

Abstract
Biosynthesis of the ribosomally synthesized and post-translationally modified peptide (RiPP), pyrroloquinoline quinone (PQQ), is initiated when precursor peptide, PqqA, is recognized and bound by the RiPP precursor peptide recognition element (RRE), PqqD, for presentation to the first enzyme in the pathway, PqqE. Unlike other RiPP-producing, post-ribosomal peptide synthesis (PRPS) pathways in which the RRE is a component domain of the first enzyme, PqqD is predominantly a separate scaffolding protein that forms a ternary complex with the precursor peptide and first tailoring enzyme. As PqqD is a stable, independent RRE, this makes the PQQ pathway an ideal PRPS model system for probing RRE interactions using NMR. Herein we present both the solution NMR structure of Methylobacterium extorquens PqqD, as well as results from 1H,15N-HSQC binding experiments that identify the PqqD residues involved in binding the precursor peptide, PqqA, and the enzyme, PqqE. The reported structural model for an independent RRE, along with the mapped binding surfaces, will inform future efforts to both understand and manipulate PRPS pathways.
Keywords: Pyrroloquinoline quinone, PQQ, RiPP biosynthesis, RiPP recognition element, PqqA, PqqD, PqqE, NMR solution structure
TOC image
RiPPs comprise a collection of diverse natural products with probiotic, as well as antibiotic, anti-cancer, and anti-viral activities.1–4 Recent genome sequencing efforts have demonstrated that RiPPs represent a major natural product biosynthesis route additional to terpenoid, alkaloid, polyketide, and non-ribosomal peptide (NRP) biosyntheses.5 In NRP biosynthesis, peptide bond formation requires multimodular “megaenzymes” in an assembly line encoded within the operon.4–6 In comparison, PRPS pathways can produce equally complex and diverse molecules from simpler operons, as they utilize ribosomal machinery to produce the precursor peptide that is subsequently modified. RiPPs can encompass 70+ amino acids, such as the putative pore-forming bacteriocin, enterocin AS-48, for which an X-ray crystal structure exists,7 or be relatively small, as with the 330 Da tricyclic o-quinone, PQQ (C14H6N2O8, IUPAC: 4,5-dihydro-4,5-dioxo-1H-pyrrolo[2,3-f] quinolone-2,7,9-tricarboxylic acid) derived from two amino acid residues (Fig. 1).8, 9 PQQ biosynthesis is part of a PRPS biosynthesis subclass that includes pantocin, mycofactocin, and thyroid hormone biosyntheses.5, 10, 11
Figure 1.
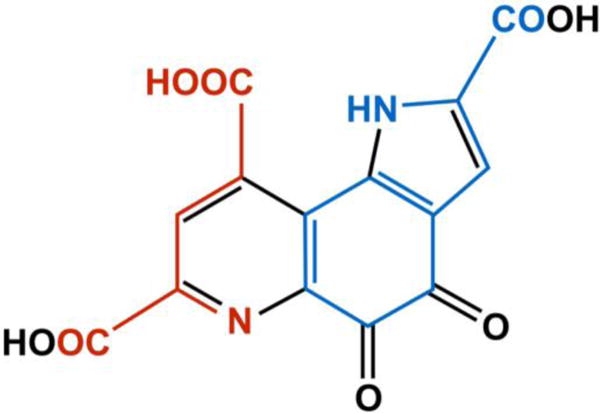
Chemical structure of PQQ. All of the carbons and nitrogens in PQQ come from a glutamate and tyrosine in an absolutely conserved EXXXY sequence located in the precursor peptide, PqqA. Atoms and bonds in red are derived from the glutamate, and those in blue are from the tyrosine. Black represents modifications during biosynthesis.
PQQ is the cofactor for a class of bacterial aldose sugar and alcohol dehydrogenases.12–15 The cofactor is synthesized outside of the dehydrogenase framework, and then non-covalently inserted into the apoenzyme.16–18 These dehydrogenases––and therefore, PQQ––give a growth advantage to the bacteria when aldose sugars and/or alcohols are available.19 The electrons generated by the reduction of PQQ to PQQH2 are fed into the constitutive electron transport chain of the organism, and ultimately generate ATP. More recently, data suggested that PQQ may be a sugar oxidoreductase coenzyme in the basidiomycete mushroom, Coprinopsis cinerea.20
The majority of PQQ-producing bacteria are Gram-negative, and include a number of human pathogens, such as Klebsiella pneumoniae and Burkholderia cenocepacia.21 Interestingly, PQQ also enhances the growth rate of some non-PQQ-producing bacteria, such as Escherichia coli, which under certain nutrient conditions express a PQQ-dependent dehydrogenase.18, 22 Therefore, PQQ is a prokaryotic vitamin.
Eukaryotes do not synthesis PQQ, however, removal of PQQ from the diets of mice and rats leads to decreased fertility, smaller litter sizes, and slowed neonatal growth.23–26 The specific mechanism(s) by which PQQ alters reproductive success are currently unknown, although data are emerging that PQQ promotes mitochondrial metabolism.27–33 As an antioxidant, PQQ can undergo approximately 20,000 redox cycles, making it, on a molar basis, 100-times more efficient than ascorbic acid as a cellular redox molecule.34–36
While biochemically intriguing in its own right, the PQQ biosynthesis pathway contains an independent RRE protein that can serve as a model system for understanding the presentation of precursor peptides by enzyme-integrated RRE domains, which are present in more than 50% of all PRPS pathways.5, 37 PQQ biosynthesis requires five to six genes encoded by the pqq operon; pqqA–E are absolutely required, while an additional gene for a putative protease, pqqF, is also often present.21, 38 PQQ biosynthesis is initiated when the precursor peptide, PqqA, is recognized and bound by PqqD for presentation to the first tailoring enzyme, PqqE, a member of the radical S-adenosyl methionine (rSAM) superfamily of enzymes that additionally contain a C-terminal Subtilosin, PQQ, Anaerobic Sulphatase (anSME), and Mycofactocin (SPASM) domain.38 PqqD is a member of the structurally conserved RREs, but is unusual in being a stand-alone peptide chaperone, as most RREs are domains within PRPS biosynthesis enzymes.37, 39 The core segment of PqqA contains an absolutely conserved EXXXY sequence, where the glutamate and tyrosine residues contain all the carbons and nitrogens comprising PQQ (Fig. 1).40, 41
This paper presents the NMR solution structure for Methylobacterium extorquens (Me) PqqD, which represents the first experimental structure of PqqD in its physiological monomeric form. Further, we identify the residues of PqqD involved in binding PqqA alone, and in binding PqqA and PqqE within the ternary complex. These findings are placed in the context of other structural work on RRE domain interactions with RiPP precursor peptides and the enzymes of which they are a part.
EXPERIMENTAL PROCEDURES
Materials
The T4 DNA ligase and all restriction enzymes were purchased from New England BioLabs (Ipswich, MA). Oligonucleotides were purchased from Eurofins (Huntsville, AL). Polymerase was obtained from Agilent (Santa Clara, CA). DNA sequencing was performed by the University of California DNA Sequencing Facility (Berkeley, CA). The precursor peptide, MePqqA (Uniprot Q49148), was chemically synthesized and purified (80% purity) by CPC Scientific (Emeryville, CA), and was used without further purification in experiments. The MePqqA peptide used in these studies did not include the N-terminal methionine, and residue 11 was changed from a cysteine to a serine to avoid inter-peptide crosslinking. The final sequence for the construct was KWAAPIVSEISVGMEVTSYESAEIDTFN (the absolutely conserved EXXXY sequence is highlighted).
Recombinant expression of 13C,15N-labeled PqqD
Although PqqD predominantly exists as a separate protein in most PQQ biosynthesis pathways, MePqqD is found N-terminally fused to MePqqC. A truncation of MePqqCD (Uniprot Q49150) containing only the MePqqD portion was constructed, as done in previous studies in which the presence of the MePqqC portion of the native polypeptide was shown to have no effect on MePqqA and MePqqE binding to MePqqD.42 The truncation sequence was derived by aligning the MePqqCD natural fusion sequence with PqqD and PqqDE sequences from 8 other bacterial species: Pseudomonas putida, Acinetobacter calcoaceticus, Klebsiella pneumoniae, Azotobacter vinelandii, Gluconobacter oxydans, Rhodopseudomonas palustris, Methylocystis sp. (containing the natural fusion, PqqDE), and Xanthomonas campestris (XcPqqD)(Clustal Omega, www.ebi.ac.uk/Tools/msa/clustalo/).43–45 The 94 residues comprising the MePqqD used in the study include an N-terminal methionine followed by 5 residues (EPTAF) representing the juncture of the linker and PqqD protein and the remaining C-terminal residues from MePqqCD. Thus, the MePqqD sequence is: MEPTAFSGSDVPRLPRGVRLRFDEVRNKHVLLAPERTFDLDDNAVAVLKLVDGRNTVSQIAQILGQTYDADPAIIEADILPMLAGLAQKRVLER (Fig. S1).
