

SUMMARY
Besides circadian rhythms, oscillations cycling with a 12h period exist. However, the prevalence, origin, regulation and function of mammalian 12h rhythms remain elusive. Utilizing an unbiased mathematical approach identifying all superimposed oscillations, we uncovered prevalent 12h gene expression and metabolic rhythms in mouse liver, coupled with a physiological 12h unfolded protein response oscillation. The mammalian 12h rhythm is cell-autonomous, driven by a dedicated 12h pacemaker distinct from the circadian clock and can be entrained in vitro by metabolic and ER stress cues. Mechanistically, we identified XBP1s as a transcriptional regulator of the mammalian 12h-clock. Down-regulation of the 12h gene expression strongly correlates with human hepatic steatosis and steatohepatitis, implying its importance in maintaining metabolic homeostasis. The mammalian 12h rhythm of gene expression also is conserved in nematodes and crustaceans, indicating an ancient origin of the 12h-clock. Our work sheds new light on how perturbed biological rhythms contribute to human disease.
eTOR blurb
Besides circadian rhythms, oscillations with ~12h period exist but their origin, regulation and function remain elusive. Zhu et al. show that a cell-autonomous 12h-clock functions independently from the circadian clock to coordinate ER and mitochondria functions. The 12h-clock is conserved in nematodes, crustaceans and mammals and likely circatidal in origin.

INTRODUCTION
In addition to the well-characterized 24h circadian rhythm, biological rhythms that cycle with shorter periods also exist (Hughes et al., 2009). For example, marine animals of the coastal regions harbor a dominant circatidal clock in tuned to the ~12h ebb and flow of the tides (Wilcockson and Zhang, 2008). A small cluster of genes cycling with a 12h period was identified in several peripheral murine tissues (Hoogerwerf et al., 2008; Hughes et al., 2009; Zhang et al., 2014). In humans, body temperature, blood pressure, cognitive performance, circulating hormones and sleep patterns all exhibit 12h rhythms (Ayala et al., 1990; Bjerner et al., 1955; Broughton and Mullington, 1992; Colquhoun et al., 1978). Altered 12h rhythms also were reported in human diseases (Haus et al., 2001; Otsuka et al., 1997). These data collectively imply that the 12h rhythm is potentially important for systemic homeostasis and its dysregulation may lead to the development and progression of metabolic diseases.
Among these 12h cycling genes, the most intriguing discovery was the identification of ~200 genes cycling with a 12h period in mouse liver (Hughes et al., 2009). However, since the COSOPT method used to identify 12h cycling transcripts in this study requires the user to pre-assign a period range (Refinetti et al., 2007); only dominant 12h genes were identified. Thus, 200 significantly underestimates the true prevalence of 12h cycling genes, as the vast majority are likely to be superimposed by other oscillations, and thus evade detection.
To be able to both comprehensively identify novel genes with superimposed 12h rhythms, we employed an unbiased mathematical approach called the “eigenvalue/pencil” method to identify all superimposed oscillations from high-resolution time series data and identified a total of 3,652 hepatic genes with 12h rhythms. These genes are enriched in endoplasmic reticulum (ER) and mitochondria metabolism and quality control pathways, indicating that the mammalian 12h clock orchestrates Coordinated Rhythms of ER and Mitochondria Actions (CREMA). We further demonstrated that a dedicated 12h pacemaker distinct from the circadian clock in mammalian cells controls the majority of 12h rhythmic gene expression and metabolism. The 12h rhythm of gene expression is cell-autonomous, can be established in vitro in response to metabolic stress and/or ER stress cues and loss of 12h cycling gene expression strongly correlates with progression to nonalcoholic fatty liver disease (NAFLD) in humans. Mechanistically, we identified the spliced form of XBP1 (XBP1s), an unfolded protein response (UPR) transcription factor, as a key transcriptional regulator of the 12h rhythm. Intriguingly, the 12h rhythm of UPR and metabolic gene expression is evolutionarily conserved in nematodes. Moreover, the 12h rhythm of mitochondrial DNA (mtDNA)-encoded gene transcription, which was previously only found in marine crustaceans harboring circatidal rhythms, also is conserved in mammals and nematodes, suggesting strongly that the 12h clock has an ancient origin.
RESULTS
The eigenvalue/pencil method identifies all superimposed biological oscillations in an unbiased manner
To comprehensively identify both dominant and superimposed biological oscillations in an unbiased manner, we utilized an eigenvalue/pencil approach that was previously developed for spectrum analysis in the digital signal processing field. Exerting no constraints on periods, it permits the unbiased identification of various superimposed periodic oscillations from a given time series dataset. We initially applied our approach to a high resolution hepatic gene expression microarray from mice kept under constant darkness (analyzed at 1h interval for 48 hours) (Hughes et al., 2009) and analyzed a total of 18,108 hepatic genes.
The vast majority of oscillations identified were circadian rhythms and oscillations that cycle at different harmonics (~12h, 8h, 4h, etc.) of the circadian rhythm (Figures 1AC, Table S1). We identified a total of 5,005 genes with circadian rhythms including all known core clock genes, which is a significant increase compared to an earlier study using the COSOPT method (Hughes et al., 2009) (Figures S1A–E). For the 3,519 newly identified circadian genes, a significant portion are superimposed by other dominant oscillations of smaller periods (Figure S1D), thus circumventing detection by the original method. Importantly, genes with superimposed circadian rhythms reveal drastically altered cycling patterns in BMAL1−/− mice (BMAL1 KO) (Yang et al., 2016) (Figure S1F), while genes without superimposed circadian rhythm exhibit similar diurnal gene expression profiles between wild-type (WT) and BMAL1 KO mice, with examples including Eif2ak3 (also known as Perk) and Gmppb (Figure S1G). Further, our method demonstrates that their unique oscillatory patterns are largely composed of three oscillations with periods of ~12h, 8h and 6h (Figure S1G). Finally, by comparing oscillations identified from different microarray probes against the same gene (Figures S2A–C), we concluded that oscillations with periods of ~12h and ~8h also are robust rhythms with potential biological significance. Taken together, these results indicate that our eigenvalue/pencil method can accurately identify superimposed oscillations from high-resolution microarray in an unbiased manner.
Figure 1. The eigenvalue/pencil method reveals prevalence of 12h hepatic mRNA oscillations. (A).
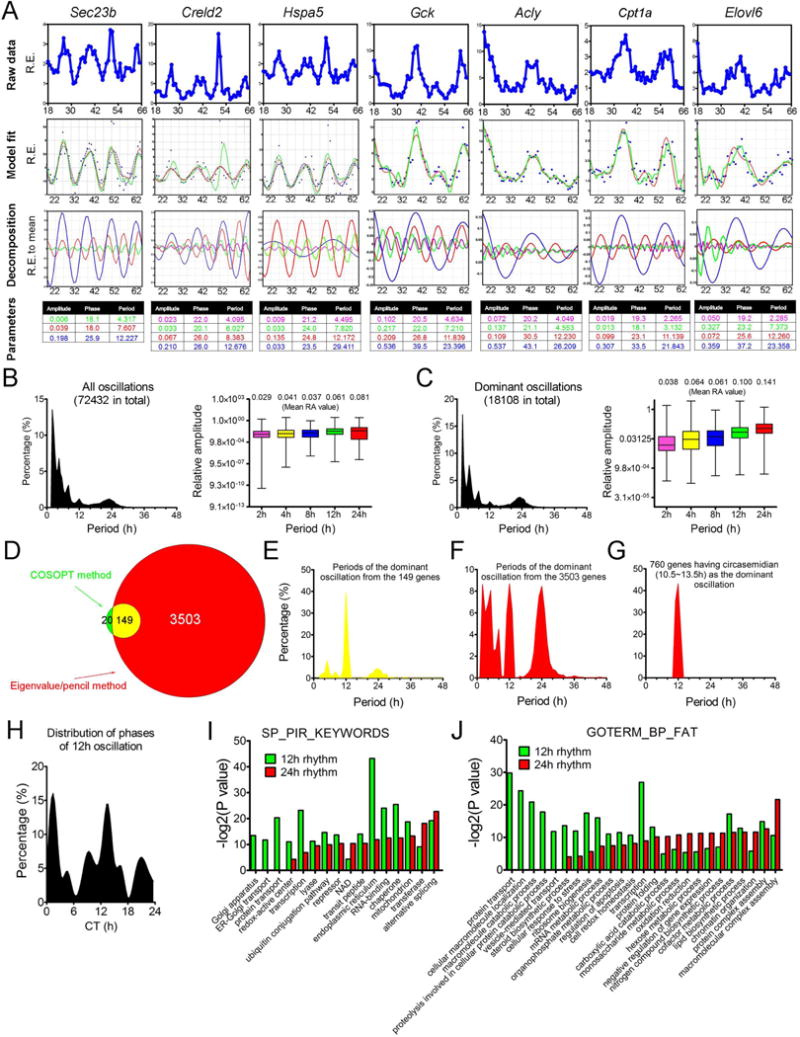
Decomposition of gene oscillations by the eigenvalue/pencil method. Top row: raw microarray data; second row: fitting two different models (5th-order approximation in dashed red and 9th-order approximation in solid green) to raw data (blue dots); third row: superimposed oscillations revealed by the 9th-order approximation; fourth row: amplitudes, phases and periods of different oscillations with the color matching the different oscillations depicted in the third row. (B–C) The distribution of periods (left) and average amplitudes (right) of all (B) or dominant (C) oscillations identified. (D) Venn diagram comparison of the number of 12h genes identified via the eigenvalue/pencil or the COSOPT method. (E–G) The distributions of periods of the dominant oscillations from the 149 commonly found 12h genes (E), the 3,503 12h genes only identified by the eigenvalue method (F) or the 760 dominant 12h genes identified by the eigenvalue method (G). (H) The distribution of phases of all 12h genes identified by the eigenvalue approach. (I–J) GO analyses revealing top-enriched SP_PIR_KEYWORDS (I) or GOTERM_BP_FAT (J) terms in all 12h and circadian genes identified by the eigenvalue approach.
Prevalent hepatic 12h mRNA oscillation of metabolism and ER stress/UPR genes
In addition to circadian genes, we identified a total of 3,652 (20%) and 6,294 (35%) genes having ~12h and 8h oscillations (Tables S1 and 2), respectively, significantly expanding their repertoires compared to the previous study (Hughes et al., 2009) (Figures 1D–G, S2D-G). Our method also outperforms JTK_CYCLE (Hughes et al., 2010), ARSER (Yang and Su, 2010) and RAIN (Thaben and Westermark, 2014) methods in robustly uncovering novel genes with superimposed 12h rhythms (Figures S2H-S and Table S3). Of these 12h and 8h genes, 760 and 1,430 of them are dominant (whose 12h or 8h amplitudes are the greatest among all identified oscillations) (Figures 1G, S2G and Table S2). Overall, the average amplitude gradually decreases with shorter periods, with those of 12h oscillations exhibiting the second largest amplitude (Figures 1B–C).
We focused on the 12h rhythms due to their unexpected prevalence. The phases of 12h rhythms are heavily restricted to dawn (CT0) and dusk (CT12) (Figure 1H), contrasting with the evenly-distributed phases of circadian rhythm (Koike et al., 2012). Gene Ontology (GO) terms preferentially associated with 12h genes are highly enriched for ER and metabolism-related biological pathways (Figures 1I,J, S2T and Table S4). While 12h are often the dominant oscillations in ER-related genes, 12h cycling metabolism genes often have superimposed circadian rhythms (Table S2). It is worth mentioning that these GO terms are not mutually exclusive and a significant number of metabolism genes are known to be involved in ER homeostasis and vice versa (Wang et al., 2011). Overall, we identified a total of 91 key ER/metabolism genes (Table S2) with 12h amplitude greater than 0.1. These results collectively demonstrate that the 12h rhythm of gene expression is far more prevalent than was initially appreciated and one of the major functions of the 12h rhythms is to coordinate ER stress/UPR with metabolism to ensure systemic homeostasis in metabolic tissues.
