

Abstract
Breast cancer among Palestinian women has lower incidence than in Europe or North America, yet is very frequently familial. We studied genetic causes of this familial clustering in a consecutive hospital-based series of 875 Palestinian patients with invasive breast cancer, including 453 women with diagnosis by age 40, or with breast or ovarian cancer in a mother, sister, grandmother, or aunt (“discovery series”); and 422 women diagnosed after age 40 and with negative family history (“older-onset sporadic patient series”). Genomic DNA from women in the discovery series was sequenced for all known breast cancer genes, revealing a pathogenic mutation in 13% (61/453) of patients. These mutations were screened in all patients and in 300 Palestinian female controls, revealing 1.0% (4/422) carriers among older, non-familial patients and two carriers among controls. The mutational spectrum was highly heterogeneous, including pathogenic mutations in eleven different genes: BRCA1, BRCA2, TP53, ATM, CHEK2, BARD1, BRIP1, PALB2, MRE11A, PTEN, and XRCC2. BRCA1 carriers were significantly more likely than other patients to have triple negative tumors (P = 0.03). The single most frequent mutation was TP53 p.R181C, which was significantly enriched in the discovery series compared to controls (P = 0.01) and was responsible for 15% of breast cancers among young onset or familial patients. TP53 p.R181C predisposed specifically to breast cancer with incomplete penetrance, and not to other Li-Fraumeni cancers. Palestinian women with young onset or familial breast cancer and their families would benefit from genetic analysis and counseling.
Keywords: breast cancer, BRCA1, BRCA2, TP53, Palestine
Among Palestinian women, the incidence of breast cancer is lower than among European and North American women, yet many Palestinian breast cancer patients have relatives who also developed the disease.[1] Familial clustering of breast cancer in an otherwise low-incidence population could reflect clustering of non-genetic risk factors, or genetic predisposition, or both. In order to evaluate the genetic contribution to breast cancer in the Palestinian population, we undertook to determine the frequency, spectrum, and consequences of damaging mutations in breast cancer genes among Palestinian patients.
Patients and Methods
Patients
Participants in the project were Palestinian women treated for primary invasive breast cancer between 2008 and 2016 at Augusta Victoria Hospital, Arab Care Hospital Ramallah, El Husseini-Beit Jala Government Hospital, or Share Zedek Medical Center. Patients consenting to participate in the study were interviewed about their family history of cancers, asked for permission to review medical records of their breast cancer diagnosis and treatment, and asked to contribute 8.5 ml peripheral blood sample for genomic DNA extraction. Controls were healthy Palestinian maternity patients from Holy Family Hospital, Bethlehem. Institutional review boards of all participating institutions approved the project.
For genomic analysis, the cohort of subjects was divided into two groups based on age at diagnosis and family history. The “discovery series” included all patients diagnosed at age 40 or younger or with family history of breast or ovarian cancer in a first or second degree relative. The “older-onset, sporadic patient series” included all other patients; i.e. those diagnosed after age 40 and with no close relative with breast or ovarian cancer.
Genomics
For patients in the discovery series, germline DNA extracted from blood was sequenced using BROCA, a targeted capture and multiplexed massively parallel sequencing panel that enables detection of all classes of mutations in all known breast cancer genes.[2, 3] Genes included in this project were ATM, BARD1, BRCA1, BRCA2, BRIP1, CHEK2, FAM175A/ABRAXAS, MRE11A, NBN, PALB2, PTEN, RAD51C, RAD51D, SLX4/FANCP, TP53, and XRCC2. Sequencing was carried out to minimum 200x coverage and reads aligned to the human reference genome (hg19). Variants were identified using GATK37 and Pindel after indel realignment and base quality recalibration, and single nucleotide variants, indels; copy number variants (CNVs) were detected and annotated as previously described.[2,3,4,5] Missense mutations were included only if previously reported with experimental evidence to be damaging to protein function. For samples with large deletions that extended beyond the genomic regions targeted by BROCA, exact breakpoints were determined by whole genome sequencing, carried out to median minimum 30-fold coverage on Illumina HiSeq X-Ten instruments (Macrogen). FASTQ reads of whole genome sequence were evaluated with MANTA-SV to identify deletion breakpoints.[6]
As damaging mutations were identified in the discovery series, the 422 subjects in the older-onset, sporadic patient series and 300 adult Palestinian controls were genotyped for each variant, either by Sanger sequencing or with SNP-Type assays (Fluidigm). For mutations occurring in more than one patient, the possibility of shared ancestry was evaluated by haplotype analysis using short tandem repeat (STR) markers flanking the mutation. Statistical comparisons were based on two-tailed chi-square tests, Fisher exact tests, or tests of the difference between independent proportions, as appropriate.
