

Abstract
Post-genomic studies continue to highlight the potential clinical importance of protein phosphorylation signaling pathways in drug discovery. Unfortunately the dynamic range and variable stoichiometry of protein phosphorylation continues to stymie efforts to achieve comprehensive characterization of the human phosphoproteome. In this study, we develop a complementary, two-stage method for enrichment of cysteine-containing phosphopeptides combined with TMT multiplex labeling for relative quantification. Use of this approach with multi-dimension fractionation in mammalian cells yielded more than 7,000 unique cys-phosphopeptide sequences, comprising 15%–20% novel phosphorylation sites. Use of our approach in combination with pharmacologic inhibitors of the mammalian target of rapamycin complex 1 and 2 (mTORC1/2) identified several putatively novel protein substrates for the mechanistic target of rapamycin (mTOR) kinase.
Keywords: cysteine-containing phosphoproteomome, quantitative proteomics, mTOR, mTORC1/2
Introduction
Reversible phosphorylation on serine, threonine, or tyrosine amino acid chains plays a key role in numerous cellular processes in normal physiology and human disease [1–7]. Signaling pathways have proven to be ‘actionable,’ serving as bio-/pharmaco-dynamic markers or therapeutic targets [8–11]. As a result of these data, significant effort has been invested to profile protein phosphorylation by mass spectrometry [12–15]. The cumulative success of these efforts is evidenced by a rapid increase in the number of phosphorylation sites annotated within various databases [16, 17]. As one example, the number of nonredundant phosphorylation sites contained in the Phospho.ELM database [http://phospho.elm.eu.org/] increased from 1,703 [18] in 2004 to 16,470 [19] in 2008, with the most recent iteration [20] (v9.0, released in September 2010) comprising 42,574 phosphorylation sites across 48 species.
The nonlinear growth of phosphorylation data combined with estimates that 30–50% of all human proteins are phosphorylated, with stoichiometry approaching 90% in some cases, suggests that continued improvements in proteomic methods will be required to achieve comprehensive coverage of the mammalian phosphorylation landscape. Further support for this hypothesis is seen in various studies reporting refinements in existing phosphoproteomic techniques [21–24], as well as novel approaches for enrichment of subsets of the phosphoproteome [25–28]. With respect to the latter, Dong et al. recently reported the use of sequential Ti4+/IMAC with thiol-activated beads to isolate cys-phosphopeptides [29]. The authors identified a greater number of cys-phosphopeptides with their two-step enrichment strategy, as compared to the use of Ti4+/IMAC alone.
In the present work we sought to extend this technique and further interrogate the landscape of cys-phosphopeptides. To improve total coverage of the cys-phosphoproteome we implemented a workflow whereby the order of enrichment was performed in both directions (e.g., cys-phos and phos-cys). This approach enabled large-scale quantification of cys-phosphopeptides, which encompassed a significant fraction of novel phosphorylation sites. We combined our approach with multiplexed stable isotope labeling to interrogate mTOR-mediated signaling in human cells. This analysis highlights several potentially new mTOR phosphorylation substrates.
Materials and Methods
TMT labeling, peptide/protein identification
Unless otherwise noted below, we used standard methods for isotope labeling, and phosphopeptide sequence assignment. Details for these protocols are provided in Supporting Information Materials and Methods.
Reagents
mTOR inhibitors, Rapamycin and Torin1, were generously provided by Nathanael Gray (Dana-Farber Cancer Institute). Sources for other supplies and reagents are as follows: Activated Thiol Sepharose 4B beads (GE Healthcare, Piscataway, NJ); Sep-pak tC18 100 mg capacity 96-well plate (tC18; Waters, Milford, MA); Thermo SOLA C18 10 mg/2 ml capacity 96-well plate (SOLA C18; Thermo Scientific, Waltham, MA); Ni:NTA magnetic agarose beads (Qiagen, Valencia, CA); Ammonium bicarbonate (AMBIC; Sigma, St. Louis, MO); Triethylammonium bicarbonate (TEAB; Sigma), Ethanol (EtOH; Sigma); ACN (Macron, Avantor Performance Materials, Center Valley, PA); tris(2-carboxyethyl)phosphine (TCEP; Sigma); S-Methyl methanethiosulfonate (MMTS; Sigma); DTT (Sigma); Iodoacetamide (IAA; Sigma); TFA (Pierce, Rockford, IL); formic acid (FA; Sigma).
Cell Line Culture and Sample Processing
All cells (K562, HeLa S3, HEK-293E) were cultured in DMEM with 10% FBS and incubated to confluency at 37°C in 5% CO2. A total cell suspension was prepared by collecting the non-adherent cells followed by removal of adherent cells by use of Versene. The cells were washed three times with cold non-sterile PBS and centrifuged at 250g. Cells were lysed in buffer containing 7.2 M Guanidine HCl, 1:100 Sigma-Aldrich phosphatase inhibitor cocktails II and III, and 100 mM AMBIC at room temperature. Protein concentration was determined by BCA analysis (Pierce). Lysates were reduced (TCEP, 10 mM, 56°C, 30 min) and alkylated (MMTS, 20 mM, room-temperature, 30 min). Next, lysates were diluted to 100 mM AMBIC, followed by addition of trypsin (Promega) at an enzyme:protein ratio of 1:50 and incubated at 37°C over-night. After digestion, samples were acidified with 10% TFA, and desalted on a tC18 plate. tC18 plate was prepared by activating with 0.1% TFA, 80% ACN and equilibrating with 0.1% TFA. Samples were loaded on the plate and washed three times with 0.1% TFA. Peptides were eluted with 0.1% TFA, 80% ACN and lyophilized by vacuum centrifugation.
Interrogation of mTOR signaling by cys-phosphoproteomics
Actively proliferating HEK-293E cells were serum starved for 4 h before treatment with either 100 nM rapamycin, 250 nM Torin1, or vehicle control (DMSO) for 1 h. mTOR was activated by treatment with 150 nM insulin for 20 min.