12h rhythms of ER/metabolism genes are independent from the circadian clock
The relationship of the 12h rhythm with circadian rhythm represents a central unanswered question. In theory, the 12h rhythm either can be established by two circadian transcription activators or repressors appearing in anti-phase (Westermark and Herzel, 2013), or can be established by ‘separate’ 12h clock machinery independent from the circadian clock. To distinguish between these two possibilities, we first examined the mathematical relationships among the three most prominent oscillations identified from each of the 91 ER/metabolism genes with strong 12h rhythms. The angles among different oscillations are approximately 90 degrees in most situations, suggesting that the 12h oscillation (and also 8 h oscillation) is largely mathematically independent from the circadian rhythm (Table S2). To verify the mathematical results empirically, we analyzed mouse hepatic RNA-Seq from WT and BMAL1 KO mice for these 91 genes under constant darkness conditions (Yang et al., 2016). We reasoned that if the 12h rhythm is independent from the circadian clock, one would expect to observe more discernible 12h rhythms of gene expression in BMAL1 KO mice. We classified these 91 genes into four clusters based upon whether the superimposed 12h rhythm is dominant or not. For the 51 dominant 12h genes, all but 7 (Figure 2A-cluster II) revealed perceptible ~12 rhythms in BMAL1 KO mice (i.e. Acly and Ddit3). For the 40 remaining 12h non-dominant genes, 17 lack superimposed circadian rhythms (Figure 2A-cluster III) and their gene expression profiles are similar between WT and BMAL1 KO mice (i.e. Eif2ak3 and Pparg). The remaining 23 genes have non-dominant 12h rhythms superimposed with both circadian rhythms and other oscillations of smaller periods (Figure 2A-cluster IV). Thus, in BMAL1 KO mice the expression profiles often exhibited 2 non-symmetrical peaks within one diurnal cycle (i.e. Fasn and Gck), consistent with the superimposition patterns of 12h rhythms and oscillations of smaller periods. As an important control, we failed to observe discernible 12h rhythms of gene expression in BMAL1 KO mice for genes lacking 12h rhythms in WT mice (Figure S3A). The independent relationship between 12h and 24h rhythms is 1) further validated in CLOCK mutant mice (Miller et al., 2007) (Figures S3B–C) and 2) supported by the spatial segregation of 12h and 24h cycling enhancer RNAs (eRNAs) genes harboring superimposed 24h and 12h mRNA oscillations (Fang et al., 2014) (Figures S3D–F). These observations demonstrate that the 12h rhythm of gene expression, especially those involved in ER/metabolic pathways, is driven by a dedicated 12h pacemaker that is distinct from the circadian clock in mammalian cells.
Figure 2. Hepatic 12h oscillations are independent from the circadian clock.
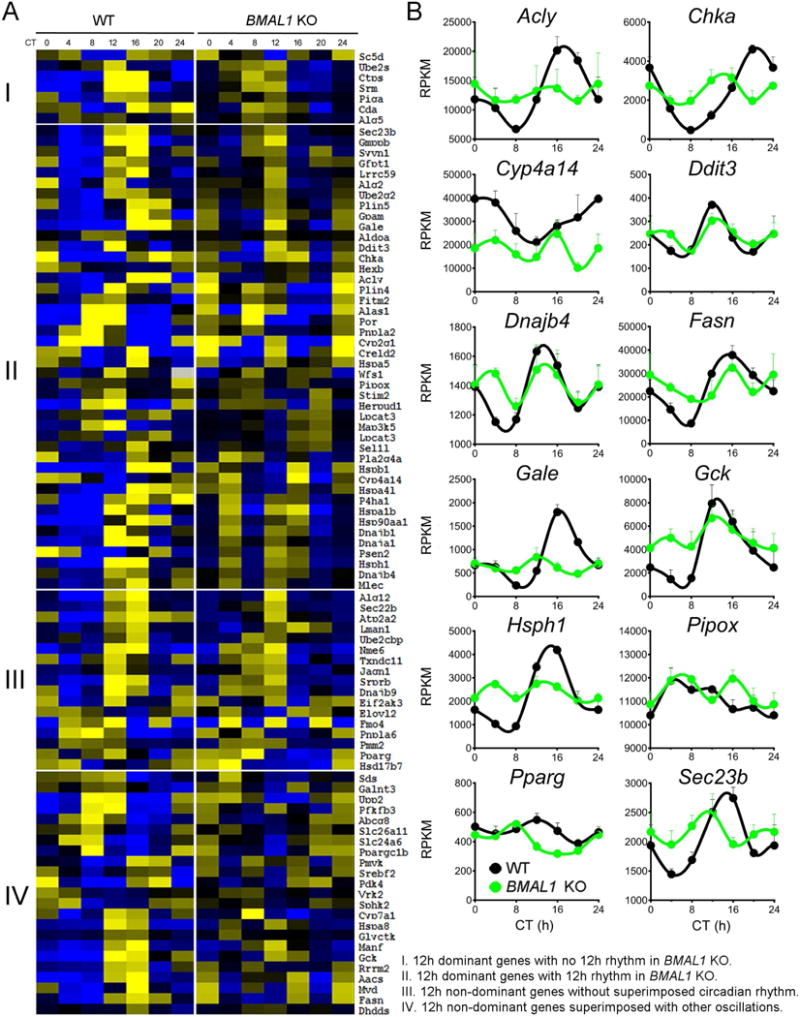
Heatmap (A) and representative RPKM normalized gene expression (B) from WT and BMAL1 KO mouse under constant darkness conditions. Data are graphed as the mean ± SEM (n = 4).
The 12h rhythm of gene expression can be established by metabolic and ER stress cues in vitro
Our findings raise an important question: Can the 12h rhythm of gene expression be found in vitro or does its establishment require systemic cues only found in vivo as previously suggested (Cretenet et al., 2010; Hughes et al., 2009)? Applying the eigenvalue/pencil method to a high-resolution microarray of NIH3T3 cells synchronized by forskolin (Hughes et al., 2009) revealed no 12h rhythms in vitro (Figure S4A). While these data may simply imply that the 12h rhythm of gene expression is not found in vitro, an alternative explanation is that forskolin is only capable of synchronizing the circadian clock but not the circadian clock-independent 12h clock; therefore the 12h rhythm is not observed in vitro in this study.
To seek evidence supporting the latter hypothesis, we searched for other chemical ‘cues’ that may synchronize the 12h clock in vitro. Since the 12h genes are enriched in UPR pathways, we speculated that treating cells transiently with an ER stress inducer tunicamycin (Tu) might establish the 12h mRNA rhythms. Of the six genes that are responsive to Tu (Figure S4B), a 2h Tu shock (25ng/ml) is sufficient to establish robust 12h mRNA oscillations in MEFs, which can persist for at least 48h after the initial induction (Figure 3A). The phase and amplitude of 12h mRNA oscillations are dependent on the dose of Tu (Figure S4E). The 12h oscillations of gene expression in vitro also are independent from the circadian clock as Bmal1 ablation fails to affect their 12h rhythms (Figure 3B). These data are consistent with the observation that a 2h Tu induction fails to establish the circadian clock in MEFs (Figure S4C).
Figure 3. 12h rhythm of gene expression can be synchronized by ER and metabolic stress cues.
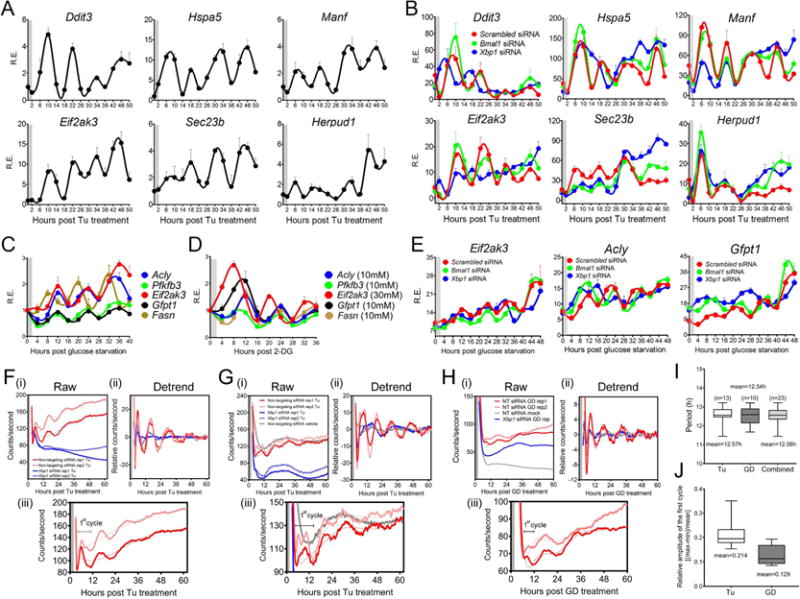
MEFs were treated with Tu (A) or were transfected with different siRNAs and treated with Tu (B) and qPCR were performed post Tu. (C–D) MEFs were glucose depleted (C) or treated with different concentrations of 2-DG (D) for 2h. (E) MEFs were transfected with different siRNAs and glucose depleted for 2h and qPCR was performed post-GD. (F–J) Real-time luminescence recording of non-targeting or Xbp1 siRNA-transfected Eif2ak3-dluc MEFs in response to vehicle (mock) control, Tu (F,G) or glucose depletion treatment (H). Raw (i), detrended (ii) and magnified graphs (iii) are provided. For Tu treatment, two independent experiments are shown. Gray boxes indicate the duration of treatment. (I) Calculated periods of Tu, GD-entrained and combined oscillations. (J) Relative amplitudes of the first cycle in Tu and GD-entrained oscillations. Data are graphed as the mean ± SEM (n=3) for qPCR.
We expanded our search for other 12h clock-synchronizing cues that are more physiologically relevant than Tu. Because of the strong enrichment of metabolic genes with 12h rhythms, we reasoned that a transient metabolic stress like glucose depletion (GD) or 2-deoxy-D-glucose (2-DG) treatment, which also leads to ER stress (Xu et al., 2005), may be sufficient to establish the 12h rhythms. As expected, a 2h GD or 2-DG treatment establishes the 12h rhythms of genes involved in glucose and fatty acid metabolism such as Gfpt1 and Acly (Figures 3C–D). GD and 2-DG also established the 12h rhythm of UPR gene Eif2ak3 (Figures 3C–D). Despite smaller amplitudes, GD also can establish circadian oscillation of core circadian clock genes Bmal1 and Rev-erbα with similar phases compared to Dex-induced rhythms (Figures S4I–J). GD-induced 12h rhythms are also independent of the circadian clock (Figures 3E, S4J).
To further confirm these qPCR results, we generated a MEF line stably expressing destabilized luciferase driven by an 1kb Eif2ak3 promoter (Eif2ak3-dluc) and subjected the cells to real-time luminescence recording. As shown in Figures 3F–H, a 2h Tu or GD treatment, but not vehicle or mock treatment, is sufficient to establish the robust ~12h rhythm of luciferase activity, with Tu being a stronger Zeitgeber than GD (Figure 3J). We calculated the period of Eif2ak3-dluc oscillation as 12.56 ± 0.10h (mean±SEM) (Figure 3I). These data further suggest that by regulating genes involved in metabolism and UPR and responding to metabolic and ER stress cues, the mammalian 12h clock functions to coordinate physiologically dynamic metabolic and ER stress rhythms.