Results
Clinical features of the patients and their tumors
The total study sample included 875 Palestinian women with a diagnosis of invasive breast cancer, including 453 women in the discovery series and 422 women in the older-onset sporadic patient series. Demographic features of the subjects are shown in Table 1. Pathology records were sought for the patients in the discovery series. Tumor stage, grade, and hormonal status were available for 61% (278/453), 36% (161/453), and 49% (220/453) of these patients, respectively. Among patients with pathology data, the distribution of tumor stage was 9% stage 1, 45% stage 2, 41% stage 3, and 5% stage 4. The distribution of tumor grade was 4% grade I, 45% grade II, and 51% grade III. Of tumors with hormone receptor profiles, 20% were triple negative (TNBC).
Table 1.
Demographic and clinical characteristics of the patients
| Characteristic | Mutation discovery cohort | General patient cohort | Total | |
|---|---|---|---|---|
| Total unrelated breast cancer patients | 453 | 422 | 875 | |
| Age at diagnosis (dx) and family history (FH) | ||||
| Dx ≤ 40 y, positive FH* | 79 | 0 | 79 | |
| Dx ≤ 40 y, negative FH | 186 | 0 | 186 | |
| Dx > 40 y, positive FH | 188 | 0 | 188 | |
| Dx > 40 y, negative FH | 0 | 422 | 422 | |
| Mean age at diagnosis (years) | 41.6 | 51.8 | 46.5 | |
| (sd ±10.0) | (sd ±8.8) | (sd ±10.7) | ||
| Residence | ||||
| West Bank | 305 | 286 | 591 | |
| Gaza | 101 | 111 | 212 | |
| East Jerusalem | 47 | 25 | 72 | |
Positive FH: breast or ovarian cancer in mother, sister, grandmother, or aunt
Burden of mutations in breast cancer genes
For the patients in the discovery series, genomic analysis using BROCA of all known breast cancer genes revealed that 13.5% (61/453) of patients carried an unambiguously damaging germline mutation in any of eleven different genes (Tables 2, S1). For BRCA1 and BRCA2, 6.8% (31/453) of patients carried a damaging mutation. Carrier frequency was highest in patients with both young onset of breast cancer and positive family history: 27% (21/79) among patients with both positive family history and young age at diagnosis, 13% (25/188) among patients with a positive family history but diagnosed after age 40, and 8.0% (15/186) among patients diagnosed by age 40 but with negative family history (P=0.0002). Carrier frequencies were highest for BRCA1 (11 patients) and BRCA2 (20), followed by TP53 (9), ATM (6), CHEK2 (5), BARD (4), BRIP1 (2), MRE11A (1), PALB2 (1), PTEN (1) and XRCC2 (1) (Figure 1). Four previously unreported variants of unknown significance were also identified, one each in ATM, BRIP1, CHEK2, and FAM175A (Table S2). Among patients for whom tumor hormone receptor status was known, 9% (4/44) of those with TNBC compared to 2% (3/176) of those with receptor positive breast cancer carried a pathogenic mutation in BRCA1 (P=0.03). The relationship between TNBC and BRCA1 mutation status is similar in this population to that observed elsewhere.[7] Across all genes, 41 different damaging mutations were identified in the mutation discovery series. Genotyping these 41 mutations in the general population series yielded 1.0% (4/422) additional mutation carriers. One mutation appeared in controls: BARD1 p.Q735X in two cancer-free women.
Table 2.
