in health and disease: infection strategies by Helicobacter pylori. Curr Top Microbiol Immunol. 2017;400:1–26. doi: 10.1007/978-3-319-50520-6_1. [DOI] [PubMed] [Google Scholar]
- 2.Mejías-Luque R, Gerhard M. Immune evasion strategies and persistence of Helicobacter pylori. Curr Top Microbiol Immunol. 2017;400:53–71. doi: 10.1007/978-3-319-50520-6_3. [DOI] [PubMed] [Google Scholar]
- 3.Salama NR, Hartung ML, Müller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol. 2013;11:385–399. doi: 10.1038/nrmicro3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Figueiredo C, Camargo CM, Leite M, Fuentes-Pananá EM, Rabkin CS, Machado JC. Pathogenesis of gastric cancer: genetics and molecular classification. Curr Top Microbiol Immunol. 2017;400:277–304. doi: 10.1007/978-3-319-50520-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cover TL, Peek RM., Jr Diet, microbial virulence, and Helicobacter pylori-induced gastric cancer. Gut Microbes. 2013;4:482–493. doi: 10.4161/gmic.26262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Backert S, Blaser MJ. The role of CagA in the gastric biology of Helicobacter pylori. Cancer Res. 2016;76:4028–4031. doi: 10.1158/0008-5472.CAN-16-1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gobert AP, Wilson KT. Human and Helicobacter pylori interactions determine the outcome of gastric diseases. Curr Top Microbiol Immunol. 2017;400:27–52. doi: 10.1007/978-3-319-50520-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Backert S, Tegtmeyer N, Fischer W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015;10:955–965. doi: 10.2217/fmb.15.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergé C, Terradot L. Structural insights into Helicobacter pylori Cag protein interactions with host cell factors. Curr Top Microbiol Immunol. 2017;400:129–147. doi: 10.1007/978-3-319-50520-6_6. [DOI] [PubMed] [Google Scholar]
- 10.Backert S, Feller SM, Wessler S. Emerging roles of Abl family tyrosine kinases in microbial pathogenesis. Trends Biochem Sci. 2008;33:80–90. doi: 10.1016/j.tibs.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Lind J, Backert S, Pfleiderer K, Berg DE, Yamaoka Y, Sticht H, et al. Systematic analysis of phosphotyrosine antibodies recognizing single phosphorylated EPIYA-motifs in CagA of Western-type Helicobacter pylori strains. PLoS ONE. 2014;9:e96488. doi: 10.1371/journal.pone.0096488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lind J, Backert S, Hoffmann R, Eichler J, Yamaoka Y, Perez-Perez GI, et al. Systematic analysis of phosphotyrosine antibodies recognizing single phosphorylated EPIYA-motifs in CagA of East Asian-type Helicobacter pylori strains. BMC Microbiol. 2016;16:201. doi: 10.1186/s12866-016-0820-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, et al. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in western and East Asian Helicobacter pylori strains. J Clin Invest. 2012;122:1553–1566. doi: 10.1172/JCI61143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014;15:306–316. doi: 10.1016/j.chom.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 16.Brandt S, Kwok T, Hartig R, König W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Backert S, Naumann M. What a disorder: proinflammatory signaling pathways induced by Helicobacter pylori. Trends Microbiol. 2010;18:479–486. doi: 10.1016/j.tim.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Posselt G, Backert S, Wessler S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun Signal. 2013;11:77. doi: 10.1186/1478-811X-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rad R, Gerhard M, Lang R, Schöniger M, Rösch T, Schepp W, et al. The Helicobacter pylori blood group antigen-binding adhesin facilitates bacterial colonization and augments a nonspecific immune response. J Immunol. 2002;168:3033–3041. doi: 10.4049/jimmunol.168.6.3033. [DOI] [PubMed] [Google Scholar]