The mammalian 12h rhythm of gene expression is cell-autonomous
We next addressed whether the observed 12h rhythm in vitro is cell autonomous. We generated another MEF line stably expressing destabilized GFP driven by the same 1kb Eif2ak3 promoter (Eif2ak3-dGFP) and subjected these cells to time-lapse microscopy in the absence of external cues. Although during the imaging, the cells were cultured in serum-free medium, we still observed cell proliferation (Figure 4), likely because the parental MEFs were previously immortalized by SV40 and resistant to serum starvation-induced cell cycle arrest (Stashi et al., 2014). For rapidly proliferating cells (doubling time <20h), we decided to quantify the combined dGFP intensities of the daughter (and granddaughter) cells arising from the same original single cell after every cell division event (termed cell lineages). After mathematically correcting for the baseline dGFP changes, we observed a robust ~12h period of dGFP oscillation in single cell lineages (Figures 4A–F, S4K, Supplemental movie 1). Intriguingly, the ~12h rhythm of dGFP oscillation in daughter cells is not reset by the cell divisions and the phases of oscillations are synchronized between the two daughter cells as well (Figures 4A–C and Supplemental movie 1), which further justifies our method of tracking the whole cell lineages following cell divisions. Of a total of 89 single cell lineage recordings, the period of most dominant Eif2ak3-dGFP oscillations centered around ~12h (Figure 4G) and the period of cell-autonomous Eif2ak3-dGFP oscillation was calculated to be 12.59 ± 0.11h (mean±SEM), consistent with population luminescence results. Further, the average amplitudes of cell-autonomous 12h Eif2ak3-dGFP oscillation is 0.2571 (Figure S4L), larger than that of Tu or GD-entrained population amplitudes. Superimposed oscillations with smaller amplitudes were also identified from single cell recordings that cycle with periods of 8.45 ± 0.08h and 6.45 ± 0.06h, respectively. Intriguingly, these periods are consistent with the decomposition results of hepatic Eif2ak3 expression in vivo (Figure S1G).
Figure 4. The mammalian 12h rhythm of gene expression is cell-autonomous.

Eif2ak3-dGFP cells were subjected to time-lapse imaging. Representative images (A,D) and quantifications of both raw and mathematically detrended data (B,C,E,F). (G) Distribution of the periods of dominant oscillations from 89 single cell recordings.
Prevalence of 12h rhythm of hepatic protein expression
In addition to the prevalence of 12h mRNA rhythms, we also identified 434 (~15%) hepatic proteins cycling with a period of ~12h by analyzing a published time series hepatic proteomics dataset (Robles et al., 2014) (Figure S5A). A limited ~35% overlap with 12h cycling mRNAs indicates that both transcriptional and post-transcriptional controls contribute to the regulation of 12h rhythms of hepatic gene expression (Table S5), which is consistent with the known limited overlap between hepatic 24h cycling mRNA and proteins (Robles et al., 2014). GO analysis of 12h cycling proteins revealed strong enrichment of ER, and intriguingly, mitochondria-associated metabolism pathways (Figures 5A, S5B–C and Table S5). In light of these findings, we coined the term Coordinated Rhythms of ER and Mitochondria Actions (CREMA) to reflect the coupled rhythmic actions of the two organelles in maintaining systemic metabolic homeostasis and stress response (Figure S6A). We validated several of their 12h rhythms by high-resolution immunoblot analysis (Figures S5D–F).
Figure 5. XBP1s transcriptionally regulates the mammalian 12h clock. (A).
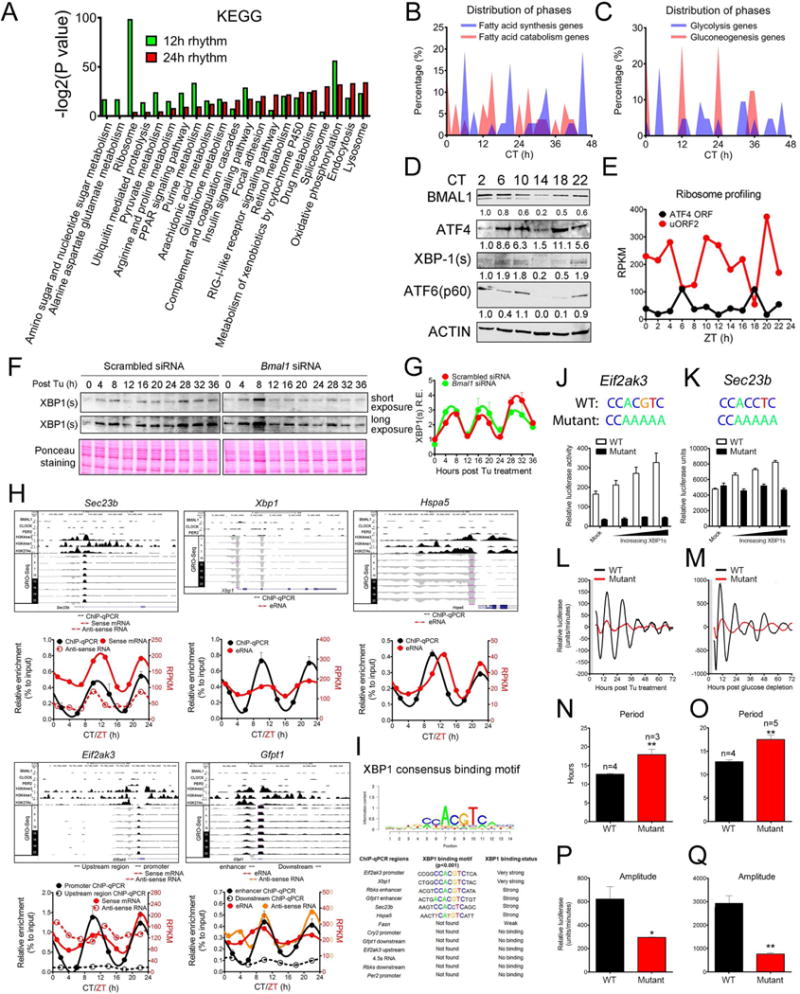
GO analysis of 12h and 24h cycling proteins (B–C) Distribution of phases for 12h cycling fatty acid (B) and glucose metabolism genes (C). (D) Immunoblot of hepatic nuclear levels of UPR TFs and BMAL1 under CT conditions. (E) Quantification of ribosome-binding intensity on hepatic ATF4 ORF or upstream open reading frame 2 (uORF2) by ribosome profiling at different ZTs. (F–G) Representative immunoblot (F) and quantification from biological duplicates (G) of wild-type or Bmal1 knockdown MEFs at different time points post Tu treatment. (H) Hepatic XBP1s binding to regulatory regions of key 12h genes under constant darkness conditions assayed by ChIP-qPCR (n=3~4) overlaid with RPKM normalized quantification of mouse hepatic RNA synthesis rate under a 12h L/D schedule calculated from a published Gro-Seq database (Fang et al., 2014) and BMAL1, CLOCK, PER2, H3K4me3, H3K4me1 and H3K27Ac cistromes (Koike et al., 2012). Amplicons of ChIP-qPCR and regions of RNA for quantification are illustrated by arrows of different colors. (I) Consensus XBP1s binding motif and recovered motifs from positive XBP1s ChIP-qPCR amplicons. (J–K) Transient luciferase assay using WT and XBP1s binding site mutant Eif2ak3-dluc (J) or Sec23b-dluc (K) vectors with increasing doses of XBP1s expression vector in MEFs (n=4). (L–Q) Real-time luminescence recording of WT and XBP1s binding site mutant stable Eif2ak3-dluc MEFs in response to Tu (L,N,P) or glucose depletion (M,O,P). Quantified period (N-O) and amplitude (P–Q) were provided. Gray boxes indicate the duration of treatment. Data are graphed as the mean ± SEM. **P<0.01.
Unlike the phases of 12h mRNA rhythms, the phases of 12h protein oscillations are distributed evenly throughout the diurnal cycle (Figures S5A, S7). However, a closer examination reveals a clear phase separation of proteins involved in opposing metabolic pathways (Figure S6A). For instance, proteins participating in FAO, OXPHOS and gluconeogenesis, show peaks at early morning (ZT0~4) and early evening (ZT12~16) while proteins involved in glycolysis and fatty acid synthesis peak in the afternoon (ZT6~10) and late night (ZT18~22) (Figures 5B–C and S7). Purine de novo synthesis proteins peak at midday (ZT4~8) and midnight (ZT16~20), preceding the two peaks of 12h mRNA transcription at ZT12 and ZT24 (Figure S6A). These results demonstrated a prevalent 12h hepatic rhythms at the protein level.
XBP1s transcriptionally regulates the 12h rhythm of gene expression
Systemic metabolism depends strongly on ER function and genetic ablation of UPR modulators severely impairs metabolic homeostasis (Bravo et al., 2013). Therefore, it is highly likely that the 12h rhythms of metabolic gene expression are in tune with the 12h oscillations of ER stress/UPR in vivo. Supporting this hypothesis, we found robust 12h oscillations of hepatic XBP1s and ATF4 (Figure 5D). Analysis of ribosome profiling data of ATF4 open reading frame usage (Janich et al., 2015) further confirmed the 12h oscillations of hepatic ATF4 translation in vivo (Figures 5E, S5G). Moreover, we observed robust 12h rhythm of XBP1s expression in Tu-synchronized wild-type and Bmal1 knock-down MEFs (Figures 5F–G), thus confirming the cell-autonomy and circadian clock-independence of the 12h UPR.
We next hypothesized that XBP1s transcriptionally regulates the 12h rhythm of gene expression via direct chromatin recruitment to regulatory regions. To test this hypothesis, we first performed ChIP-qPCR on XBP1s in livers of mice housed under constant darkness conditions. We designed 10 primer pairs targeting promoter (H3K4me3 high/H3K4me1low), enhancer (H3K4me3low/H3K4me1high/eRNA positive) or negative control regions of 7 genes exhibiting 12h rhythm of mRNA expression. In all cases, we detected robust 12h rhythm of XBP1s recruitment to either promoter or enhancer regions, with peak binding observed at CT10 and CT22 (Figures 5H, S5H), consistent with its hepatic protein expression (Figure 5D). The 12h rhythm of XBP1s binding correlated with 12h rhythm of eRNA and pre-mRNA expression (Figures 5H, S5H). Core circadian clock TF (BMAL1/CLOCK/PER2) binding sites are either absent or spatially separated from XBP1s binding sites (Figures 5H, S5H) and no XBP1s recruitment was found on the promoter regions of core clock genes (Figure S5I). In addition, we found robust XBP1 binding motifs (CCACGTC) within 500bp proximity (± 250bp) of the amplicon in 6 of 7 genes with XBP1s binding (Figure 5I). Finally, we verified the cell-autonomous and BMAL1-independent 12h rhythms of XBP1s recruitment to Sec23b, Eif2ak3 and Hspa5 gene regulatory regions in Tu-treated MEFs (Figures S5J–L). In sum, we demonstrate that a 12h rhythm of chromatin recruitment of XBP1s to 12h rhythm genes occurs in vivo and in vitro.
We next investigate whether XBP1s alters 12h rhythm of gene expression. While XBP1s overexpression leads to a dose-dependent increase of both Eif2ak3 and Sec23b promoter-driven luciferase activity, mutating the consensus XBP1s binding motif completely abolished this response (Figures 5J–K). Further, while stable Eif2ak3-dluc MEFs with WT promoter reveal robust 12h oscillation of luciferase activity in response to both Tu and GD treatments (Figures 3F–G), mutating the XBP1s binding site significantly reduced the amplitude and lengthened the period (Figures 5N–Q). Finally, siRNA-mediated knockdown of XBP1 significantly dampened the 12h rhythm of metabolic and UPR gene expression and Eif2ak3-dluc oscillation in both Tu and GD-synchronized MEFs (Figures 3B,E,F–H), yet failed to affect Dex-induced circadian oscillation of core clock genes (Figure S4I). Together, these results demonstrate that XBP1s transcriptionally regulates the 12h rhythm, but not the circadian rhythm of gene expression.
12h rhythm of hepatic metabolism is influenced by feeding behavior in mice
A 12h rhythm of hepatic metabolic gene expression should impact the 12h rhythm of hepatic metabolite oscillation. To test this hypothesis, we performed a post-hoc analysis of a published hepatic metabolomics dataset (Eckel-Mahan et al., 2013) and found ~15% (46 out 305) of the metabolites with a 12h period including various fatty acids, 2-lyso-PCs, carnitine, sphingosine, 9-HODE, succinyl-CoA, ribose and vitamin B1 (Figure 6A). The 12h rhythms of metabolites often correlate with the 12h oscillations of metabolic enzymes involved in the metabolism of these metabolites (Figures 6B, S6B–C). Of particular interest are the 12h rhythms of Lpcat3 expression and a number of 2-lyso-PCs. LPCAT3 functions to increase membrane fluidity by promoting the incorporation of polyunsaturated fatty acids into phospholipids (Figure 6B), and previous studies showed that Lpcat3 induction significantly ameliorates ER stress through regulating ER membrane fluidity in mouse liver (Rong et al., 2013). These findings again indicate that the hepatic 12h clock can coordinate ER stress with metabolism to maintain systemic homeostasis.