Cys-Phos Enrichment
Tryptic peptides were reconstituted in 100 mM AMBIC and reduced with 100 mM TCEP at 56°C. Reduced peptides were desalted by use of a tC18 plate and then dried by vacuum centrifugation. Activated thiol sepharose 4B beads were prepared by sequentially washing the dry resin four times with water and three times with 0.5 M TEAB, with final resuspension in 0.5 M TEAB. Dried peptides were reconstituted with 0.5 M TEAB and an equal volume of 4B bead suspension with vortexing at 37°C for 1 h. After incubation, the beads were washed 3 times with 0.5 M TEAB. Cysteine-containing peptides were eluted with two sequential washes of 10 mM DTT, 0.5 M TEAB for 20 min at 37°C with vortexing. Elutions were pooled and alkylated with 50 mM IAA in the dark at room temperature. After incubation, the samples were desalted as described above, but using a SOLA C18 SPE plate (10 mg/2mL capacity). Qiagen Ni:NTA magnetic agarose beads stored as a 5% suspension in 30% ethanol were used for phosphopeptide enrichment. The magnetic beads were prepared by rinsing three times with water and then incubating in 100 mM EDTA pH 8 for 30 min. Next, the beads were rinsed three times with water and incubated in 10 mM FeCl3 for 30 min. Prior to incubation with the sample peptides, the bead pellet was rinsed three times with water and once with 80% ACN, 0.1% TFA and finally resuspended with 80% ACN, 0.1% TFA. Cysteine-containing peptides enriched by use of thiol 4B beads were reconstituted in 80% ACN, 0.1% TFA and incubated with the Fe-activated magnetic agarose NTA bead suspension for 30 min. After incubation, the supernatant was retained and the bead pellet was washed three times with 80% ACN, 0.1% TFA. The bead pellet was eluted with 1.4% ammonia by weight, 3 mM EDTA and water. Elutions were pooled and dried by vacuum centrifugation prior to LC-MS/MS analysis.
Phos-Cys Enrichment
Qiagen Ni:NTA magnetic agarose beads were activated as described above. Tryptic peptides were reconstituted in 600 μL of 80% ACN, 0.1% TFA and incubated with Fe-activated magnetic agarose NTA bead suspension for 30 min. After incubation, the supernatant was retained and the bead pellet was washed three times with 80% ACN, 0.1% TFA. The bead pellet was eluted with 1.4 % ammonia by weight, 3 mM EDTA and water. Elutions were pooled, and dried by vacuum centrifugation. Dried phosphopeptides were reconstituted in 100 mM AMBIC and then reduced with 100 mM TCEP for 30 min at 56°C. Reduced peptides were desalted using a SOLA C18 plate and dried by vacuum centrifugation. Thiol sepharose 4B beads were prepared and activated as described above. Dried peptides were reconstituted with 0.5 M TEAB and an equal volume of thiol sepharose 4B bead suspension with vortexing at 37°C for 1 h. After incubation, the beads were washed three times with 0.5 M TEAB. Peptides were eluted, alkylated, desalted, and dried as described above.
Interpretation of identified phosphorylation sites
Using Python and our pep2gene algorithm [30], we parsed the ELM, CST, and HPRD databases and compiled a combined database of phosphosites (cumulative phospho-database); for each entry in this database we derived tryptic peptides for each possible source protein, and each protein-relative phosphorylation site was recorded as a previously observed or ‘known’ site. To match this combined database against our results we performed a similar operation, where for each observed phosphopeptide we generated the list of proteins and corresponding protein-relative sites contained therein. We then ran a direct site-to-site comparison for these two databases; if any protein-relative site in the set of our phosphosites matched a site in the combined database, this phosphosite was annotated as ‘known’. In an effort to further minimize false-positives, we also included in the ‘known’ category any protein-relative site in our data which matched to the combined database within ±5 amino acids, or within a longer sequence string as defined by a contiguous block of phosphorylatable (S, T, or Y) amino acids. In this way we hoped to account for potentially confounding situations in which the phosphorylation site occurred near the protein N-terminus or resided directly adjacent to other S, T, or Y residues, and hence may be more prone to mis-assignment, particularly with lower-quality MS/MS spectra.
Results
In Silico Analysis of Potential Cys-Phosphopeptides
Estimates indicate that cysteine is found in more than 90% of human proteins but only in ~25% of all tryptic peptides [31, 32], with the latter value reduced somewhat when considering the optimum molecular weight range of peptides most amenable to traditional shotgun proteomic analysis. We verified these results based on recent releases of UniProtKB/Swiss-Prot Fungi, UniProtKB/Swiss-Prot Rodent and UniProtKB/Swiss-Prot Human (released July, 2016) (Table 1A). We then extended this analysis to phosphoproteins annotated in three common databases: Phospho.ELM (ELM) [19] [http://phospho.elm.eu.org/, v9.0 downloaded June, 2016], PhosphoSitePlus (PSP) [33] [http://www.phosphosite.org/, downloaded July, 2016] and Human Protein Reference Database (HPRD) [34] [http://www.hprd.org/, downloaded June, 2016]. Assuming trypsin digestion rules and no missed cleavages for peptides consisting of 7 to 35 amino acids, an analysis of the human component of these databases reveals a similar percentage of cysteine-containing tryptic peptides (~24%, Table 1B). In contrast, only ~16% of phosphopeptides in these databases contains a cysteine residue. Although the overlap between phosphorylated and cysteine-containing peptides is relatively low, both chemical moieties can be efficiently targeted for enrichment [31, 32, 35–49]. Interestingly the laboratory of Hanfa Zou recently demonstrated that the combined enrichment of cysteine-containing and phosphorylated-peptides enabled more efficient identification of this peptide subclass as compared to untargeted or global IMAC-based strategies [29]. Inspired by these results, we sought to further extend this paradigm to improve coverage and enable quantitative interrogation of the cys-phosphoproteome.