- 20.Backert S, Clyne M, Tegtmeyer N. Molecular mechanisms of gastric epithelial cell adhesion and injection of CagA by Helicobacter pylori. Cell Commun Signal. 2011;9:28. doi: 10.1186/1478-811X-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricci V, Giannouli M, Romano M, Zarrilli R. Helicobacter pylori gamma-glutamyl transpeptidase and its pathogenic role. World J Gastroenterol. 2014;20:630–638. doi: 10.3748/wjg.v20.i3.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frees D, Brøndsted L, Ingmer H. Bacterial proteases and virulence. Subcell Biochem. 2013;66:161–192. doi: 10.1007/978-94-007-5940-4_7. [DOI] [PubMed] [Google Scholar]
- 23.Skórko-Glonek J, Figaj D, Zarzecka U, Przepiora T, Renke J, Lipinska B. The extracellular bacterial HtrA proteins as potential therapeutic targets and vaccine candidates. Curr Med Chem. 2016 (Epub ahead of print). [DOI] [PubMed]
- 24.Wessler S, Schneider G, Backert S. Bacterial serine protease HtrA as a promising new target for antimicrobial therapy? Cell Commun Signal. 2017;15:4. doi: 10.1186/s12964-017-0162-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gottesman S, Wickner S, Maurizi MR. Protein quality control: triage by chaperone and proteases. Genes Dev. 1997;11:815–823. doi: 10.1101/gad.11.7.815. [DOI] [PubMed] [Google Scholar]
- 26.Ingmer H, Brøndsted L. Proteases in bacterial pathogenesis. Res Microbiol. 2009;160:704–710. doi: 10.1016/j.resmic.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 27.Clausen T, Southan C, Ehrmann M. The HtrA family of proteases: implications for protein composition and cell fate. Mol Cell. 2002;10:443–455. doi: 10.1016/S1097-2765(02)00658-5. [DOI] [PubMed] [Google Scholar]
- 28.Kim DY, Kim KK. Structure and function of HtrA family proteins, the key players in protein quality control. J Biochem Mol Biol. 2005;38:266–274. doi: 10.5483/bmbrep.2005.38.3.266. [DOI] [PubMed] [Google Scholar]
- 29.Backert S, Boehm M, Wessler S, Tegtmeyer N. Transmigration route of Campylobacter jejuni across polarized intestinal epithelial cells: paracellular, transcellular or both? Cell Commun Signal. 2013;11:72. doi: 10.1186/1478-811X-11-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipinska B, Fayet O, Baird L, Georgopoulos CJ. Identification, characterization, and mapping of the Escherichia coli htrA gene, whose product is essential for bacterial growth only at elevated temperatures. Bacteriol. 1989;171(3):1574–1584. doi: 10.1128/jb.171.3.1574-1584.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pedersen LL, Radulic M, Doric M, Kwaik YA. HtrA homologue of Legionella pneumophila: an indispensable element for intracellular infection of mammalian but not protozoan cells. Infect Immun. 2001;69:2569–2579. doi: 10.1128/IAI.69.4.2569-2579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cortés G, de Astorza B, Benedí VJ, Albertí S. Role of the htrA gene in Klebsiella pneumoniae virulence. Infect Immun. 2002;70:4772–4776. doi: 10.1128/IAI.70.9.4772-4776.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mo E, Peters SE, Willers C, Maskell DJ, Charles IG. Single, double and triple mutants of Salmonella enterica serovar Typhimurium degP (htrA), degQ (hhoA) and degS (hhoB) have diverse phenotypes on exposure to elevated temperature and their growth in vivo is attenuated to different extents. Infect Immun. 2007;75(4):1679–1689. doi: 10.1128/IAI.01581-06. [DOI] [PubMed] [Google Scholar]
- 34.Flannagan RS, Aubert D, Kooi C, Sokol PA, Valvano MA. Burkholderia cenocepacia requires a periplasmic HtrA protease for growth under thermal and osmotic stress and for survival in vivo. Infect Immun. 2007;75:1679–1689. doi: 10.1128/IAI.01581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boehm M, Lind J, Backert S, Tegtmeyer N. Campylobacter jejuni serine protease HtrA plays an important role in heat tolerance, oxygen resistance, host cell adhesion, invasion, and transmigration. Eur J Microbiol Immunol (Bp) 2015;5:68–80. doi: 10.1556/EuJMI-D-15-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Löwer M, Weydig C, Metzler D, Reuter A, Starzinski-Powitz A, Wessler S, et al. Prediction of extracellular proteases of the human pathogen Helicobacter pylori reveals proteolytic activity of the Hp1018/19 protein HtrA. PLoS ONE. 2008;3:e3510. doi: 10.1371/journal.pone.0003510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoy B, Löwer M, Weydig C, Carra G, Tegtmeyer N, Geppert T, et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010;11:798–804. doi: 10.1038/embor.