Figure 6. 12h rhythm of mouse hepatic metabolism. (A).
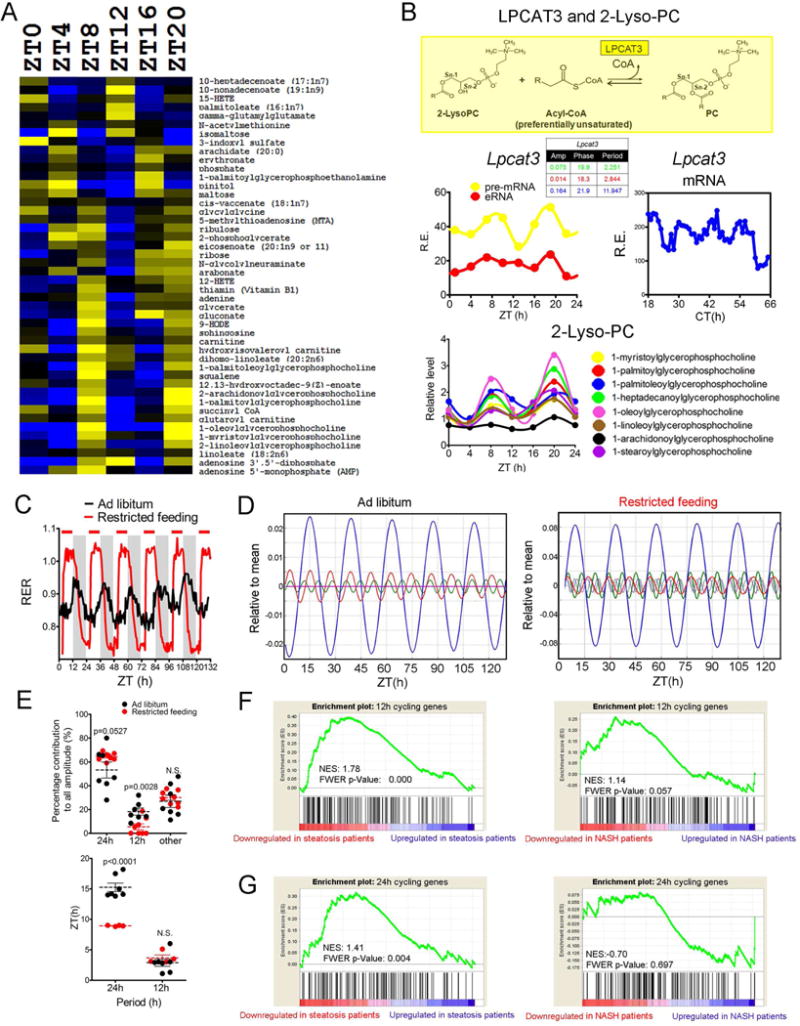
Heatmap of mouse hepatic metabolites exhibiting 12h oscillation at different ZTs. (B) 12h rhythm of Lpcat3 mRNA expression is associated with the 12h oscillation of various 2-Lyso-PC species in mouse liver. LPCAT3 catalyzes the conversion from 2-Lyso-PC to PC (top). Oscillations of Lpcat3 pre-mRNA and eRNA transcription (Fang et al., 2014) (middle left), mature Lpcat3 mRNA expression and its eigenvalue/pencil decomposition (middle right) and various 2-Lyso-PC levels (bottom). (C–E) Averaged real-time RER values of mice housed under ad libitum or restricted feeding conditions. Time of food accessibility is indicated by red bars above the RER graph for the restricted feeding group (C). Eigenvalue/pencil deconvolutions of the average RER oscillations. Blue line: 24h oscillations, red line: 12h oscillations, green line: 8h oscillations (D). The percentage contributions to the total amplitudes of RER oscillation from identified 24h, 12h and the combined other oscillations (left) and the phases of identified 24h and 12h oscillation (right) in each individual mouse (E). (F–G) GSEA analysis on 12h (F) or 24h (G) cycling genes compared with human hepatic steatosis (left) and NASH (right) microarray datasets. Normalized enrichment scores (NES) and FWER p values are shown for each analysis.
Our findings raise the intriguing possibility that other peripheral tissues possess a 12h clock that perhaps helps to synchronize 12h metabolic homeostasis. To address this hypothesis, we measured the real-time respiratory exchange ratio (RER) of mice maintained under 12h:12h light/dark condition for 5 days and applied the eigenvalue/pencil method to these data. As expected, in addition to the circadian rhythm, oscillations with smaller periods were revealed in RER with ~12h rhythms exhibiting the second largest amplitude in all 7 mice tested (Figures 6C–E, S6D–H). Daytime restricted feeding of mice from ZT3 to ZT11 significantly dampened the 12h rhythm of RER without altering their phases. but had marginal effects on the amplitude of the circadian rhythm while altering their phases by ~6h (Figures 6C–E, S6H). These effects are not due to changed total food intake between normal and restricted feeding groups (Figures S6D–F). These data suggest that the feeding behavior-dependent 12h rhythm of systemic metabolism in mice is regulated independently from the circadian rhythm.
The downregulation of 12h genes is associated with human progression to NAFLD
To determine if the 12h clock also is influential in the maintenance of systemic metabolic homeostasis in humans, we examined the gene expression profiles of the 143 genes cycling with a 12h period at both the mRNA and protein level (Table S5) in human NAFLD patients (Ahrens et al., 2013) by performing gene set enrichment analysis (GSEA) (Subramanian et al., 2005). For comparison, we performed similar analyses using another cassette of 138 genes (Table S5) that exhibit circadian rhythms at both the mRNA and protein level. As shown in Figures 6F–G, the down-regulation of 12h gene expression is strongly associated with progression to steatosis and modestly associated with non-alcoholic steatohepatitis (NASH). By contrast, the down-regulation of circadian gene expression is associated with progression to steatosis less strongly and no association was found between circadian gene expression changes and NASH. Since the 12h genes are functionally distinct from the circadian genes (Figures 1I–J), these results demonstrate that while independent from the circadian clock, the 12h clock likely plays an essential role in the maintenance of hepatic metabolic homeostasis in humans.
The 12h rhythm of mtDNA transcription is evolutionarily conserved in crustaceans and mammals
The prevalent mammalian 12h rhythm of gene expression and metabolism is reminiscent of the 12h circatidal rhythms of coastal marine animals (Wilcockson and Zhang, 2008). Recently, establishment of the circatidal clock in the crustacean E. pulchra was reported to be independent from the circadian clock (Zhang et al., 2013). In light of the newly uncovered independence of the mammalian 12h clock from the circadian clock and the fact that mammals share common marine ancestors, we initially postulate that the mammalian 12h clock evolved from the ancient circatidal clock.
Intriguingly, previous work revealed robust 12h circatidal mRNA rhythms for 10 mtDNA-encoded protein-coding genes (Mt-Nd1~6, Mt-Cox1~3 and Cytb) that encode for mitochondrial components of complexes I (NADH dehydrogenase) and IV (cytochrome c oxidase) in E. pulchra under free-running conditions (O’Neill et al., 2015). To test whether these 12h rhythms also are conserved in mammals, we performed a post-hoc analysis of two published Gro-Seq (Fang et al., 2014) and Nascent-Seq datasets (Menet et al., 2012) that measured diurnal oscillations of mouse hepatic nascent RNA transcription. We observed robust global 12h rhythms of mtDNA-encoded RNA transcription from both strands of mtDNA in both datasets, with peak transcription occurring at ZT7~8 and ZT19~20, and nadir at ZT10~12 and ZT22~ZT24 (Figures 7A, S7A). In addition to nascent RNA transcription, both mtDNA-encoded mature mRNAs and proteins reveal robust 12h rhythms (Figures S7B-C, S5C-E and Table S5). Further, nuclear-encoded subunits of electron transport chain complexes also reveal strong 12h rhythms at the protein level (Figure S5C and Table S5). Collectively, these data indicate a coordinated 12h rhythm of OXPHOS gene transcription and translation between the nucleus and mitochondria. We further found 12h rhythm of mRNA expression in mitochondrial transcription factor B1 and B2 (Tfb1m, Tfb2m) (Figure S7D). In addition to functioning as a transcription factor regulating mtDNA transcription, TFB1M also is implicated in mitochondrial protein translation (Metodiev et al., 2009). Therefore, the 12h rhythms of nuclear-encoded Tfb1m and Tfb2m gene expression is likely responsible for the 12h rhythm of transcription and translation of mtDNA-encoded genes.
Figure 7. 12h CREMA is evolutionarily conserved. (A).
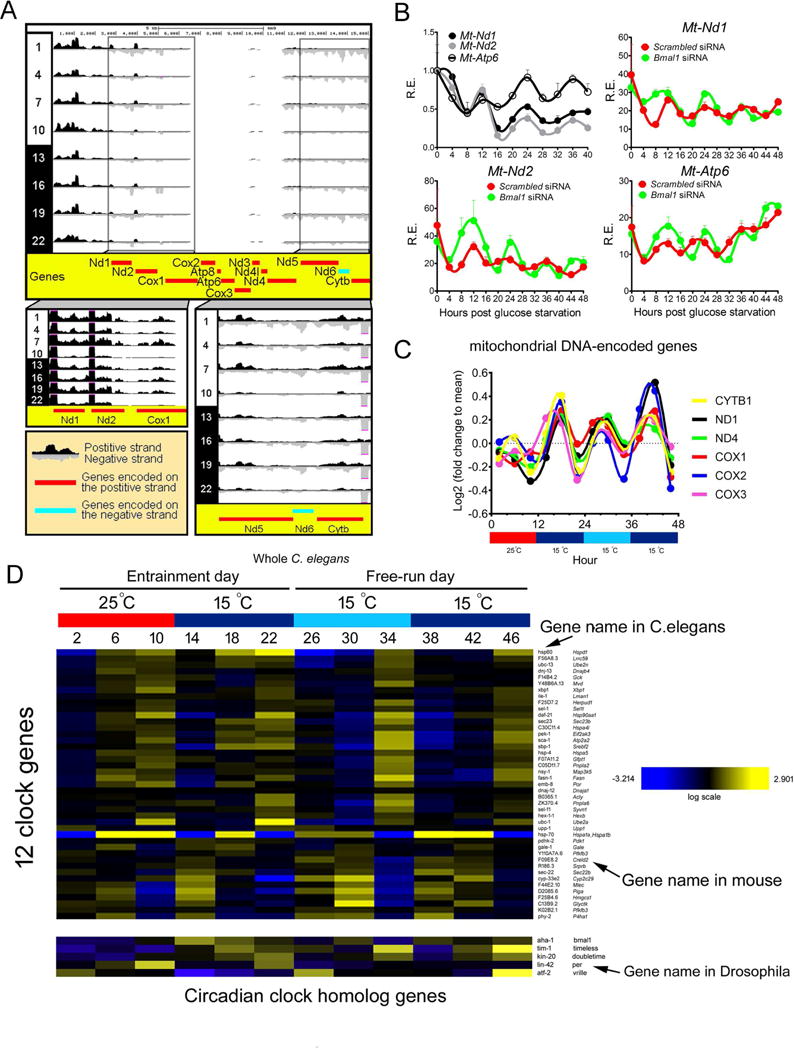
RPKM normalized quantification of mouse hepatic mtDNA-encoded RNA synthesis rate under a 12h L/D schedule calculated from published Gro-Seq. UCSC genome browser snapshot of Gro-Seq tracks across the 16.1 kb mouse mitochondrial genome. Protein-coding genes encoded on the positive (H) strand are shown in red while genes on the negative (L) strand are shown in blue. Boxes show UCSC tracks with higher magnification for specific regions. (B) MEFs were transfected with different siRNA, glucose starved and qPCR was performed post-GD. (n=3). Data are graphed as the mean ± SEM. (C–D) C.elegans are entrained under 12h warm temperature (25°C)/12h cold temperature (15°C) for 4 days before released into constant col d (15°C) condition for 24 hours and mRNA from whole organisms were extracted and subjected to microarray analysis at 4h intervals. 12h cycling of mitochondrial DNA-encoded oxidative phosphorylation genes is shown in (C). Heatmap of 12h cycling ER stress and metabolism genes as well as the lack of circadian oscillation of 24h clock genes, with C. elegans as well as mammalian/drosophila ortholog gene names are shown on the right (D).