Table 1.
(A) In silico analysis of fungi and mammalian proteomes; (B) In silico analysis of the cys-phosphoproteome based on publically available phosphoprotein databases.
| A | |||
|---|---|---|---|
| Species | Fungi | Rodent | Human |
| Total Proteins | 32,106 | 26,460 | 20,197 |
| Cysteinyl Proteins | 28,977 | 25,608 | 19,631 |
| % Cysteinyl Proteins | 90.30% | 96.80% | 97.20% |
| Total Peptides | 739,697 | 656,056 | 530,715 |
| Cysteinyl Peptides | 115,546 | 164,797 | 134,990 |
| % Cysteinyl Peptides | 15.60% | 25.10% | 25.40% |
| Cysteinyl STY-Containing Peptides | 103,600 | 146,509 | 119,506 |
| % Cysteinyl STY-Containing Peptides | 14.01% | 22.33% | 22.52% |
| B | |||||
|---|---|---|---|---|---|
| Phospho-Database | PSP | ELM | HPRD | Cumulative | % Cys |
| Total Proteins | 19,729 | 5,374 | 11,803 | 23,844 | 97.5% |
| Cysteinyl Proteins | 18,890 | 5,228 | 10,700 | 23,244 | |
| Total Peptides | 572,705 | 200,814 | 384,041 | 753,985 | 23.9% |
| Cysteinyl Peptides | 140,281 | 42,984 | 85,948 | 179,965 | |
| PhosphoPeptides | 107,981 | 13,663 | 34,816 | 142,376 | 15.6% |
| Cys-Phosphopeptides | 18,371 | 1,659 | 3,767 | 22,143 | |
Qualitative Analysis of Cys-Phosphoproteome
We enriched the cysteine peptide pool from human K562 cells by incubating tryptic peptides from whole cell lysate with thiol-activated sepharose 4B beads (Fig. 1A). Next we enriched phosphorylated cysteine-containing peptides using Fe-NTA IMAC [50] (‘Cys-Phos’ enrichment). In parallel, we reversed the order of enrichment stages on an equal aliquot of tryptic peptides (‘Phos-Cys’ enrichment). In total, across two replicate experiments we identified 1,667 unique phosphopeptides, of which 1,643 contained at least one cysteine in the sequence (Supporting Information Table 1). These cys-phosphopeptides mapped to 1,469 unique phosphosites. Similarly, the Phos-Cys approach identified 993 cys-phosphopeptides comprising 907 phosphosites. We observed an overlap of 33% across the two enrichment approaches (Fig. 1B), while replicates of each strategy (Cys-Phos and Phos-Cys) exhibited somewhat higher reproducibility (~54%) (Supporting Information Fig. 1). These data demonstrate that both capture schemes provided high efficiency (>95%) enrichment of cys-phosphopeptides and may capture somewhat complementary sets of sequences. To understand whether phosphorylation sites within these peptides had been previously identified, we queried a compendium of post-translational modifications (PTMs) on human proteins made available through the Proteome Scout web resource [51] In our initial analysis we compared phosphorylation sites in our data with those in Proteome Scout based on amino acid position relative to the protein N-terminus. To account for possible mis-assignment of specific site of phosphorylation by the search algorithm, particularly in cases where peptides contained a contiguous span of phosphorylatable S, T, or Y residues, we allowed a ‘tolerance’ of ±5 amino acids when comparing our results to phosphorylation sites in Proteome Scout. This approached suggested that our qualitative analysis resulted in the identification of 132 (7.6%) novel sites of phosphorylation. To circumvent potential confounding variables due to protein isoforms, inclusion/exclusion of signal sequences, etc. we utilized our multiplierz environment [52] to compare phosphorylation sites based on sequence alignment. We assigned phosphorylation sites in our data to one of three categories: 1) ‘Known Phosphosite,’ comprised phosphorylation sites which matched exactly based on sequence alignment to a compendia of data available through the cumulative phospho-database; 2) ‘Ambiguous Phosphosite,’ after sequence alignment, these phosphorylation sites within our data matched to within ±5 amino acids of a previously reported phosphosite; 3) ‘Novel Phosphosite,’ comprised the set of phosphorylation sites which have not been previously reported. Based on these categories, the union of our cys-phos and phos-cys peptides (Fig. 1B) comprised 70 novel phosphorylation sites (Fig. 1C).
Figure 1. Qualitative analysis of cys-phosphoproteome.
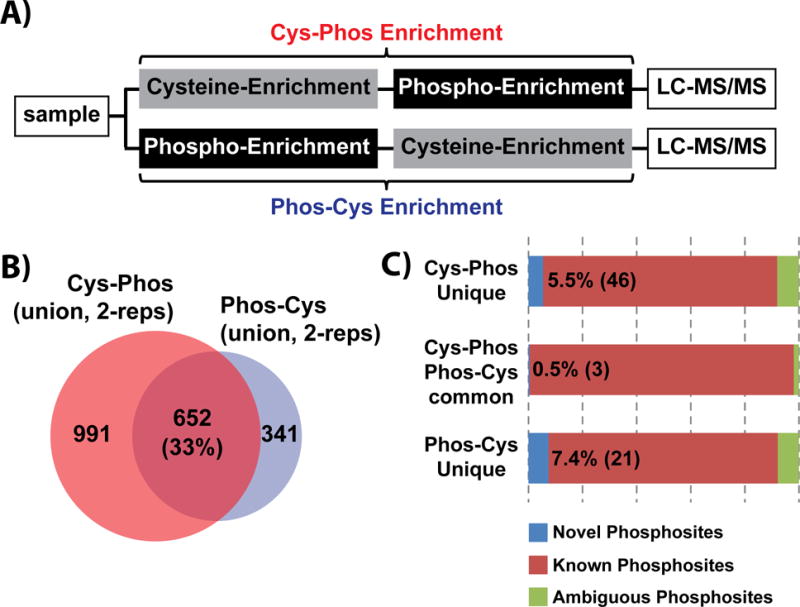
(A) Schematic workflow for cys-phosphopeptide enrichment by independent Cys-Phos and Phos-Cys approaches. (B) Number of identified cys-phosphopeptides by Cys-Phos and Phos-Cys approaches. (C) Comparison of novel phosphosites to the combined background phospho-database.