Patients with damaging mutations in the mutation discovery cohort
| Total patients | BRCA1 | BRCA2 | TP53 | ATM | BARD1 | BRIP1 | CHEK2 | MRE11A | PALB2 | PTEN | XRCC2 | Total carriers | Frequency of mutation carriers
|
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Any gene |
BRCA1 BRCA2 |
TP53 | Other gene |
||||||||||||||
| Total patients with mutations | 453 | 11 | 20 | 9 | 6 | 4 | 2 | 5 | 1 | 1 | 1 | 1 | 61 | 0.135 | 0.068 | 0.020 | 0.046 |
| Age at diagnosis and family history (FH) | |||||||||||||||||
| Dx < 40 y, positive FH* | 79 | 5 | 8 | 2 | 2 | 1 | 0 | 2 | 0 | 1 | 0 | 0 | 21 | 0.266 | 0.165 | 0.011 | 0.076 |
| Dx < 40 y, negative FH | 186 | 1 | 4 | 2 | 2 | 2 | 2 | 0 | 0 | 0 | 1 | 1 | 15 | 0.080 | 0.027 | 0.010 | 0.043 |
| Dx > 40 y, positive FH | 188 | 5 | 8 | 5 | 2 | 1 | 0 | 3 | 1 | 0 | 0 | 0 | 25 | 0.133 | 0.069 | 0.027 | 0.037 |
| Tumor hormone receptors | |||||||||||||||||
| TNBC | 44 | 4 | 3 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 8 | 0.181 | 0.159 | 0.000 | 0.023 |
| not TNBC | 176 | 3 | 7 | 2 | 3 | 2 | 1 | 2 | 0 | 1 | 0 | 1 | 22 | 0.125 | 0.057 | 0.011 | 0.057 |
| unknown | 233 | 4 | 10 | 7 | 3 | 2 | 0 | 3 | 1 | 0 | 1 | 0 | 31 | 0.133 | 0.060 | 0.030 | 0.043 |
Positive FH: breast or ovarian cancer in mother, sister, grandmother, or aunt
Figure 1.
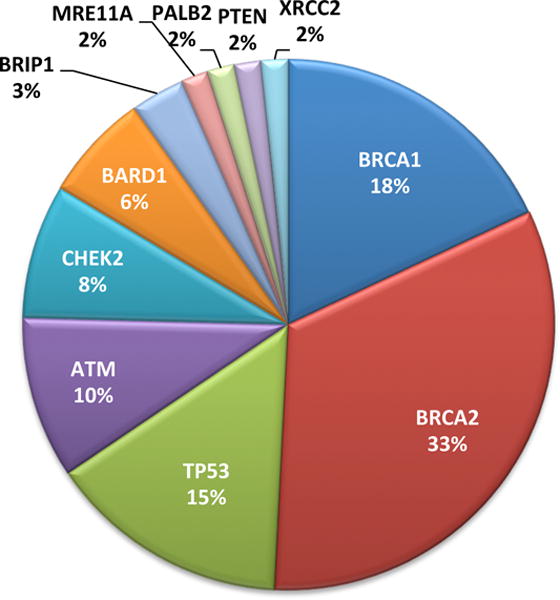
Eleven genes with damaging mutations among Palestinian patients with breast cancer diagnosed by age 40 years or with family history of breast or ovarian cancer.
Of the 41 different mutations identified in the participants, 30 were present in only one family and 11 were present in more than one apparently unrelated patient. The most frequent recurrent mutation in BRCA1 or BRCA2 was BRCA2 c.2482delGACT, originally reported from a Palestinian patient living in Saudi Arabia [8] and found in 0.7% (6/875) of all index patients. The single most frequent mutation in any gene was TP53 p.R181C, previously reported in Palestinian families [9] and found in 1.0% (9/875) of all index patients. TP53 p.R181C is described more fully below. Collectively, the 11 recurrent mutations explained 59% (36/61) of the patients with mutations in the fully evaluated discovery series. Haplotype analysis showed that recurrent mutations originated on shared ancestral haplotypes. Furthermore, of the 41 different mutations, 68% (28/41) have been reported previously and 32% (13/41) are new to this study (Table S1). Of the mutations previously reported in the literature, BRCA1 p.E1373X, BRCA2 c.2482delGACT, and TP53 p.R181C have been reported in other Palestinian families.[8,9,10] BRCA1 p.C44F and BRCA1 p.W1815X were originally described in Lebanese families.[11,12] XRCC2 p. R215X was originally described in a child with Fanconi anemia living in Saudi Arabia.[13] These mutations may be Middle Eastern “founder alleles.”