Finally, we confirmed the cell-autonomous 12h oscillation of Mt-Nd1, mt-Nd2 and Mt-Atp6 gene expression in GD-synchronized MEFs, which are also independent of Bmal1 (Figure 7B). On a final note, the calculated period value of 12.59h for the mammalian cell-autonomous Eif2ak3 oscillation is close to the reported 12.7h circatidal period in free-running E. pulchra after tidal cues entrainment (Zhang et al., 2013). These findings are striking as they indicate that the circatidal rhythm of mtDNA-encoded gene transcription in marine animals is evolutionarily conserved in mammals, thereby supporting our hypothesis that the mammalian 12h clock perhaps evolved from the circatidal clock.
The 12h rhythm of CREMA is evolutionarily conserved in both nematodes and mammals
Since the ER-related gene expression profiles were not reported in E. pulchra, it remains unclear whether the ER branch of 12h CREMA also is conserved evolutionarily in multiple species of lower organisms. To address this question, we examined a published microarray database in free-running C. elegans entrained by warm/cold temperature cycles (van der Linden et al., 2010). First, we confirmed the 12 rhythm of mtDNA-encoded OXPHOS gene transcription in nematodes (Figure 7C). Intriguingly, we further observed robust 12h mRNA rhythm of genes involved in ER stress and metabolism pathways (genes in Table S2). In fact, of all 70 genes that have orthologs in C. elegans, 43 (61%) of them show robust 12h cycling under both temperature-entrainment as well as free-run conditions (Figures 7D, S7E). These genes include C. elegans ortholog of Xbp1, Hspa1a, Eif2ak3, Sec23b, Fasn, Pfkfb3 and P4ha1, all of which have robust 12h rhythms in mouse liver (Table S2). The conservation of 12h rhythm in C. elegans is not limited to these core 12h cycling genes. In fact, of the top ten largest mammalian 12h cycling genes, seven of their C. elegans orthologs also reveal robust 12h rhythms (Figures S7F,G). Since adult C. elegans has a fixed cell number of only ~1000, the robust 12h oscillations of these genes suggest that 12h clock exists in most, if not all cells in C. elegans. More importantly, while the 12h rhythms are conserved in C. elegans, homologs of drosophila and mammalian core circadian clock as well as top 24h cycling mouse hepatic genes do not exhibit circadian rhythms under the same conditions (Figures 7D, S7H,I). The lack of circadian oscillation of canonical circadian clock genes in C. elegans have been previously reported in several studies and these genes are believed to play roles in development rather than regulating circadian rhythms in nematodes (Romanowski et al., 2014; van der Linden et al., 2010). Given that the 12h clock is conserved but not the 24h circadian clock between nematodes and mammals, this observation strongly supports our hypothesis that the 12h clock evolved ‘separately’ from the circadian clock and that the mammalian 12h clock is independent from the circadian clock.
DISCUSSION
Biological rhythms are part of ‘musica universalis’
In this study, we used a novel mathematical method to uncover all superimposed oscillations. Most of the current methodologies require the user to define a narrow period range, and then use respective algorithms to find the optimal period within that range that minimizes the p-value (Refinetti et al., 2007). These approaches are heavily biased since they presume that biological oscillations are pure rhythms with a single period. However, pure oscillation with a single frequency rarely exists in nature. Rather, physical rhythms often present as superpositions of basic waves such as the harmonic resonances found in music, light and planetary motion. Our eigenvalue/pencil method, on the other hand, does not pre-assign a period and thus permits the identification of all superimposed oscillations in an unbiased manner. Using this method, we uncovered that, just like other forms of physical oscillations, biological oscillations also are composite rhythms consisting of oscillations of various frequencies. Interestingly, these oscillations often cycle at the harmonic of the circadian rhythm, presumably to ensure that most cellular oscillations are always in sync. Therefore, it appears that harmonic resonance is a common ‘universal’ theme, a concept that was first proposed by Pythagoras as ‘musica universalis’ over two thousand years ago (Taruskin, 2008). According to this theory, all natural appearing rhythms are in essence tuned to the rhythmic movements of celestial bodies (Taruskin, 2008). While the circadian rhythm is synchronized to the 24h light/dark cycle coinciding with the Earth’s rotation, our findings suggest that the 12h clock may have evolved from the ancient circatidal clock, which is in turn entrained by the 12h tidal cues orchestrated mainly by the moon.
The cell-autonomy and independence of the mammalian 12h clock
The relationship of 12h circatidal rhythms to 24h circadian rhythms remains elusive. A previous study reported that 12h rhythm of hepatic XBP1s and a few UPR genes is under the combined regulation of the circadian clock and fasting-feeding cues and therefore is not cell-autonomous (Cretenet et al., 2010). However, our real-time luminescence and time lapse microscopy experiments strongly support the existence of a cell-autonomous mammalian 12h clock. In single cell microscopy experiment, we did observe variability of base-line GFP levels in individual Eif2ak3-dGFP recordings, which largely can be attributed to the fact that Eif2ak3 is a key sensor of ER and metabolic stress and is thus very sensitive to the subtle changes of stress in each individual cell. Since the cells are not synchronized, the metabolic rate, cell cycle phase and cell population density of/surrounding each individual cell can vary and these factors can affect the temporal baseline changes of the Eif2ak3-dGFP reporter, leading to a more “noisy’ raw data. However, after rigorous and unbiased mathematical detrending and eigenvalue/pencil decomposition, we unambiguously uncovered ~12h rhythm in ~90% of single cells recordings. Moreover, as pointed above, even the single cell Eif2ak3-dGFP oscillations are not pure oscillations, but rather superimposed oscillations with dominant periods of ~12h, and subperiods of ~8h and ~6h. Nevertheless, future studies are needed to determine whether cell-autonomous 12h rhythm also exists in other genes and cell lines.
Our study indicates that the cell-autonomous mammalian 12h rhythm is regulated independently from the circadian clock. Our finding also agrees with the recently identified independent circatidal clock in E. pulchra. Our reported conservation of 12h, but not circadian genes between C. elegans and mouse implies that while the circadian clock machinery diverged early in evolution, the 12h clock machinery remained essentially unchanged even in species as divergent as nematodes and mammals. With that said, it is important to note that we are not ruling out the possibility that the 12h rhythms of some genes may be influenced by the circadian clock and/or the effects of similar external cues. Furthermore, while our data indicates the 12h clock is independent from the circadian clock at the cellular level, they appear to be able to cross-talk at the systemic metabolic level (Hughes et al., 2012). Therefore, future studies are needed to investigate the physiologic conditions by which these distinct clocks can interact systemically in model organisms.
12h CREMA
Proteins exhibiting 12h rhythms of expression are prominently localized in both ER and mitochondria (Figure S6A), implying the coordination of these two organelles in the orchestration of 12h rhythms. This hypothesis is further supported by our data demonstrating that a 2h GD is sufficient to establish the 12h rhythms of genes involved in both UPR (Eif2ak3) and mitochondria-regulated metabolism (Mt-Nd1 and Mt-Nd2). This observation is unsurprising, as in addition to active participation of both ER and mitochondria in extensive cellular metabolism, reciprocal functional dependence of these organelles has been comprehensively demonstrated (Kornmann, 2013). The ER-mitochondria interdependence is mainly mediated via regions of the ER that are physically associated with mitochondria, termed mitochondria-associated ER membranes (MAMs). Genes encoding proteins highly enriched on the MAMs, including Ero1α, Pdia3 and Calnexin (Simmen et al., 2010), all exhibited a 12h rhythmicity of gene expression. While it remains unclear whether the physical association of the ER and mitochondria is merely a consequence or plays a causal role in the regulation of the mammalian 12h clock, our results nonetheless suggest that 12h CREMA are important for the maintenance of systemic metabolic homeostasis.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Bert W O’Malley (berto@bcm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
For mice housed under constant darkness, a cohort of 8~12 weeks old male C57BL/6J mice were entrained under a normal 12h:12h light/dark conditions for at least two weeks before released into constant darkness. Following 36 hours of constant darkness, liver tissue and plasma was harvested at 2h intervals for a total of 48h (n=3 at each CT). During all these time, the mice were fed ad libitum. For mice housed under 12h light/12 dark conditions, mice were entrained under a strict 12h light/12 dark schedule for two weeks before the experiment. After entrainment, one cohort of mice (n=7) were fed ad libitum; while the second cohort of mice (n=8) were restricted fed only during the 12h light period (ZT3 to ZT11). During these times, all the mice were still housed under the normal 12h light/12h dark schedule. The Baylor College of Medicine Institutional Animal Care and Utilization Committee approved all experiments.
Cell lines
293TN producers were purchased from System biosciences and is a derivative of HEK293T cells that express a neomycin resistance marker. 293TN cells were cultured in DMEM (4.5g/L glucose) supplemented with 10% FBS at 37 °C with 5% CO 2. The sex of this cell line was unknown.
Synchronization of MEFs
MEFs were isolated from male SRC-2fl/fl mice and immortalized by SV40 T antigen as previously described (Stashi et al., 2014). For tunicamycin treatment, MEFs were cultured in DMEM (4.5g/L glucose) supplemented with 10% FBS and treated with the indicated concentration of tunicamycin for 2h, and then washed with 1X PBS before cultured in the same medium. For glucose depletion treatment, MEFs were cultured in DMEM (10mM glucose) supplemented with 10% dialyzed FBS and then treated with DMEM (no glucose) supplemented with 10% dialyzed FBS for 2h before replaced with DMEM (10mM glucose) supplemented with 10% dialyzed FBS. For 2-DG treatment, MEFs were cultured in DMEM (10mM glucose) supplemented with 10% dialyzed FBS and then treated with different concentration of 2-DG for 2h in DMEM (10mM glucose) supplemented with 10% dialyzed FBS before replaced with DMEM (10mM glucose) supplemented with 10% dialyzed FBS. For dexamethasone treatment, MEFs were cultured in DMEM (4.5g/L glucose) supplemented with 10% FBS and treated with 100nM Dex for 30mins, and then washed with 1X PBS before cultured in the same medium. For all cell culture experiments, cells were cultured at 37 °C with 5% CO2.
METHOD DETAILS
Eigenvalue/pencil Method
Selecting hepatic genes for analysis
We initially applied our approach to a high resolution hepatic gene expression microarray dataset from mice kept under constant darkness (analyzed at 1h interval for a total of 48 hours) (Hughes et al., 2009). This microarray dataset has the highest resolution available with this degree of granularity and therefore is uniquely suitable for robustly revealing oscillations of various periods. In this study, liver RNA samples from 3~5 mice at each CT were pooled and analyzed using Affymetrix array. To select genes that are expressed in mouse liver for eigenvalue/pencil analysis, we first determined the background gene expression levels by randomly selecting 57 genes that are known to be not expressed in the liver. These genes include 45 keratin family members preferentially expressed in the skin, three probes for nestin gene preferentially expressed in the neurons and 9 genes important for T cell and mast cell development. The average expression of these 57 genes is 60, which we consider as the threshold for background. Therefore, genes whose expression is greater than 60 at at least one CT are included in the eigenvalue/pencil analysis and a total of 18,108 genes were analyzed in the end. For genes with multiple probes sets, only one probe was randomly selected and subject to the eigenvalue/pencil analysis, with exceptions for analysis described in Figure S2. The raw microarray data was normalized before the eigenvalue/pencil analysis by normalizing the probe intensity at every CT to the minimum probe value within the 48 time points so that the minimum gene expression value is always 1 for all genes. Oscillations with periods between 22~26h, 10.5~13.5h, 7~9h, 3~5h, 2~2.5h are considered to be circadian, 12h, 8h, 4h and 2h genes.