Quantitative Analysis of Cys-Phosphoproteome
We utilized TMT 6-plex reagents in conjunction with our two-stage enrichment approach to assess whether each approach (cys-phos and phos-cys) may enrich a different subset of cys-phosphopeptides (Fig. 2A). In total across duplicate analyses, we quantified 2,566 cys-phosphopeptide sequences comprising 2,241 phosphosites (Supporting Information Table 2). A histogram of peptide log2 ratios demonstrates somewhat higher abundance for peptides enriched by the Cys-Phos approach, along with a small cadre of cys-phosphopeptides which appear to be preferentially enriched by each technique (Fig. 2B).
Figure 2. Quantitative analysis of cys-phosphoproteome.
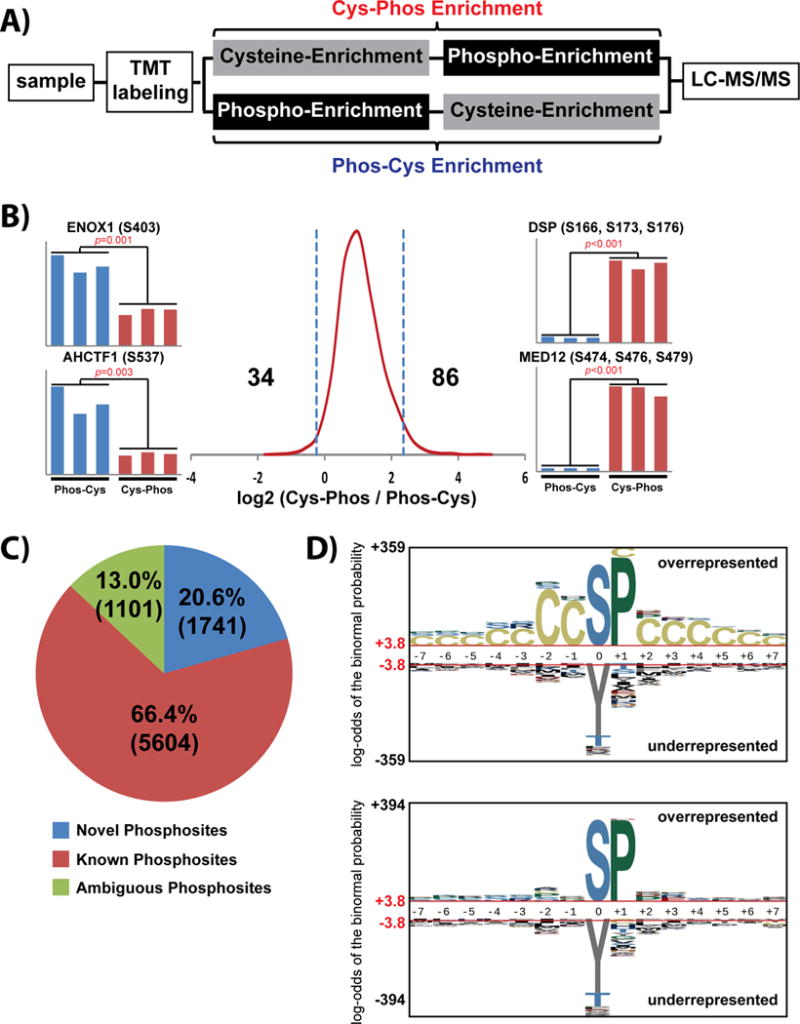
(A) Schematic workflow for quantitative profiling of cys-phosphopeptides enriched by complementary Cys-Phos and Phos-Cys approaches. (B) Histogram of log2 cys-phosphopeptide TMT reporter ion ratios. Cys-phosphopeptide ratios greater than 2 standard deviations from the median value were considered as preferentially enriched by each approach (Supporting Information Table 2). (C) Characterization of phosphorylation sites derived from cys-phosphopeptides. (D) Probabilistic sequence motif analysis of cys-phosphopeptides identified by complementary Cys-Phos and Phos-Cys approaches. Input data consisted of cys-phosphopeptides with high-confidence phosphorylation site assignment. Background data consisted of (top) the cumulative phospho-database (Supporting Information Materials and Methods) or (bottom) the subset of cys-phosphopeptides from the cumulative phospho-database
We next utilized our automated 3-dimension RP-SAX-RP platform [24] to obtain deeper coverage of the cys-phosphoproteome in HeLa cells. At a depth of 16 RP-SAX-RP fractions we quantified 10,844 cys-phosphopeptides corresponding to 8,446 phosphosites (Supporting Information Table 3). Strikingly approximately 20% of the phosphorylation sites in this analysis were novel (Fig. 2C) based on the criteria described above. To explore whether our set of cysteine-proximal phosphorylation sites comprised a specific motif we used our data as input to pLOGO [53] (Figure 2D). These results suggest a preponderance of proline-directed phosphorylation, but the uniform distribution of cysteine across the pLOGO plots indicates that cysteine itself likely does not participate in kinase substrate recognition.