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoy B, Geppert T, Boehm M, Reisen F, Plattner P, Gadermaier G, et al. Distinct roles of secreted HtrA proteases from gram-negative pathogens in cleaving the junctional protein and tumor suppressor E-cadherin. J Biol Chem. 2012;23:10115–10120. doi: 10.1074/jbc.C111.333419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boehm M, Hoy B, Rohde M, Tegtmeyer N, Bæk KT, Oyarzabal OA, et al. Rapid paracellular transmigration of Campylobacter jejuni across polarized epithelial cells without affecting TER: role of proteolytic-active HtrA cleaving E-cadherin but not fibronectin. Gut Pathog. 2012;4:3. doi: 10.1186/1757-4749-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt TP, Perna AM, Fugmann T, Boehm M, Hiss Jan, Haller S, et al. Identification of E-cadherin signature motifs functioning as cleavage sites for Helicobacter pylori HtrA. Sci Rep. 2016;6:23264. doi: 10.1038/srep23264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abfalter CM, Schubert M, Götz C, Schmidt TP, Posselt G, Wessler S. HtrA-mediated E-cadherin cleavage is limited to DegP and DegQ homologs expressed by gram-negative pathogens. Cell Commun Signal. 2016;14(1):30. doi: 10.1186/s12964-016-0153-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Backert S, Schmidt TP, Harrer A, Wessler S. Exploiting the gastric epithelial barrier: Helicobacter pylori’s attack on tight and adherens junctions. Curr Top Microbiol Immunol. 2017;400:195–226. doi: 10.1007/978-3-319-50520-6_9. [DOI] [PubMed] [Google Scholar]
- 43.Heimesaat MM, Alutis M, Grundmann U, Fischer A, Tegtmeyer N, Boehm M, et al. The role of serine protease HtrA in acute ulcerative enterocolitis and extra-intestinal immune responses during Campylobacter jejuni infection of gnotobiotic IL-10 deficient mice. Front Cell Infect Microbiol. 2014;4:77. doi: 10.3389/fcimb.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heimesaat MM, Fischer A, Alutis M, Grundmann U, Boehm M, Tegtmeyer N, et al. The impact of serine protease HtrA in apoptosis, intestinal immune responses and extra-intestinal histopathology during Campylobacter jejuni infection of infant mice. Gut Pathog. 2014;6:16. doi: 10.1186/1757-4749-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li SR, Dorrell N, Everest PH, Dougan G, Wren BW. Construction and characterization of a Yersinia enterocolitica O:8 high-temperature requirement (htrA) isogenic mutant. Infect Immun. 1996;64:2088–2094. doi: 10.1128/iai.64.6.2088-2094.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Humphreys S, Stevenson A, Bacon A, Weinhardt AB, Roberts M. The alternative sigma factor, sigmaE, is critically important for the virulence of Salmonella typhimurium. Infect Immun. 1999;67:1560–1568. doi: 10.1128/iai.67.4.1560-1568.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Purdy GE, Hong M, Payne SM. Shigella flexneri DegP facilitates IcsA surface expression and is required for efficient intercellular spread. Infect Immun. 2002;70:6355–6364. doi: 10.1128/IAI.70.11.6355-6364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson RL, Brown LL, Kirkwood-watts D, Warren TK, Lund SA, King DS, et al. Listeria monocytogenes 10403S HtrA is necessary for resistance to cellular stress and virulence. Microbiology. 2006;74:765–768. doi: 10.1128/IAI.74.1.765-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gloeckl S, Ong VA, Patel P, Tyndall JD, Timms P, Beagley KW, et al. Identification of a serine protease inhibitor which causes inclusion vacuole reduction and is lethal to Chlamydia trachomatis. Mol Microbiol. 2013;89:676–689. doi: 10.1111/mmi.12306. [DOI] [PubMed] [Google Scholar]