The truncated MepqqD gene construct was cloned into a kanamycin resistant pET28a vector with an N-terminal His6-tag (EMD Millipore). The gene was used to transform E. coli BL21 (DE3) cells for gene expression. Cells were grown aerobically in minimal media supplemented with 13C- and 15N-labeled ammonium chloride and D-glucose, respectively (Cambridge Isotope Laboratories, Tewksbury, MA). Yield for the His6-tagged, 13C-,15N-labeled MePqqD was 27 mgs per liter of culture. After immobilized metal affinity chromatography purification (nickel column), the protein was buffer exchanged into 25 mM potassium phosphate at pH 6.5 for all NMR experiments. For experiments involving the ternary complex only, the PqqD sample had been lyophilized for storage.
Recombinant expression of unlabeled PqqE
The gene encoding MePqqE (UniProt P71517) was cloned into the pET28a vector from genomic DNA using the NdeI and XhoI restriction sites. After sequence verification, the cloned gene was used to transform E. coli BL21(DE3) that also contained a suf operon plasmid, pPH149, for gene expression. The pPH149 and pqqE/pET28a-transformed cells were grown aerobically at 37 °C in TB media containing 35 g/ml chloramphenicol and 50 g/ml kanamycin. At a cell density of A600 = 0.6, 50 μM iron(III) citrate and 5 mM fumarate were added. The growth flasks were immediately stoppered to transition to anaerobic conditions. Anaerobic growth was allowed to continue for 30 minutes at 37 °C, after which the expression of His6-MePqqE was induced by adding 0.4 mM isopropyl-D-galactopyranoside. Following a 12-hour induction period at 19 °C, cells were centrifuged for 10 minutes at 6,500 rpm and then frozen. Subsequent work was performed in an anaerobic chamber. Cells were suspended in 5x (mass of cell paste) degassed 50 mM Tris buffer, pH 7.9, 50 mM imidazole, 1 mM TCEP, and 200 mM sodium chloride (lysis buffer). Cells were lysed following the manufacturer’s protocol for BugBuster (Novagen). BugBuster was supplemented with Benzonase (Novagen). Lysate was transferred into sealed tubes for centrifugation (outside the anaerobic chamber) for 15 minutes at 20,000 rpm. The sealed tubes were then moved back into the anaerobic chamber where the combined supernatants were loaded onto a 5 ml His-Trap FF column (GE Healthcare) using a peristaltic pump. The column, maintained at 22 °C, was washed first with lysis buffer to remove non-tagged protein and then with a degassed solution consisting of 300 mM imidazole, 50 mM Tris (pH 7.9), 1 mM TCEP, and 200 mM sodium chloride to elute the tagged protein. Fractions were combined, concentrated, and buffer exchanged using PD-10 columns equilibrated with degassed 50 mM Tris (pH 7.9), 100 mM sodium chloride, 5% glycerol, and 1 mM TCEP. The protein, anaerobically aliquoted and sealed in cryogenic vials, was then flash frozen in liquid nitrogen. SDS-PAGE was used to confirm protein homogeneity. His6-MePqqE yield was 18 mgs per liter of culture.
NMR Experiments
The PqqD protein was highly stable, and periodic 1H,15N-HSQCs on the single sample used for all experiments showed no discernable degradation over the multiple months of data collection. Likewise, the varying ratios of PqqD + PqqA and PqqD + PqqA + PqqE were highly stable, and a single preparation of each of these also lasted for the duration of the experiments needed for structure determination and binding studies. Protein samples were stored at 4°C when not in use.
All NMR experiments were performed using D2O matched 5 mm Shigemi microtubes. The PqqD sample contained 0.14 mM MePqqD, 1.0 mM sodium azide, and 5% D2O. The physiological stoichiometry for PqqD with PqqA is 1:1, with a dissociation constant (KD) of ~200 nM for MePqqA-PqqD.42 To determine the proper molar ratio of PqqD and PqqA for defining the residues of PqqD affected by binding PqqA, three 2D 1H,15N-HSQC spectra were acquired in ratios of 1:0, 1:0.7, and 1:1.3 of PqqD:PqqA, respectively. The 2D HSQC spectra demonstrated that at the PqqD:PqqA ratio of 1:1.3, all chemical shift changes were complete with peaks fully evolved. The PqqD + PqqA samples were made by adding 0, 5, and 10 μL of 6.4 mM PqqA, in identical buffer, to the 270 μL PqqD only sample (for the 1:0, 1:0.7, and 1:1.3 sample ratios, respectively). The starting concentration of PqqD was 0.17 mM in the presence of 1.0 mM sodium azide, and 5% D2O.
The physiological stoichiometry for PqqA with PqqE and PqqD is 1:1:1, with KDs of ~12 μM for the MePqqD-PqqE binary complex, and ~5 μM for the MePqqD-PqqA-PqqE ternary complex.42 Protein solubility was an issue with the PqqD + PqqA + PqqE sample, as PqqE tended to precipitate out of solution as the ratio of PqqE to PqqD exceeded 0.2 to 1. However, chemical shift assignments for the identification of PqqD residues involved in binding PqqE in the presence of PqqA could be ascertained using a ratio of 1:1.25:0.2 for PqqD:PqqA:PqqE. Therefore, the PqqD + PqqA + PqqE sample was made to a similar volume as the other samples, but contained 0.80 mM MePqqD (~ six times the quantity used in the previous experiments in order to increase signal-to-noise due to the weak NMR signal of the slower tumbling ternary complex), 1 mM MePqqA (25% excess over PqqD), 0.16 mM MePqqE (only 20% of PqqD), 1.0 mM sodium azide, and 5% D2O The PqqE was diamagnetic as the samples were treated aerobically throughout all experiments, as was done in the previously published work that determined KDs.42
All experiments were performed at 25°C on Bruker 850 or 900 MHz spectrometers, each equipped with 5 mm TCI CryoProbes. NMRPipe46 was used to process experimental data. Proton chemical shifts were calibrated with respect to the water signal relative to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS), and 13C and 15N chemical shifts were indirectly referenced to DSS.47 Data were analyzed leading to peak assignments using Sparky.48 The complete backbone and side chain chemical shift assignments for MePqqD have been deposited in the BioMagResBank (BMRB) database (www.bmrb.wisc.edu) with accession numbers 26634 for PqqD only, 26690 for the PqqD + PqqA complex, and 26696 for the PqqD + PqqA + PqqE ternary complex.49, 50
Binding studies (PqqD + PqqA)
As for chemical shift assignments, NMR data acquired from PqqD + PqqA were similar to that of PqqD alone. Typically, sequence-specific backbone assignments were completed using two 3D spectra, IBS_Best_HNCACB and IBS_Best_HN(CO)CACB.51 The IBS_Best_HNCACB experiment finds both intra- and inter-residue correlations, whereas the IBS_Best_HN(CO)CACB finds only inter-residue correlations. The IBS_Best version of the two experiments improves signal sensitivity. Combining these two spectra, backbone chemical shifts including 1HN, 15N, 13CA, and 13CB were assigned.49 The assigned 1HN and 15N shifts were then used to identify PqqD residues involved in the binding of PqqA. Data sets acquired from experiments using both PqqD alone and PqqD + PqqA samples were used to assign 13C´ and sidechain chemical shifts.49 Complete chemical shift assignments were obtained for both samples. Finally, an overlay of the 1H,15N-HSQC spectra for PqqD alone and PqqD + PqqA identified residues where the weighted peak shifts (Equation 1)52 were at least 1.5 standard deviations from the statistical mean for all shifts, thus indicating which of the RRE residues are involved in binding the precursor peptide.
| (1) |
Binding studies (PqqD + PqqA + PqqE)
A comparison of the 2D 1H,15N-HSQC spectra acquired from samples PqqD alone, PqqD + PqqA, and PqqD + PqqA + PqqE showed that the changes in chemical shifts for spectra from the binary complex to the ternary complex were not as significant as those from PqqD alone and PqqD + PqqA. It was not, therefore, necessary to acquire a full set of NMR data in order to assign the chemical shifts of PqqD in the ternary complex as it was for PqqD + PqqA. We were able to use the assigned chemical shifts of PqqD in the binary complex and limited NMR data for PqqD + PqqA + PqqE to assign the backbone chemical shifts of PqqD in the ternary complex. These data included a 3D IBS_Best_HNCA, a 3D HNCO, and the first 1H-13C 2D plane of an HN(CA)CO, allowing for the assignment of chemical shifts for backbone HN, N, CA, and CO.51 No experiments aimed at assigning sidechain chemical shifts were performed. A 2D 1H,15N-HSQC using the IBS_SOFAST pulse sequence was also acquired.53 The IBS_SOFAST pulse sequence enhanced signal sensitivity to about three times that of a regular HSQC, and allowed for the acquisition of a 2D HSQC with high sensitivity in just 20 minutes. Again, an overlay of the 1H,15N-HSQC spectra for PqqD alone versus that of PqqD + PqqA + PqqE identified residues where the weighted peak shifts were at least 1.5 standard deviations from the mean for all shifts, indicating RRE residues involved in binding the initial tailoring enzyme, PqqE, during peptide presentation.