Genomic deletion of the BRCA1 promoter
Whole genome sequencing of patient MK1ES revealed a 29 kb genomic deletion of the BRCA1 promoter region with breakpoints that are specific to this Palestinian family (Figure S1). In other studies from our lab, we have encountered other deletions of the BRCA1 promoter region, with breakpoints within a few kb of the breakpoints of this allele, in four other families of various European ancestries. The 35 kb genomic region that includes the BRCA1 promoter is both segmentally duplicated and densely packed with repetitive sequences. This region may be a hotspot for both germline and somatic deletion leading to loss of function of BRCA1.
Founder mutation in TP53
The mutation with the highest frequency in the cohort was TP53 p.R181C (c.541G>A) at chr17:7,578,389 (Figure 2) This missense appeared in nine of the 453 patients in the discovery series versus zero of 300 Palestinian controls (P = 0.014). The variant is classified as “likely pathogenic” on ClinVar and is not present on ExAC. Seven of the nine families with the mutation are from Hebron. Although the patients are not aware of any direct relationship among them, the mutation occurs on a haplotype of at least 2 MB shared by all nine families, indicating a common ancestor (Figures S2, S3). TP53 p.R181C was among the first inherited mutations of TP53 reported in the literature, in a family with early onset breast cancer and melanotic spindle cell cancer of the mediastinum.[14] It was described recently in a family of unspecified ancestry with breast cancer, melanoma, and renal cell cancer[15] and in other Palestinian families with breast cancer.[9] It has also been reported as a somatic mutation in a breast cancer patient with an inherited BRCA1 mutation.[16]
Figure 2.
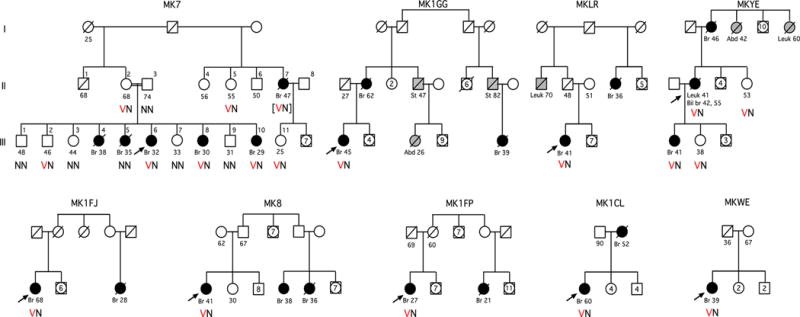
Palestinian families harboring TP53 p.R181C, illustrating cancers and genotypes of tested individuals. Black symbols indicate breast cancer and gray symbols indicate other cancers. Ages beneath the symbols indicate age at diagnosis for persons with cancer and age at most recent follow up or death for cancer-free persons. V indicates the variant allele cysteine at TP53 residue 181 and N indicates the reference allele arginine.
None of the Palestinian families that harbor TP53 p.R181C, either in this study or those previously reported, fulfill clinical criteria for Li-Fraumeni syndrome.[9,17] Of the breast cancer patients in this project with this mutation, the average age at diagnosis was 38 years, but penetrance was incomplete. In family MK7, which includes genotyped family members in three generations (Figure 2), sisters III-6, III-8, and III-10 and maternal aunt II-7 carry the mutation and were diagnosed with breast cancer at ages 29, 30, 32, and 47. However, mutation carriers II-2 and II-5 from the previous generation remain cancer-free at ages 55 and 68. Furthermore, sister III-5, diagnosed with breast cancer at age 32, does not carry the mutation. Haplotype analysis of III-5 is consistent with her carrying the maternal wild type allele of TP53 (Figure S1). Evaluation of DNA from III-5 by both BROCA and whole exome sequencing yielded genotypes consistent with her relationship in the family, but with no indication of an alternate explanation for her early onset breast cancer.
Mutations in BRCA1 and BARD1 RING domains
Three other missense mutations – BRCA1 p.H41Y, BRCA1 p.C44F, and BARD1 p.C83R – alter critical cysteine or histidine residues of the RING domains of BRCA1 and BARD1. Each of these mutations was found in one Palestinian patient, each with a family history of young onset breast cancer and/or ovarian cancer (Figure 3). None appeared in Palestinian controls. BRCA1 p.C44F has been classified as pathogenic by ClinVar, as has BRCA1 p.H41R, which did not appear among the Palestinian patients but alters same residue as BRCA1 p.H41Y. All mutations of the critical zinc-binding residues of the BRCA1 and BARD1 RING domains that have been tested experimentally have been shown to abrogate protein function, by being defective in homology-directed DNA repair, destabilizing the BRCA1-BARD1 heterodimer, and/or altering its ubiquitination function of the BRCA1/BARD1 heterodimer.[18,19,20,21] Incidence of breast and ovarian cancer in the families of carriers of these mutations is similar to that in Palestinian families with truncating mutations in BRCA1 and BARD1.