- 50.Ye M, Sharma K, Thakur M, Smith AA, Buyuktanir O, Xiang X, et al. HtrA, a temperature- and stationary phase-activated protease involved in maturation of a key microbial virulence determinant, facilitates Borrelia burgdorferi infection in mammalian hosts. Infect Immun. 2016;84(8):2372–2381. doi: 10.1128/IAI.00360-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salama NR, Shepherd B, Falkow S. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol. 2004;186:7926–7935. doi: 10.1128/JB.186.23.7926-7935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tegtmeyer N, Moodley Y, Yamaoka Y, Pernitzsch SR, Schmidt V, Traverso FR, et al. Characterisation of worldwide Helicobacter pylori strains reveals genetic conservation and essentiality of serine protease HtrA. Mol Microbiol. 2016;99(5):925–944. doi: 10.1111/mmi.13276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boneca IG, Ecobichon C, Chaput C, Mathieu A, Guadagnini S, Prévost MC, et al. Development of inducible systems to engineer conditional mutants of essential genes of Helicobacter pylori. Appl Environ Microbiol. 2008;74(7):2095–2102. doi: 10.1128/AEM.01348-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Backert S, Ziska E, Brinkmann V, Zimny-Arndt U, Fauconnier A, Jungblut PR, et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2000;2(2):155–164. doi: 10.1046/j.1462-5822.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- 55.Kwok T, Backert S, Schwarz H, Berger J, Meyer TF. Specific entry of Helicobacter pylori into cultured gastric epithelial cells via a zipper-like mechanism. Infect Immun. 2002;70(4):2108–2120. doi: 10.1128/IAI.70.4.2108-2120.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449(7164):862–866. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- 57.Jiménez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, et al. Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog. 2009;5:e1000684. doi: 10.1371/journal.ppat.1000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology. 2010;139(1):239–248. doi: 10.1053/j.gastro.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Conradi J, Huber S, Gaus K, Mertink F, Royo Gracia S, Strijowski U, et al. Cyclic RGD peptides interfere with binding of the Helicobacter pylori protein CagL to integrins αVβ3 and α5β1. Amino Acids. 2012;43:219–232. doi: 10.1007/s00726-011-1066-0. [DOI] [PubMed] [Google Scholar]
- 60.Conradi J, Tegtmeyer N, Woźna M, Wissbrock M, Michalek C, Gagell C, et al. An RGD helper sequence in CagL of Helicobacter pylori assists in interactions with integrins and injection of CagA. Front Cell Infect Microbiol. 2012;2:70. doi: 10.3389/fcimb.2012.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barden S, Lange S, Tegtmeyer N, Conradi J, Sewald N, Backert S, et al. A helical RGD motif promoting cell adhesion: crystal structures of the Helicobacter pylori type IV secretion system pilus protein CagL. Structure. 2013;21:1931–1941. doi: 10.1016/j.str.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 62.Tegtmeyer N, Lind J, Schmid B, Backert S. Helicobacter pylori CagL Y58/E59 mutation turns-off type IV secretion-dependent delivery of CagA into host cells. PLoS ONE. 2014;9(6):e97782. doi: 10.1371/journal.pone.0097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5(10):816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 64.Horton ER, Humphries JD, James J, Jones MC, Askari JA, Humphries MJ. The integrin adhesome network at a glance. J Cell Sci. 2016;129:4159–4163. doi: 10.1242/jcs.192054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li B, Huang C. Regulation of EMT by STAT3 in gastrointestinal cancer. Int J Oncol. 2017;50:753–767. doi: 10.3892/ijo.2017.3846. [DOI] [PubMed] [Google Scholar]
- 66.Saadat I, Higashi H, Obuse C, Murata-Kamiya N, Umeda M, Saito Y, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- 67.Lee DG, Kim HS, Lee YS, Cha SY, Kim S, Ota I, et al. H. pylori CagA promotes snail-mediated epithelial-mesenchymal transition by reducing GSK-3 activity. Nat Commun. 2014;5:4423. doi: 10.1038/ncomms5423. [DOI] [PubMed] [Google Scholar]