3D structure determination
To determine the three-dimensional structure of MePqqD, we utilized internuclear distances from NOE spectra, dihedral angles estimated from the chemical shifts using TALOS+, scalar 3JHNHA coupling constants from HNHA spectrum, and hydrogen bonds (Table 1).54, 55 Even though the only deposited structure for PqqD (XcPqqD, PDB ID: 3G2B) is a non-physiological dimer in which β-hairpins are exchanged between monomers, an in silico PqqD monomer model was created using PyMOL by combining the β-hairpin from one monomer with the α-helical bundle of the other (Fig. S2).56, 57 This model had a fold similar to the RRE domains of PRPS biosynthesis enzyme crystal structures, and was used to inform NOE peak assignments. The intensities of NOE cross peaks of two 3D 15N- and 13C-edited NOESY spectra were binned into three categories: strong, medium, and weak, corresponding to 1.8–3.7 Å, 1.8–5.0 Å, and 1.8–6.0 Å distance restraints, respectively. A total of 2,697 NOE-derived distances were obtained from the NMR spectra and converted into unambiguous structural restraints. On average, we obtained 28.7 unique distance restraints per residue. The chemical shifts for the 1Hα, 13Cα, 13Cβ, and 13CO resonances were used as an input for TALOS+ to predict the backbone dihedral angles ϕ and ψ. A total of 158 ϕ and ψ angle restraints were implemented in the structure calculation protocol. Three-bond scalar 3JHNHA coupling constants were obtained from the 3D HNHA experiment using J = sqrt(−Scross/Sdiagonal)/(2πδ), where the Scross and Sdiagonal are the signal intensities of cross and diagonal peaks, respectively, and δ (= 0.013 sec) is the time delay used in this experiment.55 A total of 82 3JHNHA coupling constants were entered into the structure calculation protocol.
Table 1.
Structural statistics for 20 model ensemble of MePqqD
| NMR Restraints | 2,967 |
|
| |
| Total distance restraints | 2,697 |
| Intraresidue (i-j=0) | 549 |
| sequential (|i-j|=l) | 862 |
| medium range (l<|i-j|<5) | 740 |
| long range <|i-j|5) | 546 |
|
| |
| Total dihedral angle restraints | 158 |
| φ | 79 |
| Ψ | 79 |
|
| |
| 3JHNHA coupling constants | 82 |
|
| |
| Hydrogen bonds | 30 |
|
| |
| RMSD from idealized geometry | |
| bonds (Ǻ) | 0.012 |
| angles (deg.) | 1.4 |
|
| |
| RMSD (Ǻ) | |
| all backbone atoms of residues 7–93 | 0.7 ±0.1 |
| all heavy atoms of residues 7–93 | 1.3 ± 0.2 |
| all backbone atoms of ordered residues* | 0.6 ±0.1 |
| all heavy atoms of ordered residues | 1.1 ±0.2 |
|
| |
| Ramaehandran | |
| Most favorable regions | 97.9 % |
| Allowed regions | 2.1 % |
| Disallowed regions | 0% |
Ordered residues: 7–21, 29–32, 39–61, 71–86, and 91–93.
Structure calculations were performed using XPLOR-NIH version 2.37.58, 59 As a starting structure, an extended conformation of the polypeptide with randomized dihedral angles was used. Torsion angle restrained molecular dynamics (rMD) was carried out at 3,500 K for 100 ps. Subsequently, hybrid simulated annealing and rMD calculations were performed. After several rounds of structure calculations, the conformers converged to a similar fold with ~1 Å of root mean square deviation (RMSD) for all heavy atoms. At this stage, 15 hydrogen bonds between the carbonyl and the amide groups of the backbone in the α-helices and between β-strands were identified using two criteria: donor-acceptor distances < 2.4 Å and angles < 35°. Therefore, a total of 30 hydrogen bond restraints (each hydrogen bond having two distance restraints) were included in the final stage of energy minimization. A hundred conformers were calculated, and the 20 best structures were chosen for the final analysis. During structural refinement, calculated structures and constraints were validated using the Protein Structure Validation Suite (PSVS) (http://psvs-1_5-dev.nesg.org/). The average RMSDs for all atoms and for just the α-carbons were obtained using the PyMol ‘super’ command (allowing 5 outlier rejection cycles).57 The calculations were performed using the lowest energy MePqqD model’s core fold (residues 17–90) and the XcPqqD in silico model’s core-fold (defined as residues 15–31 from one monomer and residues 34–88 from the second)(Fig. S2). The NMR solution structure for MePqqD, based on BMRB entry 26634, has been deposited in the RCSB Protein Databank (PDB) (www.rcsb.org) with the PDB ID 5SXY.60, 61 Figures containing images of protein structures were created using MacPyMOL,57 and NMR HSQC spectra were generated using Sparky.48
RESULTS
Structure of Methylobacterium extorquens PqqD
This structure represents the first detailed physiological structure of PqqD in its monomeric solution state.42, 56, 62 The restraints used in the structure determination, and comparison between the 20 lowest energy structures are given in Table 1. Labeled PqqD showed excellent 1H,15N-HSQC signal-to-noise with clear peak separation even in the most peak-dense regions of the spectra (Fig. 2). All backbone and sidechain 1H, 15N, and 13C chemical shifts were assigned with the exception of 14 (out of 27) aromatic 13C’s and 4 (out of 21) aromatic 1H’s, due to the inherent ambiguity of C-H definition within an aromatic ring.49 Based on 1Hα, 13Cα, 13Cβ, and 13CO chemical shift resonances, PqqD was predicted to contain three β-strands and three α-helices, and the top 20 (lowest energy) annealed structures display a common core containing precisely these elements (Fig. 3).
Figure 2.
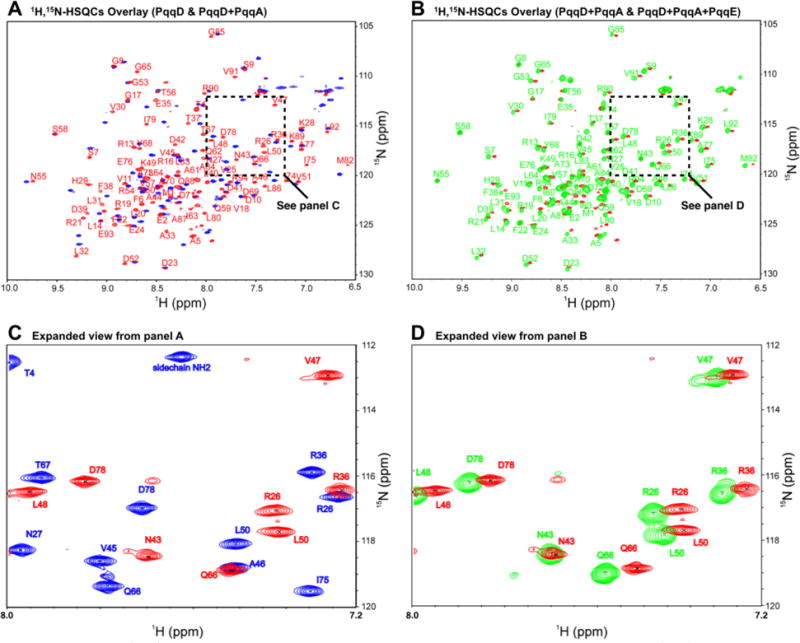
(A) 1H,15N-HSQC of PqqD alone (blue peaks) overlaid with the 1H,15N-HSQC of PqqD in complex with PqqA (red peaks). (B) 1H,15N-HSQC of PqqD in the binary complex with PqqA (red peaks) overlaid with that of PqqD in the ternary complex with PqqA + PqqE (green peaks). (C) Expanded section from panel A showing peak separation. (D) Expanded section from panel B showing peak separation.
Figure 3.