Cys-Phosphoproteomic Interrogation of mTOR Kinase Signaling
We next asked whether the capability of our cys-phosphoproteomic approach to access a unique subset of mammalian phosphorylation sites would facilitate the characterization of kinase substrates. As a proof-of-principle we chose the mammalian target of rapamycin (mTOR) kinase. The mTOR pathway regulates many major cellular processes and is implicated in an increasing number of pathological conditions, including cancer, metabolic disease, and neurodegeneration [54]. These and other results [55–57] highlight mTOR as an attractive drug target [58]. We utilized TMT labeling in conjunction with the capture of cys-phosphopeptides to monitor the response of mTOR signaling in the presence of insulin stimulation, along with the inhibitors rapamycin or Torin1 (Fig. 3A). Both compounds are active against mTORC1 while mTORC2 is only sensitive to Torin1 [59–61]. Replicate analysis by RP-SAX-RP resulted in quantification of 7,563 cys-phosphopeptides which mapped to 5,784 phosphorylation sites (Supporting Information Table 4). We compared these results to our previous analysis of mTOR signaling [57] which utilized ‘global’ phosphopeptide enrichment by use of immobilized metal affinity chromatography (Fe3+–NTA IMAC). Cys-phosphopeptides in this previous study comprised 12% of all phosphopeptides, consistent with our in silico analysis (Fig. 3B and Table 1B). From a qualitative perspective our combined, serial enrichment strategy added more than 5,300 phosphorylation sites as compared to our previous study. Moreover ~18% of the phosphosites were novel (Fig. 3B), consistent with our quantitative analysis performed in HeLa cells (Fig. 2C). We next sorted potential mTOR substrates based on phosphorylation sites which were up-regulated in response to insulin and then down-regulated (relative to insulin stimulation) following inhibitor treatment. A substrate for mTORC1 would be sensitive to both Rapamycin and Torin1 (Fig. 3C; blue points), while mTORC2 substrates would only respond to Torin1 (Fig. 3C; red points). Following these criteria we identified 45 putative protein substrates and target sites for mTORC1 or mTORC2 (Fig. 4A). Of these putative mTOR-mediated phosphorylation sites, 6 (~13%) appear to be completely novel based on the criteria described above, in addition to searches in PubMed and manual interrogation through the HPRD and Proteome Scout web resources. As a first validation step we obtained synthetic peptide analogs for three of these phosphorylation sites. Insulin receptor substrate 2 (IRS2; S518, T520) was selected as a known mTORC1 substrate and served as a positive control; in addition we confirmed two novel cys-phosphorylation sites, which may represent new mTOR substrates: myotubularin-related protein 13 (SBF2; S1847) and zinc finger and BTB domain containing 2 (ZBTB2; S473) (Fig. 4B).
Figure 3. Quantification of mTOR signaling by cys-phosphoproteomics.
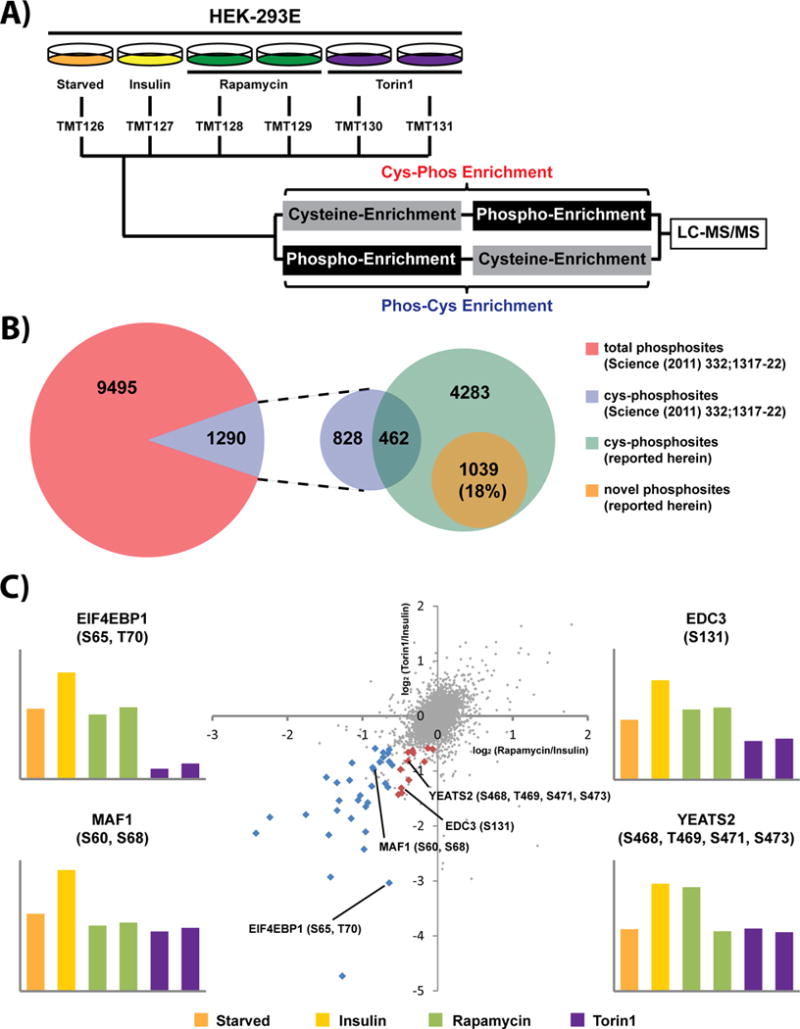
(A) mTOR was stimulated with insulin under control (DMSO) or in conjunction with Rapamycin or Torin1 treatment. (B) Comparison of phosphosites identified herein with phosphosites identified in a recent study [57] based on global IMAC enrichment. (C) Scatter plot of TMT reporter ion log2 ratios highlights phosphorylation sites likely regulated by mTOR (red and blue points).
Figure 4. Cys-phosphoproteomics identifies potential novel mTOR substrates.