The eigenvalue/pencil approach
This approach was based upon the Loewner framework originally developed for linear systems with multiple inputs and outputs and later extended to linear parametric systems (Ionita. and Antoulas., 2013; Ionita. and Antoulas., 2014). Mathematically, our method consists of the eigenvalue analysis of a pencil of matrices constructed from raw data. Specifically, the construction of a raw model of linear, time-invariant, discrete-time system driven by initial conditions is followed by dimension reduction using singular value decomposition (SVD) to identify the dominant oscillations.
First Step: We have a time series dataset expressed as h = [ℎ (1), ℎ (2) ⋯ ℎ (N)], and use it to construct a Hankel matrix as follows:
| (1) |
Wherein q ≤ k, q ≤ l. In our case, the Hankel matrix can be represented as H1,R,R+1.Then we can define the quadruple (ER, AR, BR, CR):
| (2) |
This quadruple constitutes the raw model of the data h. This model is linear, time-invariant and discrete-time:
| (3) |
wherein n = 0,1,2 ….
Second step: We use a model reduction approach to determine the dominant oscillations of the raw model. The raw model is reduced by using the SVD of H1;R;R+1. We compute the following SVDs as follows:
| (4) |
Then we choose the dimension r of the reduced system. Hence, we construct the projectors: X = u2(:,1:r), Y = v1(:,1:r); and the reduced system matrices are:
| (5) |
The associated reduced model of dimension r is:
| (6) |
Now, we have a reduced low-dimension system. We define C = Cr, B = [Er]−1Br, A = [Er]−1Ar, then the approximation
| (7) |
To gather the information of oscillations, we perform eigenvalue decomposition (EVD) of the matrix pencil (Ar Er), the same way as we perform EVD of A, then we have
| (8) |
Wherein Vr = [v1, …, vr], Λr = diag[λr, …, λr]. Then the approximation can be decomposed as
| (9) |
wherein Pi is complex amplitude of the ith oscillation. Because Pi and λi are all complex number that it can be represent as
| (10) |
wherein αi is the amplitude, σi is the decay (grow) rate, ωi is the frequency and θi is the phase of the ith oscillation.
Identifying dominant biological oscillations
Next, we used Bmal1 time-series microarray data (Hughes et al., 2009) as an example to demonstrate the application of eigenvalue/pencil to identify dominant superimposed oscillations. We calculated the oscillations of the reduced model with r= [3, 5, 7, 9], in addition to the raw model without dimension reduction. The values of pole λi under different dimension reduction (and raw model) are shown in the following table for the Bmal1 gene.
| r=3 | r=5 | r=7 | r=9 | Raw model |
|
| ||||
| 1.0012 | 1.0011 | 1.0011 | 1.0011 | 1.0053 |
| 0.9672 + 0.2522i | 0.9673 + 0.2520i | 0.9674 + 0.2519i | 0.9674 + 0.2521i | 0.9684 + 0.2599i |
| 0.9672 – 0.2522i | 0.9673 – 0.2520i | 0.9674 – 0.2519i | 0.9674 – 0.2521i | 0.9684 – 0.2599i |
| 0.8720 + 0.5137i | 0.8761 + 0.5151i | 0.8805 + 0.5150i | 0.8473 + 0.4592i | |
| 0.8720 – 0.5137i | 0.8761 – 0.5151i | 0.8805 – 0.5150i | 0.8473 – 0.4592i | |
| 0.6218 + 0.7802i | 0.6357 + 0.7685i | 0.7932 + 0.7400i | ||
| 0.6218 – 0.7802i | 0.6357 – 0.7685i | 0.7932 – 0.7400i | ||
| –0.3691 + 0.8542i | –0.3526 + 1.1144i | |||
| –0.3691 – 0.8542i | –0.3526 – 1.1144i | |||
The striking feature of this analysis is that the pole values of oscillations of large periods remain mostly unchanged as more oscillations are added by using higher reduced models (the top four oscillations revealed by 9th reduced model corresponds to T=24.65, 11.86, 7.14 and 3.17h as shown in Figure S1F). These data suggest that 9th order model is a good trade-off between the fitness of the model to the raw data and the likelihood that the oscillations of newly added smaller periods are noise rather than real biological oscillations. This assertion is further supported by the examination of superimposed oscillations from different probe sets against the same gene (Figures S2A–C). Therefore, we believe the 9th reduced model, which can identify up to four different oscillations, is the optimal model to reveal robust novel oscillations.
siRNA Transient Transfections
MEFs were transfected with 10μM of different siRNAs for 24~48 hours with Lipofectamine RNAiMAX reagents (Life technologies) per the manufacturer’s instructions. Source of siRNA are as follows: siGENOME Non-Targeting siRNA pool (Dharmacon, D-001206-13-05), siGENOME SMARTpool ARNTL (Dharmacon, L-040483-01-0005), siGENOME SMARTpool XBP1 (Dharmacon, L-040825-00-0005).
Immunoblot
Immunoblot analyses were performed as described previously (Zhu et al., 2015). Briefly, proteins separated by 4~20% gradient SDS-PAGE gels (Biorad) were transferred to nitrocellulose membranes, blocked in TBST buffer supplemented with 5% bovine serum albumin (BSA) and incubated overnight with primary antibody at 4°C. Blots were incubated with an appropriate secondary antibody coupled to horseradish peroxidase at room temperature for 1 hour, and reacted with ECL reagents per the manufacturer’s (Thermo) suggestion and detected on X-ray film by autoradiography. The antibodies used in immunoblot are provided in key resources table.
qRT-PCR
Total mRNA was isolated from murine embryonic fibroblasts (MEFs) or liver with PureLink RNA mini kit (Life Technologies) per the manufacturer’s instructions. Reverse transcription was carried out using 5μg of RNA using Superscript III (Life Technologies) per the manufacturer’s instructions. For gene expression analyses, cDNA samples were diluted 1/30-fold (for all other genes except for 18sRNA) and 1/900-fold (for 18sRNA). qPCR was performed using the Taqman or SYBR green system with sequence-specific primers and/or the Universal Probe Library (Roche). All data were analyzed with 18S or β-Actin as the endogenous control. All qPCR primer sequences are shown in Table S6.
ChIP-qPCR
Liver tissue was isolated and flash frozen at designated CTs. Chromatin was isolated using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling) and performed per the manufacturer’s suggestion. Briefly, mouse liver samples were submerged in PBS + 1% formaldehyde, cut into small (~1 mm3) pieces with a razor blade and incubated at room temperature for 20 minutes. Fixation was stopped by the addition of 0.125 M glycine (final concentration). The tissue pieces were then treated with a TissueTearer and finally spun down and washed twice in PBS. Chromatin was isolated by the addition of lysis buffer, followed by disruption with a Dounce homogenizer. Lysates were enzymatically digested with MNase and the DNA was digested to an average length of 145 bp. Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, Proteinase K and heated for reverse-crosslinking, followed by ethanol precipitation. Pellets were resuspended and the resulting DNA was quantified on a NanoDrop spectrophotometer. An aliquot of chromatin (5 g) was precleared with protein A agarose beads (Invitrogen). Genomic DNA regions of interest were isolated using 2 g of antibody. Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinking were reversed by incubation overnight at 65 °C, and ChIP DNA was pur ified by phenol-chloroform extraction and ethanol precipitation. For ChIP-qPCR on MEFs, cells were fixed with 1% formaldehyde for 20mins and ChIP was performed essentially the same way. XBP1 (M186) (Santa Cruz) and (poly6195) (Biolegend) antibodies were used for XBP1 ChIP-qPCR. All ChIP-qPCR primer sequences are shown in Table S6.
Cloning
XBP1s-pHAGE
The XBP1s overexpression plasmid is a generous gift from Dr. Xi Chen at Baylor College of Medicine and made in-house in Dr. Xi Chen’s lab.
Eif2ak3-dluc
An 1026bp long mouse Eif2ak3 promoter (−754bp to +272bp) was PCR amplified from NIH3T3 cells genomic DNA using the following primers: forward primer: ATAGAGCTCAAACCAACCGGGAGACTTAAT; reverse primer: ATACTCGAGCCAGCAGCAGGAACAGAA and subcloned into pGL4.16[luc2CP/Hygro] vector (E6711, Promega), which encodes for a destabilized luciferase gene. All sequences were confirmed by sequencing.
Eif2ak3-dluc with mutated XBP1s binding site
To mutate CCACGTC to CCAAAAA, site-directed mutagenesis was performed using the Q5® Site-Directed Mutagenesis Kit (E0554S) (NEB) with the following primers: forward primer: CAGTGGCTGAtttttGGCCGGGCAGCTC; reverse primer: CCACCTGAGTGACAGCCT per the manufacturer’s suggestion. All sequences were confirmed by sequencing.
Eif2ak3-dGFP
An 1026bp mouse Eif2ak3 promoter (−754bp to +272bp) was PCR amplified from NIH3T3 cells genomic DNA using the following primers: forward primer: ATAATCGATAAACCAACCGGGAGACTTAAT; reverse primer: ATAACTAGTCCAGCAGCAGGAACAGAA and subcloned into pGreenfire-mCMV-dscGFP-EF1-Puro lentiviral vector (TR010PA-P, System Biosciences), which encodes for a destabilized GFP gene. All sequences were confirmed by sequencing.
Sec23b-dluc
An 654bp long mouse Sec23b promoter (−513bp to +141bp) was PCR amplified from NIH3T3 cells genomic DNA using the following primers: forward primer: ATAATCGATAGCACAGAAACCACAGCA; reverse primer: ATAACTAGTTTCACCATTGCAGAGCCA and subcloned into pGL4.16[luc2CP/Hygro] vector (E6711, Promega), which encodes for a destabilized luciferase gene. All sequences were confirmed by sequencing.
Sec23b-dluc with mutated XBP1s binding site
To mutate CCACCTC to CCAAAAA, site-directed mutagenesis was performed using the Q5® Site-Directed Mutagenesis Kit (E0554S) (NEB) with the following primers: forward primer: ACTGTGAGCTttttTGGACTTGGCGGTGG; reverse primer: CGATTGGTCACCGCTCCG per the manufacturer’s suggestion. All sequences were confirmed by sequencing.
Transient Luciferase Assay
MEFs were co-transfected with internal control vector pRL-TK and Eif2ak3-dluc, Eif2ak3-dluc with mutated XBP1s binding site, Sec23b-dluc or Sec23b-dluc with mutated XBP1s binding site using Lipofetamine 2000 for 24h before transfected with increasing amount of XBP1s over-expression vector using Lipofetamine 2000 for another 24h. After that, the cells were lysed with 50μl of 1x passive lysis buffer and subject to dual-reporter luciferase assay per the manufacturer’s suggestion. The Firefly luciferase signal was normalized to that of Renilla luciferase signal.
Generation of Stable MEFs Lines
To generate Eif2ak3-dluc or Eif2ak3-dluc with mutated XBP1s binding site stably expressing MEFs, MEFs cultured in 15cm dish were transiently transfected with Eif2ak3-dluc or Eif2ak3-dluc with mutated XBP1s binding site plasmids for 48 hours. After that, cells were cultured in the presence of 200μg/ml hygromycin to select for resistant cells. To generate stable Eif2ak3-dGFP cells, Eif2ak3-dGFP lentivirus were first generated by co-transfecting 293TN producer cells with Eif2ak3-dGFP, pMD2G and psPAX2 plasmids with the ratio of 3:2:1 for 48 hours and collecting supernatant. Virus were filtered with a 0.45μM syringe filter. MEFs were subsequently infected with Eif2ak3-dGFP encoding lentivirus in the presence of 4 μg/ml polybrene for 48 hours and cultured in the presence of 4 μg/ml puromycin to select for stable cells. Cells were subject to multiple rounds of virus infections to increase the percentage of GFP positive cells.