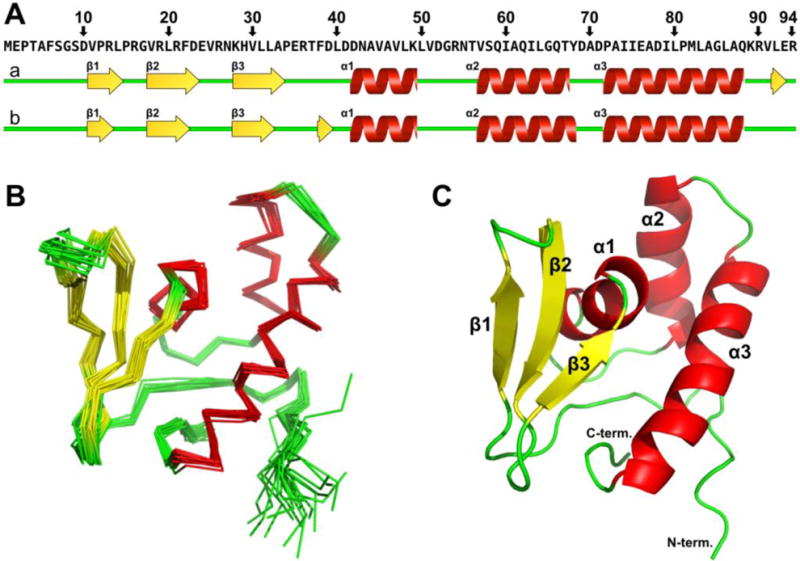
NMR structure of MePqqD. (A) Topology and location of secondary structure vs primary sequence as predicted by TALOS+ (a) and CSI2.0 (b). (B) Cα trace of the superposition of the 20 lowest energy NMR conformers. (C) Cartoon of the lowest energy conformer. α-helices, red; β-strands, yellow; loops, green.
All residues in the ensemble (20 × 94) were in favored or allowed Ramachandran regions with no residues in disallowed regions. PqqD structural order predicted by TALOS+54 and CSI2.063, 64 indicated a high level of disorder at the N-terminus (residues 1 to 7, Fig. S3). This is perhaps not surprising as the PqqD construct used in this study was derived from the natural MePqqCD fusion, and so the N-terminus in the native protein is connected to the PqqC C-terminus via a 26-residue polypeptide linker (Fig. S4). There is no evidence that PqqC and PqqD specifically interact, and so MePqqD can be viewed as “tethered” and independent of MePqqC.
PqqD residues involved in binding PqqA
PqqA must be bound to PqqD to become a substrate for the first enzyme in PQQ biosynthesis, PqqE.65 To map the binding site, NMR was used to define the PqqD residues with which PqqA interacts. 2D HSQC spectra demonstrated that at a PqqD + PqqA ratio of 1:0.7 (Fig. 4B), the chemical shifts observed were in identical positions to those for PqqD alone (Fig. 4A) and PqqD fully bound to PqqA (Fig. 4C). This indicates slow chemical exchange relative to the NMR time scale, and is consistent with the reported KD of ~200 nM.42 As such, an HSQC titration experiment to track migrating peaks was not possible, and all chemical shifts for the PqqD + PqqA sample had to be completely reassigned.
Figure 4.
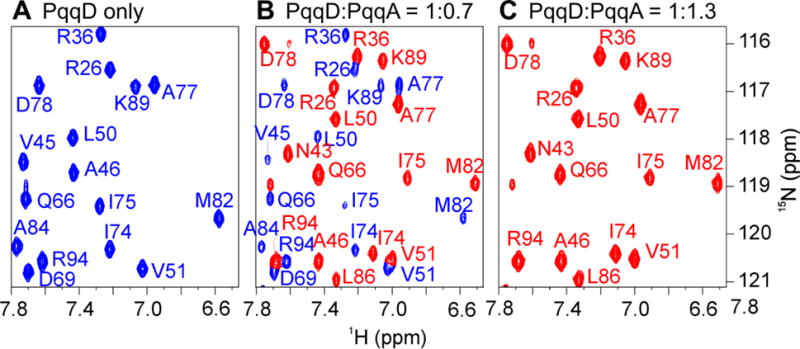
(A). 1H,15N-HSQC for PqqD alone. (B) The spectrum before saturation at a PqqD:PqqA ratio of 1:0.7. Peaks colored based on assignment to PqqD alone (panel A) or the PqqD + PqqA complex (panel C). (C) The fully evolved spectrum indicating saturation at a ratio of 1:1.3.
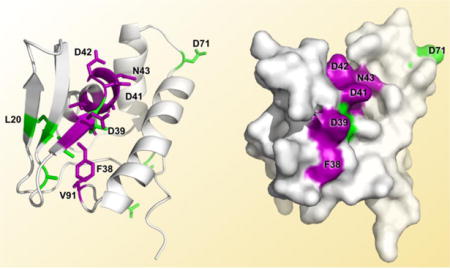
An examination of the weighted (Equation 1) 1H,15N-HSQC peak shifts ≥ 1.5σ above the mean shift between the unbound and binary complex spectra identified the following PqqD residues as involved (either directly or indirectly) in binding PqqA: Phe38*, Asp39*, Asp41*, Asp42*, Asn43*, Ala44*, Val45, Val47, Leu48, and Val91 (asterisks indicate residues ≥ 2σ above the mean shift). These residues are almost contiguous in the primary sequence (38–48), except for residue 91, and those with ≥ 2σ above the mean shift are located in the third β-strand (β3) and first α-helix (α1) (Fig. 3C) that together form a “saddle” in the structure between the β-sheet and the α-helical bundle (purple, Fig. 5).
Figure 5.
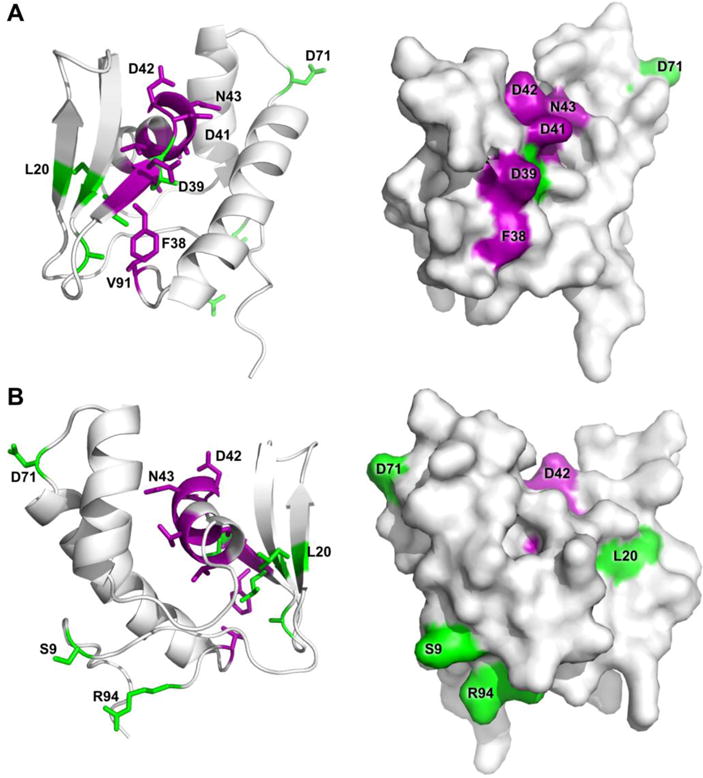
PqqD residues identified as having 1H,15N-HSQC peak shifts ≥ 1.5σ above the mean shift are indicated for the complex with PqqA (purple) along with additional residue peak shifts upon addition of PqqE to the PqqD + PqqA complex (green). Ternary complex interactions are based on chemical shifts of the backbone only, however sidechains are displayed for recognition ease. PqqD cartoon and molecular surface renderings from the same viewpoint are shown in each panel, with panel B being a 120° rotation of panel A around an axis vertical in the plane of the paper.
Additional PqqD residues perturbed when PqqE binds the binary complex, PqqD + PqqA
For the rSAM enzyme, PqqE, to act on the precursor peptide, PqqA, it must be bound to PqqD.65 PqqE is the first enzyme in PQQ biosynthesis, and it catalyzes the formation of a carbon–carbon cross-link between the Tyr and Glu of PqqA, the residues that provide most of the raw material for PQQ (Fig. 1).62, 65 Although MePqqE can form a binary complex with MePqqD in the absence of MePqqA (KD ~ 12 μM), it is the ternary complex PqqD + PqqA + PqqE that is physiologically relevant.42, 62, 65 Therefore, NMR was used to define additional residues on PqqD that were involved in binding PqqE within the context of the ternary complex. PqqE is large compared to PqqD and PqqA (MePqqE, 41.7 kDa; MePqqD, 10.4 kDa; MePqqA, 3.07 kDa), and so the ternary complex tumbles significantly more slowly than PqqD + PqqA. To overcome the resulting weaker signal-to-noise the concentration of PqqD was increased, but this led to PqqE precipitating out of solution at ratios greater than 20% PqqD. Therefore, while PqqE binding to the PqqD + PqqA complex was within the regime where an NMR titration experiment could be possible (KD ~5 μM), it was impractical.42 Even though PqqD, PqqA, and PqqE bind in a 1:1:1 ratio, PqqE at 20% the concentration of PqqD was sufficient to visualize HSQC peak shifts (Figs. 2B, 2D). Reassignment of the chemical shifts was carried out for backbone atoms only.