- 68.Zhang YM, Noto JM, Hammond CE, Barth JL, Argraves WS, Backert S, et al. Helicobacter pylori-induced posttranscriptional regulation of H-K-ATPase α-subunit gene expression by miRNA. Am J Physiol Gastrointest Liver Physiol. 2014;306:G606–G613. doi: 10.1152/ajpgi.00333.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wiedemann T, Hofbaur S, Tegtmeyer N, Huber S, Sewald N, Wessler S, et al. Helicobacter pylori CagL dependent induction of gastrin expression via a novel αvβ5-integrin-integrin linked kinase signaling complex. Gut. 2012;61:986–996. doi: 10.1136/gutjnl-2011-300525. [DOI] [PubMed] [Google Scholar]
- 70.Kim DJ, Park JH, Franchi L, Backert S, Núñez G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1β production in Helicobacter pylori infected dendritic cells. Eur J Immunol. 2013;43:2650–2658. doi: 10.1002/eji.201243281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patel SR, Smith K, Letley DP, Cook KW, Memon AA, Ingram RJ, et al. Helicobacter pylori downregulates expression of human β-defensin 1 in the gastric mucosa in a type IV secretion-dependent fashion. Cell Microbiol. 2013;15:2080–2092. doi: 10.1111/cmi.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tenguria S, Ansari SA, Khan N, Ranjan A, Devi S, Tegtmeyer N, et al. Helicobacter pylori cell translocating kinase (CtkA/JHP0940) is pro-apoptotic in mouse macrophages and acts as auto-phosphorylating tyrosine kinase. Int J Med Microbiol. 2014;304:1066–1076. doi: 10.1016/j.ijmm.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 73.Bumann D, Aksu S, Wendland M, Janek K, Zimny-Arndt U, Sabarth N, et al. Proteome analysis of secreted proteins of the gastric pathogen H. pylori. Infect Immun. 2002;70(7):3396–3403. doi: 10.1128/IAI.70.7.3396-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roure S, Bonis M, Chaput C, Ecobichon C, Mattox A, Barrière C, et al. Peptidoglycan maturation enzymes affect flagellar functionality in bacteria. Mol Microbiol. 2012;86:845–856. doi: 10.1111/mmi.12019. [DOI] [PubMed] [Google Scholar]
- 75.Tegtmeyer N, Rivas Traverso F, Rohde M, Oyarzabal OA, Lehn N, Schneider-Brachert W, et al. Electron microscopic, genetic and protein expression analyses of Helicobacter acinonychis strains from a Bengal tiger. PLoS ONE. 2013;8:e71220. doi: 10.1371/journal.pone.0071220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tegtmeyer N, Wittelsberger R, Hartig R, Wessler S, Martinez-Quiles N, Backert S. Serine phosphorylation of cortactin controls focal adhesion kinase activity and cell scattering induced by Helicobacter pylori. Cell Host Microbe. 2011;9(6):520–531. doi: 10.1016/j.chom.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 77.Boehm M, Krause-Gruszczynska M, Rohde M, Tegtmeyer N, Takahashi S, Oyarzabal OA, et al. Major host factors involved in epithelial cell invasion of Campylobacter jejuni: role of fibronectin, integrin beta1, FAK, Tiam-1, and DOCK180 in activating Rho GTPase Rac1. Front Cell Infect Microbiol. 2011;12:17. doi: 10.3389/fcimb.2011.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang XS, Tegtmeyer N, Traube L, Jindal S, Perez-Perez G, Sticht H, et al. A specific A/T polymorphism in Western tyrosine phosphorylation B-motifs regulates Helicobacter pylori CagA epithelial cell interactions. PLoS Pathog. 2015;11(2):e1004621. doi: 10.1371/journal.ppat.1004621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hirsch C, Tegtmeyer N, Rohde M, Rowland M, Oyarzabal OA, Backert S. Live Helicobacter pylori in the root canal of endodontic-infected deciduous teeth. J Gastroenterol. 2012;47:936–940. doi: 10.1007/s00535-012-0618-8. [DOI] [PubMed] [Google Scholar]
- 80.Kumar Pachathundikandi S, Brandt S, Madassery J, Backert S. Induction of TLR-2 and TLR-5 expression by Helicobacter pylori switches cagPAI-dependent signaling leading to the secretion of IL-8 and TNF-α. PLoS ONE. 2011;6:e19614. doi: 10.1371/journal.pone.0019614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.