Real-time Luminescence Assay
Stable Eif2ak3-dluc or Eif2ak3-dluc with mutated XBP1s binding site MEFs were cultured in DMEM (4.5g/L glucose) supplemented with 10% FBS and treated with 25ng/ml of Tu (high glucose DMEM with 10% FBS) or glucose depletion (high glucose DMEM with 10% dialyzed FBS) for 2h before subjected to real-time luminescence assay using a Lumicycle (Actimetrics) as previously described (Zhu et al., 2015). Briefly, after Tu or GD treatment, MEFs were washed with 1x PBS and cultured with DMEM (4.5g/L glucose) supplemented with 0.1 mM Luciferin and 10mM HEPES buffer in 35 mm tissue culture dishes in the absence of serum and transferred immediately to Lumicycle for real-time luminescence analysis. Periods of oscillation were identified by embedded Periodogram function. For siRNA treated MEFs, MEFs were transfected with non-targeting or Xbp1 siRNA for 48 hours before subject to Tu or GD shock and real-time luminescence assay as described above.
Time-lapse Microscopy
Time-lapse microscopy was performed on Eif2ak3-dGFP cells using IncuCyte® Live Cell Analysis System (Essen Bioscience). Eif2ak3-dGFP was subject to imaging using 300ms integration time at 30min interval. During the imaging, the cells were cultured in serum free medium. GFP intensity for single cells (cell lineages) was subsequently quantified automatically by automated image processing software CellProfiler (version 2.2.0) (Carpenter et al., 2006). We quantified a total of 89 single cell/cell lineages, collected from 12 different fields of views from 6 different wells from three independent imaging experiments and dominant and superimposed oscillations were subsequently identified by the eigenvalue/pencil approach. The CellProfiler pipeline used for identification, quantification and tracking of single cells as well as raw images are available upon request. The videos were generated using VirtualDubMod 1.5.10 (https://sourceforge.net/projects/virtualdubmod/files/) using indicated frame per second (fps). The raw data were subject to polynomial detrend (n=4~7) and eigenvalue/pencil method were applied to uncover top superimposed oscillations from mathematically detrended data.
Measurement of Plasma Glucose and Insulin
Plasma glucose level was measured by a glucometer and plasma insulin level was measured by a commercial insulin ELISA kit (EZRMI-13K, Millipore) per the manufacturer’s instructions.
Metabolic Profiling Using CLAMS
Calorimetry (Columbus Instruments) was used for real time measuring of Respiratory Exchange Ratio (RER). Mice fed ad libitum (n=7 biological replicates) or restricted fed during the day (ZT3 to ZT11) (n=8 biological replicates) were acclimated to the chambers for at least one week and food intake was monitored for 5 days under a 12h light/dark cycle. Data were collected for VO2 consumed and VCO2 released and RER was calculated as VCO2/VO2.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Analysis by ARSER, RAIN and JTK_CYCLE
To compare our eigenvalue/pencil method with other methods commonly used to identify cycling transcripts in the circadian field, we analyzed the same 18,108 hepatic genes to uncover 12h cycling genes using three separate methods: ARSER (Yang and Su, 2010), RAIN (Thaben and Westermark, 2014) and JTK_CYCLE (Hughes et al., 2010) with respective algorithms described in their publications. However, unlike our eigenvalue/pencil method, which does not require a pre-assignment of periods range, all three methods need the user to assign a period range. For identifying novel ~12h cycling genes, we used a loose criteria of period range from 10 to 14h. We additionally used two different p value cut-offs (p<0.01 and p<0.05) for selecting high confidence ~12h cycling genes. The detailed results are provided in Table S3. The data analysis was performed in Bioconductor (3.4) for RAIN (http://www.bioconductor.org/packages/release/bioc/html/rain.html) and in MetaCycle for both JTK_CYCLE and ARSER (https://github.com/gangwug/MetaCycle).
Gene Ontology Analysis
DAVID (Huang da et al., 2009) (https://david.ncifcrf.gov) was used to perform Gene Ontology analysis. Briefly, gene names were first converted to DAVID-recognizable IDs using Gene Accession Conversion Tool. The updated gene list was then subject to GO analysis using Mus musculus as background and with Functional Annotation Chart function. KEGG_PATHWAY, SP_PIR_KEYWORDS, GOTERM_BP_FAT and GOTERM_CC_FAT were used as GO categories for all GO analysis. Only GO terms with p value smaller than 0.05 were included for further analysis.
Post hoc Analysis of Hepatic SILAC Data
The time series hepatic mass spectrometry-based proteomics data from mice kept under constant darkness for 45h was previously published (n=3 triplicates at each CT) (Robles et al., 2014). We first normalized the raw data by calculating the log2 (fold change over mean) at each CT for each sample and then averaged the normalized values of the three replicates at each CT. Upon examining the normalized data, we observed noticeable baseline changes in most reported protein oscillations. Therefore, we then subjected the normalized data to polynomial detrend (n=3). Due to the relatively poor resolution of the dataset (at 3h interval), we decided against the use of our eigenvalue/pencil approach but rather employed a more simplified but well-established CircWave software to only characterize proteins cycling with a dominant 12h period (Keller et al., 2009). The normalized and detrended dataset was subject to CircaWave analysis (CircWave batch 3.3). A less stringent cutoff of p value=0.1 was used because we reason since most 12h rhythms of gene expression are superimposed by other oscillations; a less stringent criterion will be more likely to identify 12h cycling proteins with asymmetrical 12h peaks caused by the superimposition. The log2 normalized and detrended data for proteins with 12h oscillations are provided in Table S5. For simplicity, only the mean values at each CT are provided. Heatmaps were generated by Gene Cluster 3.0 and TreeView 3.0 alpha 3.0 on polynomial detrended data.
Post hoc Analysis of Hepatic Metabolomic Data
The time series hepatic metabolomics data from mice entrained under 12h light/12 dark schedule for 24h was previously published (n=5 biological replicates at each CT) (Eckel-Mahan et al., 2013). Only the mean value of each metabolite was reported in the original study. Like the SILAC dataset, due to the poor resolution and short duration of the dataset (at 4h interval for a total of only 24h), we again employed CircWave method to only characterize metabolites cycling with a dominant 12h period. For this analysis, a cutoff of p value=0.1 was also used. Heatmaps were generated by Gene Cluster 3.0 and TreeView 3.0 alpha 3.0.
Post hoc Analysis of C.elegans Microarray
In this study (van der Linden et al., 2010), C. elegans were entrained by either 12h light/12 dark or 12h warm temperature (25 °C)/12h c old temperature (15 °C) conditions. We specifically analyzed the microarray data under 25 °C/15 °C entrained as well as 15 °C free-run conditions. The raw microarray data reported in experiment 3 was polynomial detrended (n=2) before subject to CircWave analysis to identify dominant 12h cycling genes (p≤0.1). The C. elegans ortholog of mammalian genes were identified using NCBI AceView tool (https://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/index.html). Heatmaps were generated by Gene Cluster 3.0 and TreeView 3.0 alpha 3.0 on polynomial detrended data.
Gene Set Enrichment Analysis
Gene Set Enrichment Analysis (GSEA) was performed using GSEA 2.0. Gene sets of top ~12h and ~24h cycling genes (at both mRNA and protein level) were provided in Table S5. Human NASH and hepatic steatosis microarray data were obtained from published database (Ahrens et al., 2013). 1000 permutations were performed using phenotype permutation option. Weighted and signal2noise were selected for Enrichment Statistic and Metric for ranking genes, respectively. Normalized Enrichment Score (NES) and FWER p value were provided for each analysis.
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism 7 software. All data were tested for normal distribution of variables. All normally distributed data were displayed as means ± standard error of the mean (SEM) unless otherwise noted. Measurements between two groups were performed with an unpaired Student’s t test.
DATA AND SOFTWARE AVAILABILITY
Raw and Cell Profiler-processed images for single cell Eif2ak3-dGFP recordings were deposited into Mendeley Data with URL: https://data.mendeley.com/datasets/wk8sfwbfw4/draft?a=468fb576-a664-45ea-a370-34e95f8cab73
Supplementary Material
Table S1. Eigenvalue/pencil decomposition of all 18,108 genes using the 9th reduced model, related to Figure 1: Period, decay rate (smaller than 1 indicates decay, larger than 1 indicates growth), phase and amplitude for each oscillation were provided as well as the mean value. Relative amplitude was calculated by normalizing the absolute amplitude to the mean.
Table S2. All and dominant circadian, ~12h and ~8h genes revealed by the eigenvalue/pencil approach, related to Figure 1 and 2: Period, decay rate, phase and amplitude for each oscillation were provided as well as the mean value. Relative amplitude was calculated by normalizing the absolute amplitude to the mean.
Sheet 1. All genes with superimposed circadian rhythm.
Sheet 2. Genes with dominant circadian rhythm.
Sheet 3. All genes with superimposed ~12h rhythm.
Sheet 4. Genes with dominant ~12 rhythm.
Sheet 5. All genes with superimposed ~8h rhythm.
Sheet 6. Genes with dominant ~8h rhythm.
Sheet 7. Top 12h cycling ER and metabolism-related genes with relative Amp>0.1 Sheet 8. Mathematical degrees calculated among the three most prominent oscillations from the 91 ER/metabolism genes by the eigenvalue/pencil method.
Table S3. Applying the RAIN, JTK_CYCLE and ARSER methods to the 18,108 hepatic genes to uncover the ~12h cycling genes, related to Figure 1: Raw data showing the different parameters associated with the three methods as well as the p value.
Sheet 1. Genes analyzed by the RAIN method.
Sheet 2. Genes analyzed by the JTK_CYCLE method. Sheet 3. Genes analyzed by the ARSER method.
Table S4. KEGG GO terms of all circadian and ~12h genes, related to Figure 1.
Sheet 1. KEGG GO analysis of ~12h genes.
Sheet 2. KEGG GO analysis of circadian genes.
Table S5. Prevalent 12h rhythm of hepatic protein expression, related to Figure 5.
Sheet 1. Log2 mean normalized level of proteins with a 12h period in mouse liver recalculated from a published SILAC database (Robles et al., 2014).
Sheet 2. The genes list of cycling 12h proteins as well as genes with both 12h rhythms at the mRNA and protein level.
Sheet 3. GO analysis of the 143 12h mRNA/protein cycling genes.
Sheet 4. 138 genes with ~24h rhythmicity at both mRNA and protein level.
Table S6. Primer sequences for ChIP-qPCR and qRT-PCR in this study, related to STAR METHODS.
Sheet 1. Primer sequences for ChIP-qPCR.
Sheet 2. Primer sequences for qRT-PCR.
Movie S1, related to Figure 4A: Time lapse microscopy of Eif2ak3-dGFP MEFs. This movie corresponds to cell lineage 1 in Figure 4A. The movie was generated with 24fps.
Highlights.
Hepatic 12h rhythms are prevalent in ER and mitochondria homeostasis genes.
Cell-autonomous 12h rhythms are independent of the circadian clock.
The 12h rhythm of gene expression is transcriptionally regulated by XBP1s.
Mammalian 12h rhythms of gene expression are conserved in crustaceans and nematodes.
Acknowledgments
We greatly thank Naomi Gonzalez at BCM for technical support and Dr. Xi Chen at BCM for suggestions and comments on XBP1-related research as well as providing the XBP1s overexpression construct. This research was supported by grants from the NIH (U24 DK097748 and R01 HD07857 to B.W.O.) and by the Brockman Foundation to B.W.O. and C.C.D. In addition, this research also was supported by grants from the Center for Advancement of Science in Space, Peter J. Fluor Family Fund, Philip J. Carroll, Jr. Professorship, Joyce Family Foundation to C.C.D and NSF Grant CCF-1320866, as well as DFG (German Science Foundation) Grant AN-693/1-1 to A.C.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS:
B.Z. and A.C.A. designed research; C.C.D. and B.W.O. supervised the project; B.Z. performed experiments; Q.Z. and A.C.A performed mathematical analyses; B.Z. analyzed data; E.M. assisted in the time lapse microscope experiment; Y.P. generated the Eif2ak3-dGFP cell line and B.Z., Q.Z., B.Y., A.C.A., C.C.D. and B.W.O wrote the paper.