1H,15N-HSQC peak shifts ≥ 1.5σ above the mean shift between the binary complex and ternary complex spectra indicated that the additional PqqD residues involved in binding PqqE in the presence of PqqA are: Ser9*, Leu20, Leu31*, Ala33*, Leu40, Asp71, and Arg94* (asterisks indicate residues ≥ 2σ above the mean shift) (Fig. 5). The two most N-terminal β-strands and residues in the N-terminal and C-terminal tails of the polypeptide are interacting (directly or indirectly) with PqqE. Interestingly, Leu40 that showed no peak shift in the PqqD + PqqA complex, but lies between the β-strand and α-helix to which PqqA binds, now becomes perturbed in the ternary complex (green, Fig. 5). Whether this is a direct interaction, or an indirect effect mediated through PqqA, is currently unknown.
DISCUSSION
The monomeric NMR structure of MePqqD represents the first detailed, physiologically-relevant structure of PqqD
Size exclusion chromatography, along with small angle x-ray scattering (SAXS) experiments and now NMR, show that PqqD is physiologically monomeric.42, 62 The MePqqD NMR solution structure closely resembles the in silico models created to model the SAXS data, and to show that PqqD likely belongs to the “prevalent peptide-binding fold” in PRPS biosynthesis pathways.37, 42 These in silico models, and the model used to assist with NOE peak assignments, were informed by an earlier x-ray structure (PDB ID 3G2B) showing the protein as a dimer.56 The XcPqqD dimerization observed in the structure was likely due to the high protein concentration required for crystallization.56 While the secondary structure is largely intact in the XcPqqD crystal structure, the first two β-strands from the β-sheet subdomain of one monomer have switched with the equivalent subdomain portion of the second monomer. Interestingly, the subdomain-swapped elements from the XcPqqD crystal dimer retain very similar packing between the α-helices and β-sheet as observed in the MePqqD monomer (Fig. S2). The RMSD for alpha carbons of the XcPqqD core fold (β1–β2 from one monomer and β3, α1–α3 from the second) and the MePqqD core fold was 1.72 Å2. This indicates remarkable fold similarities between the NMR structure for MePqqD and the subdomain-swapped monomer from the XcPqqD crystal structure.
The β-strands and α-helices in MePqqD also closely match the constituent RRE folds seen in three PRPS biosynthesis enzymes for which crystal structures in complex with peptide exist; Lactococcus lactis NisB (PDB ID 4WD9), a lanthipeptide dehydratase in the biosynthesis pathway of nisin;66 Lyngbya sp. PCC-8106 LynD (PDB ID 4V1T), a fused cyclodehydratase involved in cyanobactin biosynthesis;67, 68 and E. coli MccB (PDB ID 3H9Q), an adenylating enzyme in the microcin C7 pathway,69 (Fig. 6).
Figure 6.
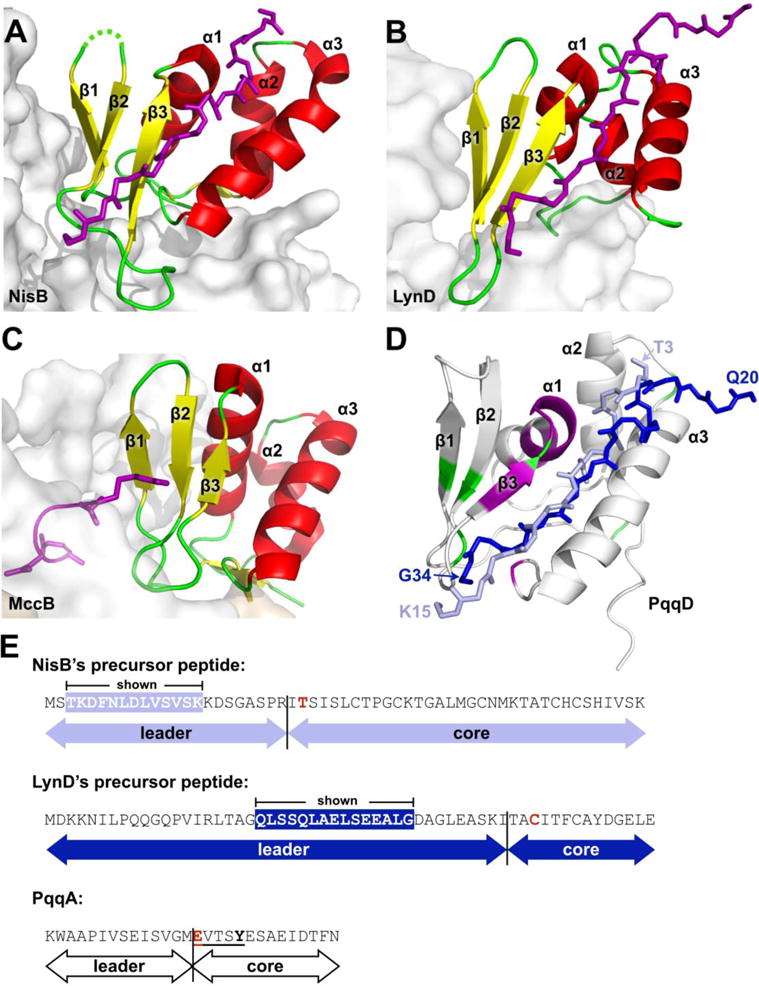
Comparison of PqqD to RREs from other PRPS enzymes in complex with peptide. (A) NisB, PDB 4WD9 (residues 142–223), (B) LynD, PDB 4V1T (residues 1–81), and (C) MccB, PDB 3H9J (residues 1–78). Constituent RREs with bound peptides are shown as cartoon and colored by secondary structure with bound precursor peptides drawn as stick and colored purple. The enzyme of which the RRE is a part is shown as a gray molecular surface. (D) PqqD residues perturbed by binding PqqA and the rSAM enzyme, PqqE, are colored purple and green, respectively. The peptides of NisB and LynD crystal structures are overlaid based on superposition of the RREs with PqqD (NisB peptide, light blue; LynD peptide, dark blue). (E) Precursor peptide sequence comparison between NisB, LynD, and PqqD. The peptide residues observed in the NisB and LynD crystal structures are white in colored boxes. The most proximal residue that is post-translationally modified is colored red.
PqqA binding to PqqD
Chemical shift perturbations observed during substrate binding can be caused directly by non-covalent interactions with either solvent molecules or binding partners, or indirectly by alterations to the conformational environment of residues not in direct contact. Comparing the residues that showed the greatest chemical shifts in the PqqD + PqqA 1H,15N-HSQC with the peptide positions in the NisB, LynD, and MccB crystal structures, PqqA appears to bind to a similar region on the surface of PqqD as the peptides of NisB and LynD (Fig. 6D).66, 70 In the case of MccB, the substrate, MccA, is a heptapeptide, one of the shortest RiPP substrates known, and the short length does not enable substantial interactions with the MccB RRE (Fig. 6C).69 As such, the MccB–MccA interaction is likely atypical with contact limited to one end of the RRE β-sheet, principally through the side chain of MccA Arg2. In the cases of NisB and LynD, the peptide forms an additional β-strand to the core β-sheet with the expected main chain hydrogen bonds to β3, and this constitutes the defining, major interaction with the RRE. In the case of PqqD, the β3 strand chemical shifts are significantly perturbed, suggesting that PqqA may also form a β-strand with the PqqD β-sheet. The residues prior to those within the β-strand adopt different conformations in the NisB and LynD peptides (Fig. 6). Despite this, both the NisB and LynD peptide side chains in this region interact with the N-terminal α3 helix side chains, and primarily involve bulky hydrophobic residues, such as Leu, Tyr, and Phe. However, it should be noted that the first four ordered residues of the LynD peptide are in a substantial crystal contact that may influence the conformer observed in this particular case. Although the binding of PqqA does not lead to large chemical shift perturbations in the α3 helix of PqqD, it is likely that there are interactions between these two elements. Interestingly, residues at the N-terminus of the α1 helix of PqqD show large chemical shift changes when complexed to PqqA (Fig. 5). This could suggest that PqqA folds over the saddle between the β2 strand and α2 helix of PqqD in a direct interaction, but equally the effect could indirectly arise from the interaction between PqqA and the β3 strand of PqqD, which is immediately N-terminal to the α1 helix.