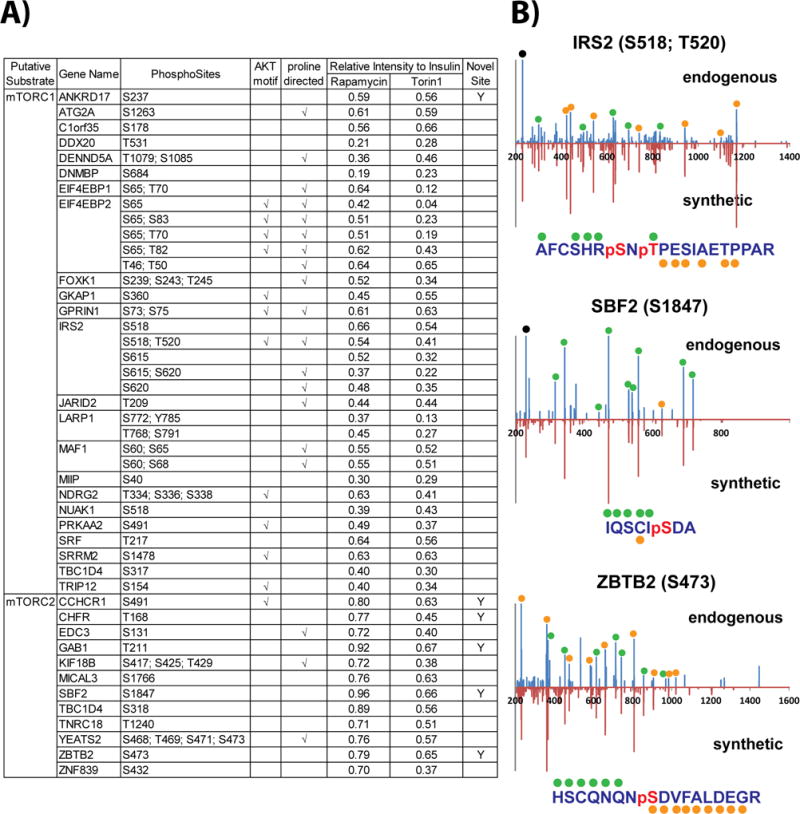
(A) List of putative substrate candidates for mTORC1 or mTORC2. (B) Mirror plots for MS/MS spectra of endogenous (top) and synthetic (bottom) cys-phosphopeptides provide analytical validation for sequence assignment and site of phosphorylation for two potential mTOR substrates. Green and orange glyphs above and below amino acid sequence indicate specific b- and y-type ions represented in the MS/MS spectra.
Discussion
Phosphoproteomic approaches are uniquely positioned to characterize signaling pathways associated with human kinases, including the identification of direct phosphorylation substrates and distal targets. These data may be suitable for use as pharmacodynamics readouts or as molecular signatures to support early detection, patient stratification, or therapeutic strategy. Despite the potential impact of these data in translational studies, comprehensive phosphoproteomic analysis remains out of reach. Estimates suggest that the human proteome may contain between 100,000 and 500,000 phosphorylation sites [26, 62, 63] on proteins which comprise multiple isoforms, splice products, etc., giving rise to a labyrinthine collection of phosphopeptides. The uneven distribution of phosphorylation on serine, threonine, and tyrosine [2, 64], along with recent studies which revealed heretofore unappreciated phosphorylation on histidine side chains, represents an additional hurdle [65]. Use of combined cys-phos and phos-cys enrichment consistently provided data comprising ~20% novel phosphorylation sites, suggesting that our method can be used in concert with existing enrichment strategies to provide deeper phosphoproteome coverage.
We used cys-phosphoproteomics to interrogate insulin-mediated mTOR signaling. TMT reporter ion ratios highlight 35 proteins whose phosphorylation patterns are consistent with potential roles in the mTORC1/2 pathways. This subset further comprises proteins which may represent underappreciated areas of mTOR biology (e.g., FOXK1, NDRG2, and CCHCR1). In addition we identified 25 cysteine-proximal phosphorylation sites contained within sequences which are consistent with other known mTOR substrates. Finally we identified 6 previously undescribed phosphorylation sites which may represent new mTOR target sites; analytical validation of two of these novel sites, S1847 on SBF2 and S473 on ZBTB2, highlight the capability of cys-phosphoproteomics to reveal new phosphorylation targets in human signaling pathways.
Supplementary Material
Significance of the study.
In this study we develop an orthogonal two-stage method to enrich cysteine-containing phosphopeptides (cys-phosphopeptides). Used in combination with TMT isotope labeling, this method enables quantification of >7,000 cys-phosphopeptides, which typically comprise some 20% novel phosphorylation sites. We utilized cys-phosphoproteomics to identify several putatively novel protein substrates of the mTOR kinase.
Acknowledgments
This work was supported by the National Cancer Institute (CA188881), the Tina Brozman Foundation, and the Strategic Research Initiative at the Dana-Farber Cancer. The authors thank Kristen Naegle (Washington University) for useful discussions and guidance with Proteome Scout.
Footnotes
The authors have no conflict of interest related to the work described herein.