Figure 3.
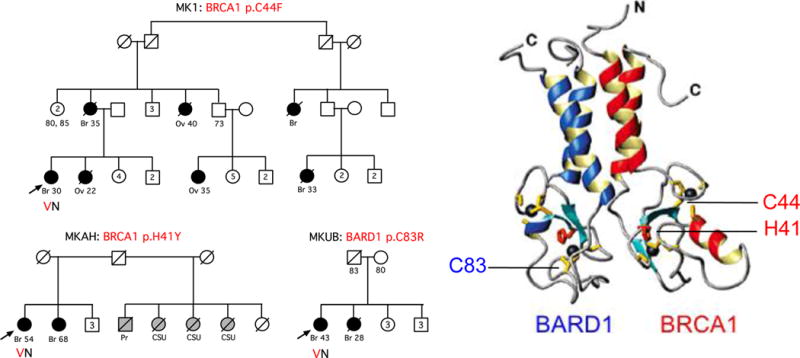
Families with mutations in the RING domains of BRCA1 and BARD1. Heterodimer structure is from reference 21.
Discussion
The study of inherited breast cancer among Palestinian patients offers insights into mutation profiles in a population not previously studied in this way, reveals new features of inherited missense mutations of TP53, and suggests a contribution to cancer control in this region.
The profile of damaging mutations in breast cancer genes among Palestinian patients is noteworthy in its combination of two features: a high level of genetic heterogeneity and a major role for recurrent or founder alleles. Heterogeneity is reflected in the 40 different mutations found among 61 mutation carriers. The importance of recurrent or founder alleles is reflected in the observation that 11 of these 40 mutations explain more than half (36/61) of all patients with mutations from the discovery series. This combination is characteristic of many European countries.[22] In the world generally, the proportion of a disease in any one geographic area explained by founder alleles is likely to decrease over the next generation as young people migrate worldwide and take their alleles with them. Fortunately, given the ease with which all mutations in all breast cancer genes can now be identified, the role of founder alleles is historically interesting but of diminishing clinical importance.
The high frequency of TP53 p.R181C among Palestinian breast cancer patients offers an opportunity to better understand inherited TP53 missense alleles. We agree with the suggestion[9] that TP53 p.R181C is a hypomorphic TP53 mutation primarily increasing risk of breast cancer. TP53 p.R181C is significantly more frequent among familial and young onset Palestinian breast cancer cases than among Palestinian controls, and is responsible for 15% of breast cancer in this group of patients. This mutation therefore has a substantial impact on breast cancer incidence among Palestinian women. On the other hand, penetrance is clearly incomplete, both in the families in this study and those reported previously. Too few families have yet been evaluated to have a robust estimate of risk, but those studied so far suggest that breast cancer risk by age 60 among carriers of this mutation may be between 50% and 75%.