PqqE binding to the PqqD–PqqA complex
For NisB, LynD, and MccB, the enzyme portion of the RRE-enzyme superstructure interacts with the RRE domain such that the β3-α1 and α2-α3 loops and the associated ends of those secondary structures are solvent exposed (Fig. 6A–C). Results from the PqqD + PqqA + PqqE binding experiments indicate that PqqD positioning relative to PqqE is similar to the position seen in the RRE-enzyme superstructures of NisB, LynD, and MccB (Fig. 6). RiPP precursor peptides are bipartite, with an N-terminal leader sequence involved in RRE-to-precursor peptide recognition, and a C-terminal core sequence that contains the residues ultimately comprising the final RiPP natural product.5 Since the C-terminal core portion of PqqA must insert into the active site of PqqE, it is likely that the peptide is oriented such that the N-terminal end is close to the α2–α3 loop and the C-terminal end of the leader of PqqA lies at the β2–β3 loop, as observed in NisB and LynD. While the NisB and LynD leader sequences of the respective precursor peptides contain 23 and 43 residues, the peptide segments for which electron density was observed and modeled were only 13 and 15 residues long, respectively, and represent only the residues critical to recognition (Fig. 6E).70, 71 In both cases, the amino acids bound to the RRE domain are significantly upstream from the start of the core peptide that is acted on by the enzyme (8 residues in NisB, and 9 residues in LynD). In both these cases, the active sites are relatively distant from the last ordered residue of the precursor peptide bound by the RRE (LynD, > 25Å; NisB, > 30Å). In contrast, the entire leader of PqqA is a similar length to the ordered peptide observed in the LynD and NisB crystal structures, with only 14 residues N-terminal to the Glu residue that is cross-linked to Tyr through the action of PqqE.65 These observations suggest that the majority of the PqqA leader is likely bound to PqqD, placing the PqqA EXXXY core sequence very close to the RRE with few intervening residues such that the interaction between PqqD and the PqqE active site may be quite intimate. PqqE is a rSAM enzyme, and the chemistry must be sequestered from solvent.72 Although the structure of PqqE is currently not known, an additional function of PqqD may be to act as a plug to prevent quenching of the radical by solvent during turnover.72 This is in contrast to LynD and NisB, both dehydratases, where reaction byproducts need to exit the active site (ADP/phosphate and glutamate, respectively), and the RRE domains are more distant.66, 70 This additional function may be specific to RiPPs whose biosynthesis involves rSAM enzymes, suggesting that even if the RRE is part of the polypeptide of the rSAM enzyme, it may require more mobility than the RRE domains of NisB, LynD, and MccB. Supporting the hypothesis that PqqD acts as a plug is the presence of a long linker (26 residues) seen in the MePqqCD natural fusion (Fig. S4).
It should be noted that the NMR experiments with PqqE were conducted aerobically, which means that the active site and SPASM domain Fe-S clusters were oxidized, and probably partially occupied; even the anaerobically isolated PqqE contains incomplete clusters that need to be reconstituted.65 As well as the [4Fe-4S] cluster of the rSAM active site, the PqqE SPASM domain contains a [4Fe-4S] and a second cluster that may be a [2Fe-2S].65, 73 Electron paramagnetic resonance data indicate that the binding of PqqD affects at least one of the [4Fe-4S] clusters, which is consistent with the hypothesis that PqqD must bind in close proximity to the active site.62
It is becoming clear that rSAM-SPASM enzymes are widespread (~14,000 annotations in the Interpro sequence database), particularly in association with RiPPs. The prevalence of rSAM enzymes in RiPP biosynthesis (both with and without SPASM domains) is likely linked to the inherent ability of radicals to initiate peptide chemistry. Bioinformatics suggests that ~ 50% of all RiPP rSAM-SPASM enzymes have an identifiable N-terminal RRE domain as part of the polypeptide, although in mycofactocin biosynthesis, like PQQ, the RRE is present as a separate protein, MftB.42, 74, 75 The chemistry catalyzed by these enzymes includes carbon-carbon bond formation (PqqE,65 StrB76), oxidative decarboxylation (MftC74, 75), and thioether bond formation (AlbA,77, 78 SkfB,79, 80 ThnB,81 SCIFF maturase82). The final RiPP products are chemically diverse with a wide-range of physiological functions; antibiotic (thurincin H, subtilosin A), growth regulation (sporulation killing factor), signaling (streptide), and redox cofactors (PQQ). Unfortunately, the only crystal structure of a rSAM-SPASM enzyme is that of anSME, which is not part of a RiPP pathway but activates a sulfatase under anaerobic conditions through post-translational modification.83 Therefore, the structural details of how peptide substrates are presented to the prevalent rSAM-SPASM enzymes found within diverse RiPP biosynthesis pathways are currently unknown. This work provides the first insight by identifying residues in PqqD that interact with PqqE, and showing that the binding surface overlaps with that of PqqA.
In conclusion, the first detailed monomeric structure of PqqD is presented. Mapping of chemical shifts perturbed by binding of PqqA, the precursor peptide, demonstrate that the binding mode of the PqqA leader peptide is likely similar to that observed in crystal structures of the unrelated dehydratases, LynD and NisB, utilizing a cleft formed between the α3 helix and β3 strand. The length of the ordered leader peptide bound to the RREs of LynD and NisB suggests that the core EXXXY residues of PqqA lie adjacent to the PqqD bound peptide. This suggests that the PqqE active site lies very close to PqqD in the ternary complex, and so PqqD may play the additional role of sequestering the PqqE radical intermediate from solvent. Structural work to precisely define these interactions is currently underway.
Supplementary Material
Figure S1: Sequence alignment of PqqDs. Figure S2: Relationship between MePqqD and the crystal structure of XcPqqD. Figure S3: TALOS+ and CSI2.0 plots for MePqqD. Figure S4: Sequence alignments to identify linker in the natural MePqqCD fusion. (PDF URL)
Acknowledgments
Data were collected at the Minnesota NMR Center, University of Minnesota. Funding for NMR instrumentation was provided by the Office of the Vice President for Research, the Medical School, the College of Biological Science, NIH, NSF, and the Minnesota Medical Foundation. Computer resources were provided by the Computational Genetics Laboratory of the University of Minnesota Supercomputing Institute.
Funding: This work was supported by NIH grants GM-66569 (C.M.W.) and GM-118117 (J.P.K.).
ABBREVIATIONS USED
- PQQ
pyrroloquinoline quinone
- PQQH2
the reduced form of PQQ
- RiPP
ribosomally produced post-translational modified peptide (refers to the product, not the pathway)
- RRE
RiPP recognition element (the protein or protein domain that binds the precursor peptide)
- PRPS
post-ribosomal peptide synthesis (refers to the pathway, not the product)
- rSAM
radical S-adenosyl-L-methionine enzyme
- SPASM
an acronym for an auxiliary extension associated with some rSAM enzymes, comes from subtilosin A, pyrroloquinoline quinone, anaerobic sulfatase, and mycofactocin
- NRP
non-ribosomally produced
Footnotes
Supporting information
Supporting Information is available free of charge at <website URL> at DOI: <DOI>.
The authors declare that they have no competing financial interests.
References
- 1.Rucker R, Chowanadisai W, Nakano M. Potential physiological importance of pyrroloquinoline quinone. Altern Med Rev. 2009;14:268–277. [PubMed] [Google Scholar]
- 2.Ohwada K, Takeda H, Yamazaki M, Isogai H, Nakano M, Shimomura M, Fukui K, Urano S. Pyrroloquinoline quinone (PQQ) prevents cognitive deficit caused by oxidative stress in rats. J Clin Biochem Nutr. 2008;42:29–34. doi: 10.3164/jcbn.2008005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi O, Kim J, Kim JG, Jeong Y, Moon JS, Park CS, Hwang I. Pyrroloquinoline quinone is a plant growth promotion factor produced by Pseudomonas fluorescens B16. Plant Physiol. 2008;146:657–668. doi: 10.1104/pp.107.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McIntosh JA, Donia MS, Schmidt EW. Ribosomal peptide natural products: bridging the ribosomal and nonribosomal worlds. Nat Prod Rep. 2009;26:537–559. doi: 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Goransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Muller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Sussmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marahiel MA. Working outside the protein-synthesis rules: insights into non-ribosomal peptide synthesis. J Pept Sci. 2009;15:799–807. doi: 10.1002/psc.1183. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez-Barrena MJ, Martinez-Ripoll M, Galvez A, Valdivia E, Maqueda M, Cruz V, Albert A. Structure of bacteriocin AS-48: from soluble state to membrane bound state. J Mol Biol. 2003;334:541–549. doi: 10.1016/j.jmb.2003.09.060. [DOI] [PubMed] [Google Scholar]