References
- 1.Olsen JV, Blagoev B, Gnad F, Macek B, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 2.Hunter T. The Croonian Lecture 1997. The phosphorylation of proteins on tyrosine: its role in cell growth and disease. Philos Trans R Soc Lond B Biol Sci. 1998;353:583–605. doi: 10.1098/rstb.1998.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goss VL, Lee KA, Moritz A, Nardone J, et al. A common phosphotyrosine signature for the Bcr-Abl kinase. Blood. 2006;107:4888–4897. doi: 10.1182/blood-2005-08-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng R, Small D. Mutant FLT3 signaling contributes to a block in myeloid differentiation. Leuk Lymphoma. 2005;46:1679–1687. doi: 10.1080/10428190500261740. [DOI] [PubMed] [Google Scholar]
- 5.Iacob RE, Pene-Dumitrescu T, Zhang J, Gray NS, et al. Conformational disturbance in Abl kinase upon mutation and deregulation. Proc Natl Acad Sci U S A. 2009;106:1386–1391. doi: 10.1073/pnas.0811912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manning BD. Challenges and opportunities in defining the essential cancer kinome. Sci Signal. 2009;2:pe15. doi: 10.1126/scisignal.263pe15. [DOI] [PubMed] [Google Scholar]
- 7.Gu TL, Nardone J, Wang Y, Loriaux M, et al. Survey of activated FLT3 signaling in leukemia. PLoS One. 2011;6:e19169. doi: 10.1371/journal.pone.0019169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kubo M, Nakamura M, Tasaki A, Yamanaka N, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004;64:6071–6074. doi: 10.1158/0008-5472.CAN-04-0416. [DOI] [PubMed] [Google Scholar]
- 9.Al-Hussaini H, Subramanyam D, Reedijk M, Sridhar SS. Notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther. 2011;10:9–15. doi: 10.1158/1535-7163.MCT-10-0677. [DOI] [PubMed] [Google Scholar]
- 10.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 11.Salavert F, Hidago MR, Amadoz A, Cubuk C, et al. Actionable pathways: interactive discovery of therapeutic targets using signaling pathway models. Nucleic Acids Res. 2016;44:W212–216. doi: 10.1093/nar/gkw369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Resing KA, Ahn NG. Protein phosphorylation analysis by electrospray ionization-mass spectrometry. Methods Enzymol. 1997;283:29–44. doi: 10.1016/s0076-6879(97)83005-4. [DOI] [PubMed] [Google Scholar]
- 13.Quadroni M, James P. Phosphopeptide analysis. EXS. 2000;88:199–213. doi: 10.1007/978-3-0348-8458-7_13. [DOI] [PubMed] [Google Scholar]
- 14.Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 15.Steen H, Jebanathirajah JA, Rush J, Morrice N, Kirschner MW. Phosphorylation analysis by mass spectrometry: myths, facts, and the consequences for qualitative and quantitative measurements. Mol Cell Proteomics. 2006;5:172–181. doi: 10.1074/mcp.M500135-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Savitski MM, Lemeer S, Boesche M, Lang M, et al. Confident phosphorylation site localization using the Mascot Delta Score. Mol Cell Proteomics. 2011;10:M110 003830. doi: 10.1074/mcp.M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 18.Diella F, Cameron S, Gemund C, Linding R, et al. Phospho ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinformatics. 2004;5:79. doi: 10.1186/1471-2105-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diella F, Gould CM, Chica C, Via A, Gibson TJ. Phospho ELM: a database of phosphorylation sites–update 2008. Nucleic Acids Res. 2008;36:D240–244. doi: 10.1093/nar/gkm772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dinkel H, Chica C, Via A, Gould CM, et al. Phospho ELM: a database of phosphorylation sites–update 2011. Nucleic Acids Res. 2011;39:D261–267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002;20:301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 22.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2199–2204. doi: 10.1073/pnas.0611217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ficarro SB, Zhang Y, Carrasco-Alfonso MJ, Garg B, et al. Online nanoflow multidimensional fractionation for high efficiency phosphopeptide analysis. Mol Cell Proteomics. 2011;10:O111 011064. doi: 10.1074/mcp.O111.011064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersson L, Porath J. Isolation of phosphoproteins by immobilized metal (Fe3+) affinity chromatography. Anal Biochem. 1986;154:250–254. doi: 10.1016/0003-2697(86)90523-3. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Zha X, Tan Y, Hornbeck PV, et al. J Biol Chem. 2002;277:39379–39387. doi: 10.1074/jbc.M206399200. [DOI] [PubMed] [Google Scholar]
- 27.Kinoshita E, Yamada A, Takeda H, Kinoshita-Kikuta E, Koike T. Novel immobilized zinc(II) affinity chromatography for phosphopeptides and phosphorylated proteins. J Sep Sci. 2005;28:155–162. doi: 10.1002/jssc.200401833. [DOI] [PubMed] [Google Scholar]
- 28.Thingholm TE, Larsen MR. The use of titanium dioxide micro-columns to selectively isolate phosphopeptides from proteolytic digests. Methods Mol Biol. 2009;527:57–66, xi. doi: 10.1007/978-1-60327-834-8_5. [DOI] [PubMed] [Google Scholar]
- 29.Dong M, Bian Y, Dong J, Wang K, et al. Selective Enrichment of Cysteine-Containing Phosphopeptides for Subphosphoproteome Analysis. J Proteome Res. 2015;14:5341–5347. doi: 10.1021/acs.jproteome.5b00830. [DOI] [PubMed] [Google Scholar]
- 30.Askenazi M, Marto JA, Linial M. The complete peptide dictionary–a meta-proteomics resource. Proteomics. 2010;10:4306–4310. doi: 10.1002/pmic.201000270. [DOI] [PubMed] [Google Scholar]
- 31.Liu T, Qian WJ, Chen WN, Jacobs JM, et al. Improved proteome coverage by using high efficiency cysteinyl peptide enrichment: the human mammary epithelial cell proteome. Proteomics. 2005;5:1263–1273. doi: 10.1002/pmic.200401055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin D, Li J, Slebos RJ, Liebler DC. Cysteinyl peptide capture for shotgun proteomics: global assessment of chemoselective fractionation. J Proteome Res. 2010;9:5461–5472. doi: 10.1021/pr1007015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, et al. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37:D767–772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clements A, Johnston MV, Larsen BS, McEwen CN. Fluorescence-based peptide labeling fractionation strategies for analysis of cysteine-containing peptides. Anal Chem. 2005;77:4495–4502. doi: 10.1021/ac050247k. [DOI] [PubMed] [Google Scholar]