SUPPLEMENTAL INFORMATION:
Supplemental materials can be found within the article online and include 7 Figures, 6 Tables and 1 Movie.
References
- Ahrens M, Ammerpohl O, von Schonfels W, Kolarova J, Bens S, Itzel T, Teufel A, Herrmann A, Brosch M, Hinrichsen H, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013;18:296–302. doi: 10.1016/j.cmet.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Ayala DE, Hermida RC, Garcia L, Iglesias T, Lodeiro C. Multiple component analysis of plasma growth hormone in children with standard and short stature. Chronobiol Int. 1990;7:217–220. doi: 10.3109/07420529009056977. [DOI] [PubMed] [Google Scholar]
- Bjerner B, Holm A, Swensson A. Diurnal variation in mental performance; a study of three-shift workers. British journal of industrial medicine. 1955;12:103–110. doi: 10.1136/oem.12.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol. 2013;301:215–290. doi: 10.1016/B978-0-12-407704-1.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton R, Mullington J. Circasemidian sleep propensity and the phase-amplitude maintenance model of human sleep/wake regulation. Journal of sleep research. 1992;1:93–98. doi: 10.1111/j.1365-2869.1992.tb00017.x. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun WP, Paine MW, Fort A. Circadian rhythm of body temperature during prolonged undersea voyages. Aviation, space, and environmental medicine. 1978;49:671–678. [PubMed] [Google Scholar]
- Cretenet G, Le Clech M, Gachon F. Circadian clock-coordinated 12 Hr period rhythmic activation of the IRE1alpha pathway controls lipid metabolism in mouse liver. Cell Metab. 2010;11:47–57. doi: 10.1016/j.cmet.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, Dilag-Penilla SA, Dyar KA, Baldi P, Sassone-Corsi P. Reprogramming of the circadian clock by nutritional challenge. Cell. 2013;155:1464–1478. doi: 10.1016/j.cell.2013.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang B, Everett LJ, Jager J, Briggs E, Armour SM, Feng D, Roy A, Gerhart-Hines Z, Sun Z, Lazar MA. Circadian enhancers coordinate multiple phases of rhythmic gene transcription in vivo. Cell. 2014;159:1140–1152. doi: 10.1016/j.cell.2014.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haus E, Dumitriu L, Nicolau GY, Bologa S, Sackett-Lundeen L. Circadian rhythms of basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), insulin-like growth factor-1 (IGF-1), insulin-like growth factor binding protein-3 (IGFBP-3), cortisol, and melatonin in women with breast cancer. Chronobiol Int. 2001;18:709–727. doi: 10.1081/cbi-100106083. [DOI] [PubMed] [Google Scholar]
- Hoogerwerf WA, Sinha M, Conesa A, Luxon BA, Shahinian VB, Cornelissen G, Halberg F, Bostwick J, Timm J, Cassone VM. Transcriptional profiling of mRNA expression in the mouse distal colon. Gastroenterology. 2008;135:2019–2029. doi: 10.1053/j.gastro.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, Hogenesch JB. Harmonics of circadian gene transcription in mammals. PLoS genetics. 2009;5:e1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JB, Kornacker K. JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. Journal of biological rhythms. 2010;25:372–380. doi: 10.1177/0748730410379711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hong HK, Chong JL, Indacochea AA, Lee SS, Han M, Takahashi JS, Hogenesch JB. Brain-specific rescue of Clock reveals system-driven transcriptional rhythms in peripheral tissue. PLoS genetics. 2012;8:e1002835. doi: 10.1371/journal.pgen.1002835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita AC, Antoulas AC. Parametrized model reduction in the Loewner Framework. In: Quarteroni A, Rozza G, editors. Reduced Order Methods for Modeling and Computational Reduction. (Springer); 2013. [Google Scholar]
- Ionita AC, Antoulas AC. Data-Driven Parametrized Model Reduction in the Loewner Framework. SIAM Journal on Scientific Computing. 2014;36:A984–A1007. [Google Scholar]
- Janich P, Arpat AB, Castelo-Szekely V, Lopes M, Gatfield D. Ribosome profiling reveals the rhythmic liver translatome and circadian clock regulation by upstream open reading frames. Genome research. 2015;25:1848–1859. doi: 10.1101/gr.195404.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Mazuch J, Abraham U, Eom GD, Herzog ED, Volk HD, Kramer A, Maier B. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci U S A. 2009;106:21407–21412. doi: 10.1073/pnas.0906361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B. The molecular hug between the ER and the mitochondria. Current opinion in cell biology. 2013;25:443–448. doi: 10.1016/j.ceb.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Menet JS, Rodriguez J, Abruzzi KC, Rosbash M. Nascent-Seq reveals novel features of mouse circadian transcriptional regulation. eLife. 2012;1:e00011. doi: 10.7554/eLife.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metodiev MD, Lesko N, Park CB, Camara Y, Shi Y, Wibom R, Hultenby K, Gustafsson CM, Larsson NG. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 2009;9:386–397. doi: 10.1016/j.cmet.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, Antoch MP, Walker JR, Esser KA, Hogenesch JB, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci U S A. 2007;104:3342–3347. doi: 10.1073/pnas.0611724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill JS, Lee KD, Zhang L, Feeney K, Webster SG, Blades MJ, Kyriacou CP, Hastings MH, Wilcockson DC. Metabolic molecular markers of the tidal clock in the marine crustacean Eurydice pulchra. Current biology: CB. 2015;25:R326–327. doi: 10.1016/j.cub.2015.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka K, Cornelissen G, Halberg F. Circadian rhythmic fractal scaling of heart rate variability in health and coronary artery disease. Clinical cardiology. 1997;20:631–638. doi: 10.1002/clc.4960200710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refinetti R, Lissen GC, Halberg F. Procedures for numerical analysis of circadian rhythms. Biological rhythm research. 2007;38:275–325. doi: 10.1080/09291010600903692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles MS, Cox J, Mann M. In-vivo quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. PLoS genetics. 2014;10:e1004047. doi: 10.1371/journal.pgen.1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanowski A, Garavaglia MJ, Goya ME, Ghiringhelli PD, Golombek DA. Potential conservation of circadian clock proteins in the phylum Nematoda as revealed by bioinformatic searches. PLoS One. 2014;9:e112871. doi: 10.1371/journal.pone.0112871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013;18:685–697. doi: 10.1016/j.cmet.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmen T, Lynes EM, Gesson K, Thomas G. Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM) Biochimica et biophysica acta. 2010;1798:1465–1473. doi: 10.1016/j.bbamem.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stashi E, Lanz RB, Mao J, Michailidis G, Zhu B, Kettner NM, Putluri N, Reineke EL, Reineke LC, Dasgupta S, et al. SRC-2 is an essential coactivator for orchestrating metabolism and circadian rhythm. Cell reports. 2014;6:633–645. doi: 10.1016/j.celrep.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taruskin R. Music in the Western World: A History in Documents (2d ed.) Choral Journal. 2008;48:68–70. [Google Scholar]
- Thaben PF, Westermark PO. Detecting rhythms in time series with RAIN. Journal of biological rhythms. 2014;29:391–400. doi: 10.1177/0748730414553029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden AM, Beverly M, Kadener S, Rodriguez J, Wasserman S, Rosbash M, Sengupta P. Genome-wide analysis of light- and temperature-entrained circadian transcripts in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000503. doi: 10.1371/journal.pbio.1000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Eno CO, Altman BJ, Zhu Y, Zhao G, Olberding KE, Rathmell JC, Li C. ER stress modulates cellular metabolism. The Biochemical journal. 2011;435:285–296. doi: 10.1042/BJ20101864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark PO, Herzel H. Mechanism for 12 hr rhythm generation by the circadian clock. Cell reports. 2013;3:1228–1238. doi: 10.1016/j.celrep.2013.03.013. [DOI] [PubMed] [Google Scholar]
- Wilcockson D, Zhang L. Circatidal clocks. Current biology : CB. 2008;18:R753–R755. doi: 10.1016/j.cub.2008.06.041. [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Chen L, Grant GR, Paschos G, Song WL, Musiek ES, Lee V, McLoughlin SC, Grosser T, Cotsarelis G, et al. Timing of expression of the core clock gene Bmal1 influences its effects on aging and survival. Science translational medicine. 2016;8:324ra316. doi: 10.1126/scitranslmed.aad3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Su Z. Analyzing circadian expression data by harmonic regression based on autoregressive spectral estimation. Bioinformatics. 2010;26:i168–174. doi: 10.1093/bioinformatics/btq189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Hastings MH, Green EW, Tauber E, Sladek M, Webster SG, Kyriacou CP, Wilcockson DC. Dissociation of circadian and circatidal timekeeping in the marine crustacean Eurydice pulchra. Current biology : CB. 2013;23:1863–1873. doi: 10.1016/j.cub.2013.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A. 2014;111:16219–16224. doi: 10.1073/pnas.1408886111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Gates LA, Stashi E, Dasgupta S, Gonzales N, Dean A, Dacso CC, York B, O’Malley BW. Coactivator-Dependent Oscillation of Chromatin Accessibility Dictates Circadian Gene Amplitude via REV-ERB Loading. Mol Cell. 2015 doi: 10.1016/j.molcel.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Eigenvalue/pencil decomposition of all 18,108 genes using the 9th reduced model, related to Figure 1: Period, decay rate (smaller than 1 indicates decay, larger than 1 indicates growth), phase and amplitude for each oscillation were provided as well as the mean value. Relative amplitude was calculated by normalizing the absolute amplitude to the mean.
Table S2. All and dominant circadian, ~12h and ~8h genes revealed by the eigenvalue/pencil approach, related to Figure 1 and 2: Period, decay rate, phase and amplitude for each oscillation were provided as well as the mean value. Relative amplitude was calculated by normalizing the absolute amplitude to the mean.
Sheet 1. All genes with superimposed circadian rhythm.
Sheet 2. Genes with dominant circadian rhythm.
Sheet 3. All genes with superimposed ~12h rhythm.
Sheet 4. Genes with dominant ~12 rhythm.
Sheet 5. All genes with superimposed ~8h rhythm.
Sheet 6. Genes with dominant ~8h rhythm.
Sheet 7. Top 12h cycling ER and metabolism-related genes with relative Amp>0.1 Sheet 8. Mathematical degrees calculated among the three most prominent oscillations from the 91 ER/metabolism genes by the eigenvalue/pencil method.
Table S3. Applying the RAIN, JTK_CYCLE and ARSER methods to the 18,108 hepatic genes to uncover the ~12h cycling genes, related to Figure 1: Raw data showing the different parameters associated with the three methods as well as the p value.
Sheet 1. Genes analyzed by the RAIN method.
Sheet 2. Genes analyzed by the JTK_CYCLE method. Sheet 3. Genes analyzed by the ARSER method.
Table S4. KEGG GO terms of all circadian and ~12h genes, related to Figure 1.
Sheet 1. KEGG GO analysis of ~12h genes.
Sheet 2. KEGG GO analysis of circadian genes.
Table S5. Prevalent 12h rhythm of hepatic protein expression, related to Figure 5.
Sheet 1. Log2 mean normalized level of proteins with a 12h period in mouse liver recalculated from a published SILAC database (Robles et al., 2014).
Sheet 2. The genes list of cycling 12h proteins as well as genes with both 12h rhythms at the mRNA and protein level.
Sheet 3. GO analysis of the 143 12h mRNA/protein cycling genes.
Sheet 4. 138 genes with ~24h rhythmicity at both mRNA and protein level.
Table S6. Primer sequences for ChIP-qPCR and qRT-PCR in this study, related to STAR METHODS.
Sheet 1. Primer sequences for ChIP-qPCR.
Sheet 2. Primer sequences for qRT-PCR.
Movie S1, related to Figure 4A: Time lapse microscopy of Eif2ak3-dGFP MEFs. This movie corresponds to cell lineage 1 in Figure 4A. The movie was generated with 24fps.