- 8.Westerling J, Frank J, Duine JA. The prosthetic group of methanol dehydrogenase from Hyphomicrobium X: electron spin resonance evidence for a quinone structure. Biochem Biophys Res Commun. 1979;87:719–724. doi: 10.1016/0006-291x(79)92018-7. [DOI] [PubMed] [Google Scholar]
- 9.Duine JA, Frank J., Jr The prosthetic group of methanol dehydrogenase. Purification and some of its properties. Biochem J. 1980;187:221–226. doi: 10.1042/bj1870221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khaliullin B, Aggarwal P, Bubas M, Eaton GR, Eaton SS, Latham JA. Mycofactocin biosynthesis: modification of the peptide MftA by the radical S-adenosylmethionine protein MftC. FEBS Lett. 2016;590:2538–2548. doi: 10.1002/1873-3468.12249. [DOI] [PubMed] [Google Scholar]
- 11.Haft DH. Bioinformatic evidence for a widely distributed, ribosomally produced electron carrier precursor, its maturation proteins, and its nicotinoprotein redox partners. BMC Genomics. 2011;12:21. doi: 10.1186/1471-2164-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toyama H, Chistoserdova L, Lidstrom ME. Sequence analysis of pqq genes required for biosynthesis of pyrroloquinoline quinone in Methylobacterium extorquens AM1 and the purification of a biosynthetic intermediate. Microbiology. 1997;143:595–602. doi: 10.1099/00221287-143-2-595. [DOI] [PubMed] [Google Scholar]
- 13.Goodwin PM, Anthony C. The biochemistry, physiology and genetics of PQQ and PQQ-containing enzymes. Adv Microb Physiol. 1998;40:1–80. doi: 10.1016/s0065-2911(08)60129-0. [DOI] [PubMed] [Google Scholar]
- 14.Duine JA. The PQQ story. J Biosci Bioeng. 1999;88:231–236. doi: 10.1016/s1389-1723(00)80002-x. [DOI] [PubMed] [Google Scholar]
- 15.Anthony C. The quinoprotein dehydrogenases for methanol and glucose. Arch Biochem Biophys. 2004;428:2–9. doi: 10.1016/j.abb.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 16.Meulenberg JJ, Sellink E, Loenen WA, Riegman NH, van Kleef M, Postma PW. Cloning of Klebsiella pneumoniae pqq genes and PQQ biosynthesis in Escherichia coli. FEMS Microbiol Lett. 1990;59:337–343. doi: 10.1016/0378-1097(90)90244-k. [DOI] [PubMed] [Google Scholar]
- 17.Meulenberg JJ, Sellink E, Riegman NH, Postma PW. Nucleotide sequence and structure of the Klebsiella pneumoniae pqq operon. Mol Gen Genet. 1992;232:284–294. doi: 10.1007/BF00280008. [DOI] [PubMed] [Google Scholar]
- 18.Velterop JS, Sellink E, Meulenberg JJM, David S, Bulder I, Postma PW. Synthesis of pyrroloquinoline quinone in-vivo and in-vitro and detection of an intermediate in the biosynthetic pathway. J Bacteriol. 1995;177:5088–5098. doi: 10.1128/jb.177.17.5088-5098.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ameyama M, Matsushita K, Shinagawa E, Hayashi M, Adachi O. Pyrroloquinoline quinone: excretion by methylotrophs and growth stimulation for microorganisms. BioFactors. 1988;1:51–53. [PubMed] [Google Scholar]
- 20.Matsumura H, Umezawa K, Takeda K, Sugimoto N, Ishida T, Samejima M, Ohno H, Yoshida M, Igarashi K, Nakamura N. Discovery of a eukaryotic pyrroloquinoline quinone-dependent oxidoreductase belonging to a new auxiliary activity family in the database of carbohydrate-active enzymes. PloS one. 2014;9:e104851. doi: 10.1371/journal.pone.0104851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen YQ, Bonnot F, Imsand EM, RoseFigura JM, Sjolander K, Klinman JP. Distribution and properties of the genes encoding the biosynthesis of the bacterial cofactor, pyrroloquinoline quinone. Biochemistry. 2012;51:2265–2275. doi: 10.1021/bi201763d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sode K, Ito K, Witarto AB, Watanabe K, Yoshida H, Postma P. Increased production of recombinant pyrroloquinoline quinone (PQQ) glucose dehydrogenase by metabolically engineered Escherichia coli strain capable of PQQ biosynthesis. J Biotechnol. 1996;49:239–243. doi: 10.1016/0168-1656(96)01540-4. [DOI] [PubMed] [Google Scholar]
- 23.Kasahara T, Kato T. Nutritional biochemistry: A new redox-cofactor vitamin for mammals. Nature. 2003;422:832. doi: 10.1038/422832a. [DOI] [PubMed] [Google Scholar]
- 24.Killgore J, Smidt C, Duich L, Romero-Chapman N, Tinker D, Reiser K, Melko M, Hyde D, Rucker RB. Nutritional importance of pyrroloquinoline quinone. Science. 1989;245:850–852. doi: 10.1126/science.2549636. [DOI] [PubMed] [Google Scholar]
- 25.Steinberg F, Stites TE, Anderson P, Storms D, Chan I, Eghbali S, Rucker R. Pyrroloquinoline quinone improves growth and reproductive performance in mice fed chemically defined diets. Exp Biol Med. 2003;228:160–166. doi: 10.1177/153537020322800205. [DOI] [PubMed] [Google Scholar]
- 26.Steinberg FM, Gershwin ME, Rucker RB. Dietary pyrroloquinoline quinone: growth and immune response in BALB/c mice. J Nutr. 1994;124:744–753. doi: 10.1093/jn/124.5.744. [DOI] [PubMed] [Google Scholar]
- 27.Bauerly K, Harris C, Chowanadisai W, Graham J, Havel PJ, Tchaparian E, Satre M, Karliner JS, Rucker RB. Altering pyrroloquinoline quinone nutritional status modulates mitochondrial, lipid, and energy metabolism in rats. PloS one. 2011;6:e21779. doi: 10.1371/journal.pone.0021779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bauerly KA, Storms DH, Harris CB, Hajizadeh S, Sun MY, Cheung CP, Satre MA, Fascetti AJ, Tchaparian E, Rucker RB. Pyrroloquinoline quinone nutritional status alters lysine metabolism and modulates mitochondrial DNA content in the mouse and rat. Biochim Biophys Acta. 2006;1760:1741–1748. doi: 10.1016/j.bbagen.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Chowanadisai W, Bauerly KA, Tchaparian E, Wong A, Cortopassi GA, Rucker RB. Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1alpha expression. J Biol Chem. 2010;285:142–152. doi: 10.1074/jbc.M109.030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris CB, Chowanadisai W, Mishchuk DO, Satre MA, Slupsky CM, Rucker RB. Dietary pyrroloquinoline quinone (PQQ) alters indicators of inflammation and mitochondrial-related metabolism in human subjects. J Nutr Biochem. 2013;24:2076–2084. doi: 10.1016/j.jnutbio.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 31.Stites T, Storms D, Bauerly K, Mah J, Harris C, Fascetti A, Rogers Q, Tchaparian E, Satre M, Rucker RB. Pyrroloquinoline quinone modulates mitochondrial quantity and function in mice. J Nutr. 2006;136:390–396. doi: 10.1093/jn/136.2.390. [DOI] [PubMed] [Google Scholar]
- 32.Singh AK, Pandey SK, Saha G, Gattupalli NK. Pyrroloquinoline quinone (PQQ) producing Escherichia coli Nissle 1917 (EcN) alleviates age associated oxidative stress and hyperlipidemia, and improves mitochondrial function in ageing rats. Exp Gerontol. 2015;66:1–9. doi: 10.1016/j.exger.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Meruvu S, Bedi YS, Chau J, Arguelles A, Rucker R, Choudhury M. Pyrroloquinoline quinone increases the expression and activity of Sirt1 and -3 genes in HepG2 cells. Nutr Res. 2015;35:844–849. doi: 10.1016/j.nutres.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 34.Fluckiger R, Paz M, Mah J, Bishop A, Gallop PM. Characterization of the glycine-dependent redox-cycling activity in animal fluids and tissues using specific inhibitors and activators: evidence for presence of PQQ. Biochem Biophys Res Commun. 1993;196:61–68. doi: 10.1006/bbrc.1993.2216. [DOI] [PubMed] [Google Scholar]
- 35.Fluckiger R, Paz MA, Gallop PM. Redox-cycling detection of dialyzable pyrroloquinoline quinone and quinoproteins. Methods Enzymol. 1995;258:140–149. doi: 10.1016/0076-6879(95)58043-3. [DOI] [PubMed] [Google Scholar]
- 36.Stites TE, Mitchell AE, Rucker RB. Physiological importance of quinoenzymes and the o-quinone family of cofactors. J Nutr. 2000;130:719–727. doi: 10.1093/jn/130.4.719. [DOI] [PubMed] [Google Scholar]
- 37.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat Chem Biol. 2015;11:564–570. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klinman JP, Bonnot F. Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem Rev. 2014;114:4343–4365. doi: 10.1021/cr400475g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oman TJ, van der Donk WA. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat Chem Biol. 2010;6:9–18. doi: 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Houck DR, Hanners JL, Unkefer CJ. Biosynthesis of pyrroloquinoline quinone. 1. identification of biosynthetic precursors using C-13 labeling and nmr-spectroscopy. J Am Chem Soc. 1988;110:6920–6921. [Google Scholar]
- 41.Houck DR, Hanners JL, Unkefer CJ. Biosynthesis of pyrroloquinoline quinone. 2. biosynthetic assembly from glutamate and tyrosine. J Am Chem Soc. 1991;113:3162–3166. [Google Scholar]