TP53 p.R181C is located in the TP53 DNA-binding domain, a region of the protein that harbors mutations causing Li-Fraumeni syndrome. However, families with this mutation do not fulfill Li-Fraumeni syndrome criteria. These observations are consistent with the complex physiological and biochemical consequences of this mutation with respect to carcinogenesis. Mutant TP53 p.R181C protein almost completely retains the capacity to suppress proliferation and to transcriptionally activate TP53 target genes p21 and mdm2.[23] On the other hand, TP53 p.R181C fails to transactivate pro-apoptotic genes BAX and IGFBP-3, and induction of apoptosis is only 30–40% that of wild-type levels.[23]
Studies at the National Institutes of Health of persons heterozygous for TP53 p.R181C demonstrated that mitochondrial respiration in myoblasts was significantly higher among mutation carriers than among their relatives without the mutation, and that mutation carriers had higher levels of mitochondrial respiratory proteins.[15] TP53 is known to regulate mitochondrial biogenesis through mitochondrial transcription factor A (TFAM) and synthesis of cytochrome c oxidase 2 (SCO2). Both TFAM and SCO2 were present at higher levels in myoblasts of carriers of TP53 p.R181C or the Li-Fraumeni mutation TP53 p.R273H, compared to myoblasts of relatives with wildtype alleles.[15] These observations suggested that carriers of TP53 p.R181C have increased capacity for oxidative phosphorylation, potentially providing cancer cells with survival and proliferative advantages.[15]
With respect to its role in the Palestinian population, it has been suggested that TP53 p.R181C is reminiscent of the TP53 p.R337H mutation in Brazil.[9, 25]. Women who carry TP53 p.R337H are at increased risk of breast cancer, but penetrance of breast cancer is incomplete, and most families with this mutation do not fulfill Li-Fraumeni syndrome criteria. In Southern Brazil the population prevalence of TP53 p.R337H is 0.3%, where it results in a high rate of adrenocortical tumors in children.[24] However, TP53 p.R337H is associated with a lower penetrance of other cancers than are conventional TP53 mutations.[25] More TP53 mutations with effects such as those of R181C and R337H are likely to be encountered as gene panel testing becomes widely used and TP53 analysis is no longer limited to rare families fulfilling Li-Fraumeni syndrome criteria or to women diagnosed with breast cancer before age 35.[26]
Finally, the frequency of mutations in breast cancer genes among Palestinian patients suggests public health actions that could be life saving. Among patients with either young onset breast cancer or a family history of breast or ovarian cancer, the proportion carrying a mutation in BRCA1 or BRCA2 was 7%, the proportion carrying TP53 p.R181C was 2%, and the proportion carrying a pathogenic mutation in any breast cancer gene was 13%. Knowledge of a patient’s genotype both informs her own treatment and enables testing with appropriate follow-up to be offered to her female relatives.[27] Given the efficiency and widespread availability of testing for all classes of mutations in all known breast cancer genes, we suggest that all young or familial patients be offered this service.
Supplementary Material
Novelty and impact.
In this first systematic survey of inherited breast cancer in Palestine, we discovered that most Palestinian breast cancer patients have a relevant family history and/or were diagnosed at age 40 or younger, and that among familial and young onset patients, 13% carry a germ-line pathogenic mutation in one of eleven breast cancer genes. Most mutations were in BRCA1 or BRCA2, but the single most frequent mutation was TP53 p.R181C, which predisposes specifically to breast cancer.
Acknowledgments
We thank Dr. Abdel Razzaq Salhab and Romooz Ayish of Augusta Victoria Hospital, Dr. Riyad Shraim and Alaa Sarahne of El Hussein-Beit Jala Government Hospital, and our colleagues at Arab Care Hospital in Ramallah and at Share Zedek Medical Center in Jerusalem, all of whom welcomed us to their hospitals and introduced us to their patients. We thank the patients and their families for their enthusiastic cooperation with our project. This project was supported by the Breast Cancer Research Foundation and by NIH grants R35CA197458 and R01CA175716.
References
- 1.Aghassi-Ippen M, Green MS, Shohat T. Familial risk factors for breast cancer among Arab women in Israel. Eur J Cancer Prev. 2002;11:327–31. doi: 10.1097/00008469-200208000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Walsh T, Lee MK, Casadei S, et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA. 2010;107:12629–33. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:18032–7. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abel HJ, Duncavage EJ, Becker N, et al. SLOPE: a quick and accurate method for locating non-SNP structural variation from targeted next-generation sequence data. Bioinformatics. 2010;26:2684–8. doi: 10.1093/bioinformatics/btq528. [DOI] [PubMed] [Google Scholar]