- 36.Dai J, Wang J, Zhang Y, Lu Z, et al. Enrichment and identification of cysteine-containing peptides from tryptic digests of performic oxidized proteins by strong cation exchange LC and MALDI-TOF/TOF MS. Anal Chem. 2005;77:7594–7604. doi: 10.1021/ac0506276. [DOI] [PubMed] [Google Scholar]
- 37.Chen SH, Hsu JL, Lin FS. Fluorescein as a versatile tag for enhanced selectivity in analyzing cysteine-containing proteins/peptides using mass spectrometry. Anal Chem. 2008;80:5251–5259. doi: 10.1021/ac800436j. [DOI] [PubMed] [Google Scholar]
- 38.Raftery MJ. Enrichment by organomercurial agarose and identification of cys-containing peptides from yeast cell lysates. Anal Chem. 2008;80:3334–3341. doi: 10.1021/ac702539q. [DOI] [PubMed] [Google Scholar]
- 39.Shiu HY, Chan TC, Ho CM, Liu Y, et al. Chemistry. 2009;15:3839–3850. doi: 10.1002/chem.200800669. [DOI] [PubMed] [Google Scholar]
- 40.Palani A, Lee JS, Huh J, Kim M, et al. Selective enrichment of cysteine-containing peptides using SPDP-functionalized superparamagnetic Fe(3)O(4)@SiO(2) nanoparticles: application to comprehensive proteomic profiling. J Proteome Res. 2008;7:3591–3596. doi: 10.1021/pr800067x. [DOI] [PubMed] [Google Scholar]
- 41.Giron P, Dayon L, David F, Sanchez JC, Rose K. Enrichment of N-terminal cysteinyl-peptides by covalent capture. J Proteomics. 2009;71:647–661. doi: 10.1016/j.jprot.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 42.Gygi SP, Rist B, Griffin TJ, Eng J, Aebersold R. Proteome analysis of low-abundance proteins using multidimensional chromatography and isotope-coded affinity tags. J Proteome Res. 2002;1:47–54. doi: 10.1021/pr015509n. [DOI] [PubMed] [Google Scholar]
- 43.Gygi SP, Rist B, Gerber SA, Turecek F, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 44.Wang S, Regnier FE. Proteomics based on selecting and quantifying cysteine containing peptides by covalent chromatography. J Chromatogr A. 2001;924:345–357. doi: 10.1016/s0021-9673(01)00961-x. [DOI] [PubMed] [Google Scholar]
- 45.Liu T, Qian WJ, Strittmatter EF, Camp DG, 2nd, et al. High-throughput comparative proteome analysis using a quantitative cysteinyl-peptide enrichment technology. Anal Chem. 2004;76:5345–5353. doi: 10.1021/ac049485q. [DOI] [PubMed] [Google Scholar]
- 46.Liu T, Qian WJ, Camp DG, Smith RD., 2nd The use of a quantitative cysteinyl-peptide enrichment technology for high-throughput quantitative proteomics. Methods Mol Biol. 2007;359:107–124. doi: 10.1007/978-1-59745-255-7_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansen KC, Schmitt-Ulms G, Chalkley RJ, Hirsch J, et al. Mass spectrometric analysis of protein mixtures at low levels using cleavable 13C-isotope-coded affinity tag and multidimensional chromatography. Mol Cell Proteomics. 2003;2:299–314. doi: 10.1074/mcp.M300021-MCP200. [DOI] [PubMed] [Google Scholar]
- 48.Zhou H, Ranish JA, Watts JD, Aebersold R. Quantitative proteome analysis by solid-phase isotope tagging and mass spectrometry. Nat Biotechnol. 2002;20:512–515. doi: 10.1038/nbt0502-512. [DOI] [PubMed] [Google Scholar]
- 49.Li S, Zeng D. CILAT–a new reagent for quantitative proteomics. Chem Commun (Camb) 2007:2181–2183. doi: 10.1039/b700109f. [DOI] [PubMed] [Google Scholar]
- 50.Ficarro SB, Adelmant G, Tomar MN, Zhang Y, et al. Magnetic bead processor for rapid evaluation and optimization of parameters for phosphopeptide enrichment. Anal Chem. 2009;81:4566–4575. doi: 10.1021/ac9004452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matlock MK, Holehouse AS, Naegle KM. ProteomeScout: a repository and analysis resource for post-translational modifications and proteins. Nucleic Acids Res. 2015;43:D521–530. doi: 10.1093/nar/gku1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parikh JR, Askenazi M, Ficarro SB, Cashorali T, et al. multiplierz: an extensible API based desktop environment for proteomics data analysis. BMC Bioinformatics. 2009;10:364. doi: 10.1186/1471-2105-10-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Shea JP, Chou MF, Quader SA, Ryan JK, et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nat Methods. 2013;10:1211–1212. doi: 10.1038/nmeth.2646. [DOI] [PubMed] [Google Scholar]
- 54.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 56.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsu PP, Kang SA, Rameseder J, Zhang Y, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smolewski P. Recent developments in targeting the mammalian target of rapamycin (mTOR) kinase pathway. Anticancer Drugs. 2006;17:487–494. doi: 10.1097/00001813-200606000-00001. [DOI] [PubMed] [Google Scholar]
- 59.Huang S, Houghton PJ. Mechanisms of resistance to rapamycins. Drug Resist Updat. 2001;4:378–391. doi: 10.1054/drup.2002.0227. [DOI] [PubMed] [Google Scholar]
- 60.Huang S, Bjornsti MA, Houghton PJ. Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther. 2003;2:222–232. doi: 10.4161/cbt.2.3.360. [DOI] [PubMed] [Google Scholar]
- 61.Thoreen CC, Kang SA, Chang JW, Liu Q, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mitchell P. Proteomics retrenches. Nat Biotechnol. 2010;28:665–670. doi: 10.1038/nbt0710-665. [DOI] [PubMed] [Google Scholar]
- 63.Lemeer S, Heck AJ. The phosphoproteomics data explosion. Curr Opin Chem Biol. 2009;13:414–420. doi: 10.1016/j.cbpa.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 64.Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A. 1980;77:1311–1315. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Srivastava S, Panda S, Li Z, Fuhs SR, et al. Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. Elife. 2016;5 doi: 10.7554/eLife.16093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.