- 42.Latham JA, Iavarone AT, Barr I, Juthani PV, Klinman JP. PqqD is a novel peptide chaperone that forms a ternary complex with the radical S-adenosylmethionine protein PqqE in the pyrroloquinoline quinone biosynthetic pathway. J Biol Chem. 2015;290:12908–12918. doi: 10.1074/jbc.M115.646521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li WZ, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N, Lopez R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015;43:W580–584. doi: 10.1093/nar/gkv279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 47.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 48.Goddard TD, K DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 49.Evans RL, 3rd, Latham JA, Klinman JP, Wilmot CM, Xia Y. (1)H, (13)C, and (15)N resonance assignments and secondary structure information for Methylobacterium extorquens PqqD and the complex of PqqD with PqqA. Biomol NMR Assign. 2016;10:385–389. doi: 10.1007/s12104-016-9705-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Kent Wenger R, Yao H, Markley JL. BioMagResBank. Nucleic Acids Res. 2008;36:D402–408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schanda P, Van Melckebeke H, Brutscher B. Speeding up three-dimensional protein NMR experiments to a few minutes. J Am Chem Soc. 2006;128:9042–9043. doi: 10.1021/ja062025p. [DOI] [PubMed] [Google Scholar]
- 52.Gustavsson M, Traaseth NJ, Karim CB, Lockamy EL, Thomas DD, Veglia G. Lipid-mediated folding/unfolding of phospholamban as a regulatory mechanism for the sarcoplasmic reticulum Ca2+-ATPase. J Mol Biol. 2011;408:755–765. doi: 10.1016/j.jmb.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gal M, Kern T, Schanda P, Frydman L, Brutscher B. An improved ultrafast 2D NMR experiment: towards atom-resolved real-time studies of protein kinetics at multi-Hz rates. J Biomol NMR. 2009;43:1–10. doi: 10.1007/s10858-008-9284-9. [DOI] [PubMed] [Google Scholar]
- 54.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS plus : a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 56.Tsai TY, Yang CY, Shih HL, Wang AH, Chou SH. Xanthomonas campestris PqqD in the pyrroloquinoline quinone biosynthesis operon adopts a novel saddle-like fold that possibly serves as a PQQ carrier. Proteins. 2009;76:1042–1048. doi: 10.1002/prot.22461. [DOI] [PubMed] [Google Scholar]
- 57.Chen ZW, Datta S, Dubois JL, Klinman JP, Mathews FS. Mutation at a strictly conserved, active site tyrosine in the copper amine oxidase leads to uncontrolled oxygenase activity. Biochemistry. 2010;49:7393–7402. doi: 10.1021/bi100643y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor-NIH for NMR molecular structure determination. Prog Nucl Mag Res Sp. 2006;48:47–62. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 59.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 60.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 62.Wecksler SR, Stoll S, Iavarone AT, Imsand EM, Tran H, Britt RD, Klinman JP. Interaction of PqqE and PqqD in the pyrroloquinoline quinone (PQQ) biosynthetic pathway links PqqD to the radical SAM superfamily. Chem Commun. 2010;46:7031–7033. doi: 10.1039/c0cc00968g. [DOI] [PubMed] [Google Scholar]
- 63.Wishart DS, Sykes BD. The C-13 chemical-shift index - a simple method for the identification of protein secondary structure using C-13 chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 64.Wishart DS, Sykes BD. Chemical-shifts as a tool for structure determination. Nucl Magn Reson Pt C. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- 65.Barr I, Latham JA, Iavarone AT, Chantarojsiri T, Hwang JD, Klinman JP. Demonstration that the radical S-adenosylmethionine (SAM) enzyme PqqE catalyzes de novo carbon-carbon cross-linking within a peptide substrate PqqA in the presence of the peptide chaperone PqqD. J Biol Chem. 2016;291:8877–8884. doi: 10.1074/jbc.C115.699918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ortega MA, Hao Y, Zhang Q, Walker MC, van der Donk WA, Nair SK. Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB. Nature. 2015;517:509–512. doi: 10.1038/nature13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, Ravel J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc Natl Acad Sci USA. 2005;102:7315–7320. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McIntosh JA, Lin ZJ, Tianero MDB, Schmidt EW. Aestuaramides, a natural library of cyanobactin cyclic peptides resulting from isoprene-derived claisen rearrangements. ACS Chem Biol. 2013;8:877–883. doi: 10.1021/cb300614c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Regni CA, Roush RF, Miller DJ, Nourse A, Walsh CT, Schulman BA. How the MccB bacterial ancestor of ubiquitin E1 initiates biosynthesis of the microcin C7 antibiotic. EMBO J. 2009;28:1953–1964. doi: 10.1038/emboj.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koehnke J, Mann G, Bent AF, Ludewig H, Shirran S, Botting C, Lebl T, Houssen WE, Jaspars M, Naismith JH. Structural analysis of leader peptide binding enables leader-free cyanobactin processing. Nat Chem Biol. 2015;11:558–563. doi: 10.1038/nchembio.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kluskens LD, Kuipers A, Rink R, de Boef E, Fekken S, Driessen AJM, Kuipers OP, Moll GN. Post-translational modification of therapeutic peptides by NisB, the dehydratase of the lantibiotic nisin. Biochemistry. 2005;44:12827–12834. doi: 10.1021/bi050805p. [DOI] [PubMed] [Google Scholar]
- 72.Shisler KA, Broderick JB. Emerging themes in radical SAM chemistry. Curr Opin Struct Biol. 2012;22:701–710. doi: 10.1016/j.sbi.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saichana N, Tanizawa K, Pechousek J, Novak P, Yakushi T, Toyama H, Frebortova J. PqqE from Methylobacterium extorquens AM1: a radical S-adenosyl-l-methionine enzyme with an unusual tolerance to oxygen. J Biochem. 2016;159:87–99. doi: 10.1093/jb/mvv073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bruender NA, Bandarian V. The radical S-adenosyl-L-methionine enzyme MftC catalyzes an oxidative decarboxylation of the C-terminus of the MftA peptide. Biochemistry. 2016;55:2813–2816. doi: 10.1021/acs.biochem.6b00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khaliullin B, Aggarwal P, Bubas M, Eaton GR, Eaton SS, Latham JA. Mycofactocin biosynthesis: modification of the peptide MftA by the radical S-adenosylmethionine protein MftC. FEBS lett. 2016;590:2538–2548. doi: 10.1002/1873-3468.12249. [DOI] [PubMed] [Google Scholar]
- 76.Schramma KR, Bushin LB, Seyedsayamdost MR. Structure and biosynthesis of a macrocyclic peptide containing an unprecedented lysine-to-tryptophan crosslink. Nat Chem. 2015;7:431–437. doi: 10.1038/nchem.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Benjdia A, Guillot A, Lefranc B, Vaudry H, Leprince J, Berteau O. Thioether bond formation by SPASM domain radical SAM enzymes: C-alpha H-atom abstraction in subtilosin A biosynthesis. Chem Commun. 2016;52:6249–6252. doi: 10.1039/c6cc01317a. [DOI] [PubMed] [Google Scholar]
- 78.Flühe E, Knappe TA, Gattner MJ, Schafer A, Burghaus O, Linne U, Marahiel MA. The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat Chem Biol. 2012;8:350–357. doi: 10.1038/nchembio.798. [DOI] [PubMed] [Google Scholar]
- 79.Bruender NA, Bandarian V. SkfB abstracts a hydrogen atom from C-alpha on SkfA to initiate thioether cross-link formation. Biochemistry. 2016;55:4131–4134. doi: 10.1021/acs.biochem.6b00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Flühe L, Burghaus O, Wieckowski BM, Giessen TW, Linne U, Marahiel MA. Two [4Fe-4S] clusters containing radical SAM enzyme SkfB catalyze thioether bond formation during the maturation of the sporulation killing factor. J Am Chem Soc. 2013;135:959–962. doi: 10.1021/ja310542g. [DOI] [PubMed] [Google Scholar]
- 81.Wieckowski BM, Hegemann JD, Mielcarek A, Boss L, Burghaus O, Marahiel MA. The PqqD homologous domain of the radical SAM enzyme ThnB is required for thioether bond formation during thurincin H maturation. FEBS Lett. 2015;589:1802–1806. doi: 10.1016/j.febslet.2015.05.032. [DOI] [PubMed] [Google Scholar]
- 82.Bruender NA, Wilcoxen J, Britt RD, Bandarian V. Biochemical and spectroscopic characterization of a radical S-adenosyl-L-methionine enzyme involved in the formation of a peptide thioether cross-link. Biochemistry. 2016;55:2122–2134. doi: 10.1021/acs.biochem.6b00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goldman PJ, Grove TL, Sites LA, McLaughlin MI, Booker SJ, Drennan CL. X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine posttranslational modification. Proc Natl Acad Sci USA. 2013;110:8519–8524. doi: 10.1073/pnas.1302417110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Sequence alignment of PqqDs. Figure S2: Relationship between MePqqD and the crystal structure of XcPqqD. Figure S3: TALOS+ and CSI2.0 plots for MePqqD. Figure S4: Sequence alignments to identify linker in the natural MePqqCD fusion. (PDF URL)