- 5.Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics. 2011;12:184. doi: 10.1186/1471-2164-12-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen X, Schulz-Trieglaff O, Shaw R, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 7.kConFab Investigators. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: a large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res. 2014;16:3419. doi: 10.1186/s13058-014-0474-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El-Harith el-HA, Abdel-Hadi MS, Steinmann D, Dork T. BRCA1 and BRCA2 mutations in breast cancer patients from Saudi Arabia. Saudi Med J. 2002;23:700–4. [PubMed] [Google Scholar]
- 9.Zick A, Kadouri L, Cohen S, et al. Recurrent TP53 missense mutation in cancer patients of Arab descent. Fam Cancer. 2017;16:295–301. doi: 10.1007/s10689-016-9951-z. [DOI] [PubMed] [Google Scholar]
- 10.Kadouri L, Bercovich D, Elimelech A, et al. A novel BRCA1 mutation in Arab kindred from east Jerusalem with breast and ovarian cancer. BMC Cancer. 2007;7:14. doi: 10.1186/1471-2407-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jalkh N, Nassar-Slaba J, Chouery E, et al. Prevalence of BRCA1 and BRCA2 mutations in familial breast cancer patients in Lebanon. Hered Cancer Clin Pract. 2012;10:7. doi: 10.1186/1897-4287-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Saghir NS, Zgheib NK, Assi HA, et al. BRCA1 and BRCA2 mutations in ethnic Lebanese Arab women with high hereditary risk breast cancer. Oncologist. 2015;20:357–64. doi: 10.1634/theoncologist.2014-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamseldin HE, Elfaki M, Alkuraya FS. Exome sequencing reveals a novel Fanconi group defined by XRCC2 mutation. J Med Genet. 2012;49:184–6. doi: 10.1136/jmedgenet-2011-100585. [DOI] [PubMed] [Google Scholar]
- 14.Sidransky D, Tokino T, Helzlsouer K, et al. Inherited p53 gene mutations in breast cancer. Cancer Res. 1992;52:2984–6. [PubMed] [Google Scholar]
- 15.Wang PY, Ma W, Park JY, et al. Increased oxidative metabolism in the Li–Fraumeni Syndrome. New Engl J Med. 2013;368:1027–32. doi: 10.1056/NEJMoa1214091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crook T, Brooks LA, Crossland S, et al. p53 mutation with frequent novel codons but not a mutator phenotype in BRCA1 and BRCA2-associated breast tumours. Oncogene. 1998;17:1681–9. doi: 10.1038/sj.onc.1202106. [DOI] [PubMed] [Google Scholar]
- 17.Bougeard G, Renaux-Petel M, Flaman JM, et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol. 2015;33:2345–52. doi: 10.1200/JCO.2014.59.5728. [DOI] [PubMed] [Google Scholar]
- 18.Lee C, Banerjee T, Gillespie J, et al. Functional analysis of BARD1 missense variants in homology-directed repair of DNA double strand breaks. Hum Mutat. 2015;36:1205–14. doi: 10.1002/humu.22902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abkevich V, Zharkikh A, Deffenbaugh AM, et al. Analysis of missense variation in human BRCA1 in the context of interspecific sequence variation. J Med Genet. 2004;41:492–507. doi: 10.1136/jmg.2003.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Enigma Consortium: Evidence-based network for the interpretation of germline mutant alleles. doi: 10.1002/humu.21628. Available at: https://enigmaconsortium.org (accessed December 2016) [DOI] [PMC free article] [PubMed]
- 21.Brzovic PS, Rajagopal P, Hoyt DW, et al. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001;8:833–7. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 22.Janavičius R. Founder BRCA1 and BRCA2 mutations in Europe: implications for hereditary breast-ovarian cancer prevention and control. EPMA J. 2010;1:397–412. doi: 10.1007/s13167-010-0037-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith PD, Crossland S, Parker G, et al. Novel p53 mutants selected in BRCA-associated tumours that dissociate transformation suppression from other wild-type p53 functions. Oncogene. 1999;18:2451–9. doi: 10.1038/sj.onc.1202565. [DOI] [PubMed] [Google Scholar]
- 24.Custódio G, Parise GA, Kiesel Filho N, et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J Clin Oncol. 2013;31:2619–26. doi: 10.1200/JCO.2012.46.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giacomazzi J, Graudenz MS, Osorio CA, et al. Prevalence of the TP53 p.R337H mutation in breast cancer patients in Brazil. PLoS One. 2014;9:e9989. doi: 10.1371/journal.pone.0099893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Genetic/familial high-risk assessment: Breast and ovarian cancer, v 1.2017. doi: 10.6004/jnccn.2021.0001. https://www.nccn.org/professionals/physician_gls/recently_updated.asp (accessed Dec 2016) [DOI] [PubMed]
- 27.National Cancer Institute. Genetics of breast and gynecologic cancers (PDQ) Available at: http://www.cancer.gov/types/breast/hp/breast-ovarian-genetics-pdq (accessed 17 Dec 2016)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.